

Abstract
Although previous studies demonstrated beneficial effects of estrogen on cardiovascular function, the Women's Health Initiative has reported an increased incidence of coronary heart disease and stroke in postmenopausal women taking hormone replacement therapy (HRT). The objective of the present study was to identify a molecular mechanism whereby estrogen, a vasodilatory hormone, could possibly increase the risk of cardiovascular disease. Isometric contractile force recordings were performed on endothelium-denuded porcine coronary arteries, while molecular and fluorescence studies identified estrogen signaling molecules in coronary smooth muscle. Estrogen (1-1000nM) relaxed arteries in an endothelium-independent fashion; however, when arteries were pretreated with agents to uncouple NO production from nitric oxide synthase (NOS), estrogen contracted coronary arteries with an EC50 of 7.3 ± 4nM. Estrogen-induced contraction was attenuated by reducing superoxide (O2−). Estrogen-stimulated O2− production was detected in NOS-uncoupled coronary myocytes. Interestingly, only the Type 1 NOS isoform (nNOS) was detected in myocytes, making this protein a likely target mediating both estrogen-induced relaxation and contraction of endothelium-denuded coronary arteries. Estrogen-induced contraction was completely inhibited by 1μM nifedipine or 10μM indomethacin, indicating involvement of dihydropyridine-sensitive calcium channels and contractile prostaglandins. We propose that a single molecular mechanism can mediate the dual and opposite effect of estrogen on coronary arteries: by stimulating Type 1 (n)NOS in coronary arteries, estrogen produces either vasodilation via NO or vasoconstriction via superoxide.
Keywords: estrogens, nitric oxide, coronary circulation, smooth muscle
1. Introduction
The Women’s Health Initiative (WHI) trials indicate that postmenopausal hormone replacement therapy (HRT) bears a significant health risk, including a 29% increase in coronary heart disease (CHD) and a 41% increase in stroke (30). However, these findings are in stark contrast to prior basic research studies demonstrating clear beneficial effects of estrogen on the cardiovascular and other systems, which had lead to HRT becoming the second-most prescribed medication in the United States. Despite the fact that estrogen produces favorable effects on plasma lipid profiles, increases fibrinolysis, and acts as a vasodilator to lower systemic blood pressure and increase organ blood flow (21, 33, 37), HRT has been increasingly discontinued in light of recent clinical trials. The obvious paradox is: How could a hormone that produces such an array of salutary effects promote significant pathology and disease? Despite continued investigation, the cellular/molecular basis for these contradictory (and confusing) responses to estrogen continues to elude explanation.
Many effects of estrogen are mediated by nitric oxide (NO) via stimulation of nitric oxide synthase (NOS). It is generally believed that the primary target of estrogen in the cardiovascular system is Type 3 NOS expressed in endothelial cells (eNOS), yet there are also endothelium-independent effects of estrogen on blood vessels or single vascular smooth muscle (VSM) cells mediated by NO (8, 11, 24, 38, 39). Interestingly, VSM cells express aromatase (i.e., estrogen synthase), indicating that estrogen may act acutely in a paracrine or even autocrine manner within the vascular wall (16). Thus, direct action of estrogen on VSM might actually be more important in older women because the vascular endothelium becomes increasingly dysfunctional with age (10, 12, 41). Nonetheless, estrogen effects on NO production in VSM cells are often ignored. Our present findings now provide evidence for a mechanism of estrogen action that could, for the first time, shed significant light upon the controversy that is estrogen and HRT. We propose that both beneficial and harmful effects of estrogen can be mediated by the same biological target, Type 1 (neuronal) NOS (nNOS), expressed in VSM cells. Depending on the microenvironment of this enzyme, estrogen exhibits a “yin-yang” influence on cellular function producing either vasodilation via NO or vasoconstriction via superoxide (O2−, a powerful oxidant which can further decrease NO bioavailability). Thus, by acting on a single molecular target, estrogen can produce completely opposite effects in vascular and other target tissues, and could thereby induce either physiological or pathophysiological responses.
Methods
Supply of coronary arteries
We obtained fresh porcine hearts from local abattoirs. We generally obtain hearts from castrated male pigs because females are typically conserved for breeding, but have observed no sexual dimorphism in responses of arteries to estrogen (11, 38). The left anterior descending (LAD) coronary artery was excised immediately and placed into ice-cold Krebs-Henseleit buffer solution of the following composition (mM): 122 NaCl, 4.7 KCl, 15.5 NaHCO3, 1.2 KH2PO4, 1.2 MgCl2, 1.8 CaCl2, 11.5 glucose, pH 7.2. Arteries were kept on ice during transport to the laboratory.
Tension studies of intact coronary arteries
Arteries were placed under a dissecting microscope, and excess fat and connective tissue wre removed in ice-cold buffer solution. Two to four 5 mm rings were obtained from each LAD, and prepared for isometric contractile force recordings as described previously (38). In order to control for possible indirect effects of endothelium-derived vasoactive factors, the endothelium was removed physically by rubbing the intimal surface and tested by observing the absence of acetylcholine-induced relaxation. Rings were mounted on two triangular tissue supports, with one support fixed to stationary glass rod, and the other attached to a force-displacement transducer. Isometric contractions or relaxations were recorded on a PC computer using MacLab software. The tissue bathing solution was the modified Krebs-Henseleit buffer described above. The solution was oxygenated continuously (95% O2 - 5% CO2) and maintained at 37°C. Coronary ring preparations were equilibrated for 90 min under an optimal resting tension of 2.0 grams, and fresh bath solution was added to the tissue chamber every 30 min to prevent accumulation of metabolic end products. After the initial equilibration, preparations were exposed to maximally effective concentrations of a contractile agonist, e.g., PGF2α, to insure stabilization of the muscles. Pharmacological inhibitors were allowed to equilibrate with the arteries for at least 30 min prior to measurement of a complete estrogen concentration-response relationship (1-1000 nM). The effects of 17α-estradiol, an “inactive” estrogen, were determined to control for possible vehicle artifact. Steroid vehicle was usually 50-75% ethanol, and this stock was diluted to a concentration of no more than 0.1% in the vessel chamber.
Cell culture
Human coronary artery smooth muscle cells were purchased from Clonetics/Cambrex Corp., and were grown in phenol red-free smooth muscle growth medium with 5% FBS as described previously (39). Steroid hormones and growth factors were removed from FBS by charcoal-stripping. Only short-term cultures (passage 3-5) were employed.
Western blotting
Arteries were obtained and prepared as described above. The endothelium was removed from some of the arteries by gentle rubbing of the luminal surface with a cotton swab. Tissues were stored in liquid nitrogen until immunoblots were to be performed. Frozen tissues were pooled and pulverized (Fisher Scientific). Protein concentrations were determined by BIO-RAD DC protein assay. ADP-sepharose beads were employed to affinity extract NOS proteins from lysates (3). Purified positive control proteins (nNOS and iNOS; pituitary extracts) were purchased from Transduction Laboratories. Proteins were separated on SDS-polyacrylamide gels using a Mini Protean II (Bio-Rad) gel kit according to the manufacturer’s instructions. Proteins were then transferred to Hybond ECL membrane (Amersham Pharmacia Biotech) using a Mini-Trans-Blot Electrophoretic Transfer Cell (Bio-Rad) at 100V for 1 hr. Blots were blocked with 5% non-fat milk overnight at 4°C. The membrane was then rinsed with Tris-buffered saline TWEEN (TBST) three times for 15min and two times for 5 min. Blots were then probed with primary antibodies (nNOS, eNOS, or iNOS; 1:1000; BD Transduction Labs) in TBST containing 1% non-fat milk protein for 1 hr. After washing, the membrane was then incubated with anti-rabbit IgG conjugated to HRP (horseradish peroxidase) and visualized with an enhanced chemiluminescence system (Amersham).
Fluorescence studies
The cell-permeable form of the O2− fluorescence indicator dihydroethidium (DHE) was employed to examine production of O2− within coronary myocytes. Cells were loaded with the fluorescent indicator during a 45-minute incubation with 2μM DHE (diluted from a 10mM stock in DMSO) in Krebs solution (described above) at 37°C. After incubation, cells were washed with Krebs solution, and placed on the stage of a Zeiss confocal laser microscope. A perfusion chamber was created by the addition of silicon grease to form in- and outflow chambers at each end of coverslips inverted onto spacers glued to a slide. Drugs were then added to the cells and fluorescence was measured. Cell imaging was conducted on a Zeiss 510 NLO laserscanning microscope operating in the confocal mode with a ×40 0.85 numerical aperture objective as described previously (20). Quantitative analysis of confocal images was under the control of Zeiss PHYSIOLOGY software.
Statistical Analysis
All data were expressed as the mean ± S.E. Statistical significance between two groups was evaluated by Student’s t test for paired data. Comparison between multiple groups was made by the One-Way Analysis of Variance test, with a post hoc Tukey test performed for differences among data groups. A probability of less than 0.05 was considered to indicate a significant difference.
RESULTS
As we and others had demonstrated previously (8, 24, 38), under normal conditions 17β-estradiol produces an endothelium-independent, concentration-dependent relaxation of precontracted arteries (Figure. 1A). For example, 300nM 17β-estradiol induced an average relaxation of 41.6 ± 2.5% in endothelium-denuded coronary arteries precontracted with 10μM prostaglandin (PG)F2α (n=4). But in contrast to this well known effect, when arteries were pretreated (20-30 min) with 100μM of an inhibitory L-arginine analog, e.g., NG-monomethyl-L-arginine (L-NMMA), Nω-nitro-L-arginine methyl ester (L-NAME), or Nω-propyl-L-arginine (N-PLA), to “uncouple” NO production from NOS and reduce NO bioavailability, estrogen now induced a concentration-dependent contraction of endothelium-denuded coronary arteries (Figure 1B). Treating “NOS-uncoupled” arteries with the same concentrations of 17α-estradiol (a biologically less active steroid) had no effect on tension (n=3; Figure 1B), thus controlling for potential effects of vehicle (0.05% ethanol) or possible nonspecific steroid effects. After estrogen-induced coronary contraction had reached maximum levels, we found that reducing O2− reversed this response. Estrogen-induced contraction was reversed by either 1mM tempol (superoxide dismutase mimetic, 70.3 ± 9%; n=4; Figure 1B) or 10mM tiron (O2− scavenger, 70.0 ± 9.5%; n=8; data not shown), suggesting that O2− mediated the contractile response of estrogen.
Figure 1.
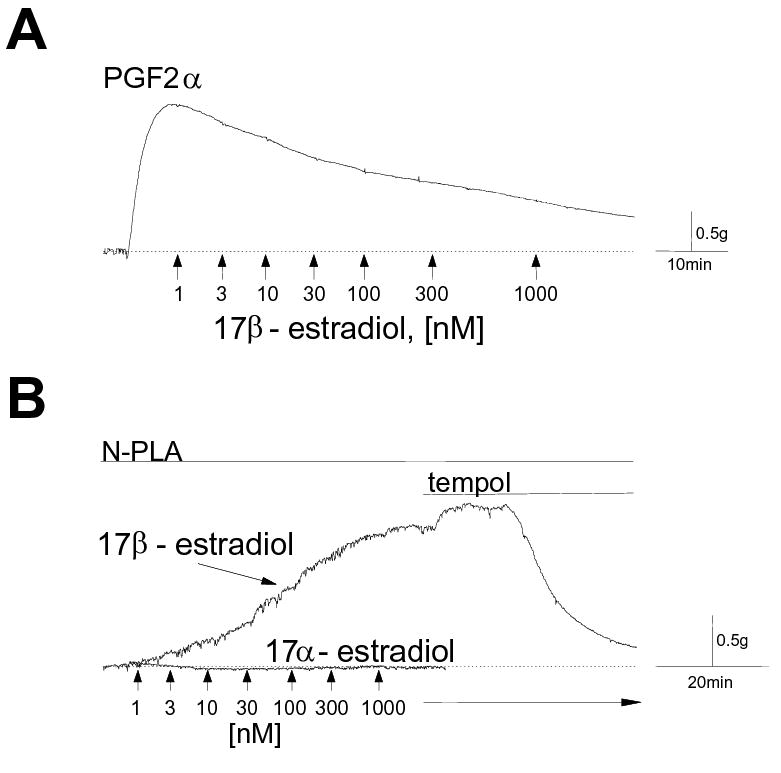
Estrogen relaxes or contracts coronary arteries. Typical tracings from isometric contractile force recordings of endothelium-denuded porcine coronary arteries in response to cumulative addition of estrogen (1-1000nM). Periods of drug exposure are indicated by lines above the traces. Baseline tension is indicated by the dotted line. (A) Estrogen relaxes precontracted coronary arteries. Arteries were exposed to 10μM prostaglandin F2α (PGF2α; 10min), and the relaxation responses to increasing concentrations of 17β-estradiol were measured. (B) Estrogen contracts NOS-uncoupled coronary arteries. Arteries were pretreated with 10-100μM Nω-propyl-L-arginine (N-PLA, 30-45 min) to reduce NO bioavailability. Cumulative addition of 17β-estradiol produced a concentration-dependent contraction, whereas 17α-estradiol had no effect over the same range of concentrations. Tempol (1mM), a superoxide dismutase mimetic, reversed the contractile effect of estrogen.
The dependency of estrogen-induced vascular contraction on O2− was further demonstrated by observing no response to estrogen when O2− accumulation was prevented in coronary arteries. As predicted, estrogen had no effect on “NOS-uncoupled” arteries that had been pretreated with either 1mM tempol (0% increase in tension, n=3, data not shown) or 10mM tiron (0% increase in tension, n=4; Figure 2A). In these same arteries, however, further addition of 10μM PGF2α induced a strong O2−-independent contraction (Figure 2A). In sum, these experiments indicate that estrogen can either relax or contract coronary arteries depending on whether NOS activity is coupled or uncoupled to NO production, with either NO or O2−, respectively, exerting the predominate influence. A summary concentration-response relationship for estrogen-induced coronary artery contraction is illustrated in Figure 2B, as is the total lack of a contractile effect when O2− accumulation is prevented (i.e., in the presence of 10mM tiron). The calculated EC50 for the contractile effect of estrogen is 7.3 ± 4 nM (n=10).
Figure 2.
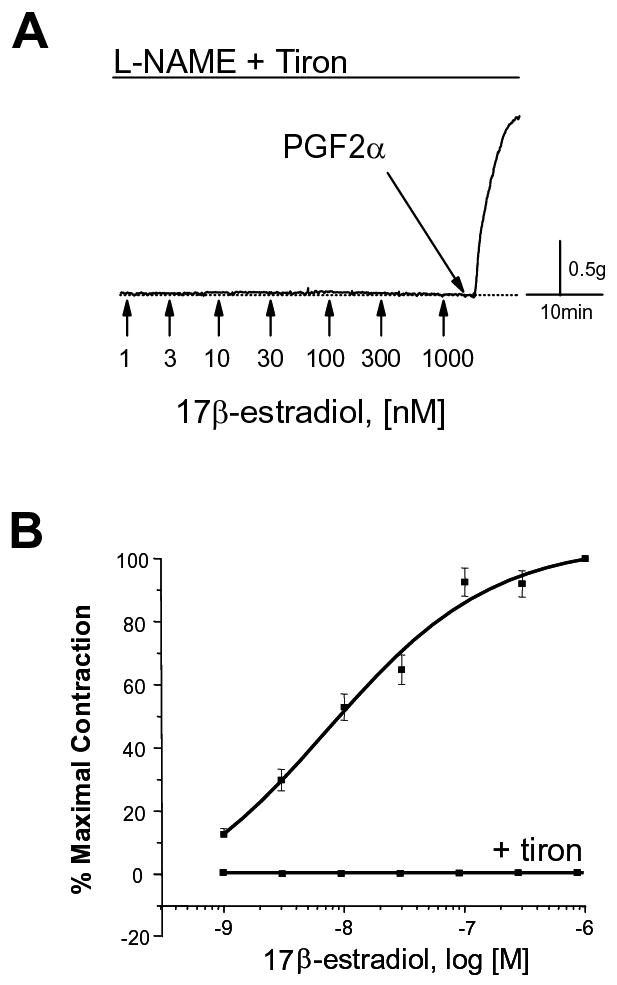
Estrogen-induced coronary artery contraction involves production of superoxide (O2−). (A) Scavenging O2− prevents the contractile effect of estrogen. Porcine coronary arteries were treated with 10μM N-nitro-L-arginine methyl ester (L-NAME, 30-45 min). Arteries were also pretreated (30 min) with tiron (10mM, O2− scavenger), as indicated. In the presence of L-NAME and tiron, estrogen failed to elicit a contractile response; however, subsequent addition of 10μM PGF2α induced an immediate cont raction of the artery. (B) Average concentration-response relationship for estrogen-induced coronary artery contraction. Each point represents the mean of 10 experiments ± S.E. in the absence (upper curve) or presence (lower curve) of 10mM tiron.
These pharmacological studies on coronary arteries indicate that O2− mediates the ability of estrogen to contract VSM when NO bioavailability is reduced. Additional evidence for this novel mechanism was obtained by observing estrogen-stimulated O2− fluorescence in isolated human coronary artery smooth muscle cells (HCASMC). Single HCASMC were loaded with the cell-permeable fluorescent probe dihydroethidium (DHE), which is commonly used to detect generation of O2− in VSM (9, 22). The ability of estrogen to stimulate O2− production in coronary myocytes is illustrated in Figure 3. Baseline fluorescence, before and 10 min after addition of 100μM L-NMMA, is illustrated in panel A, and indicates no spontaneous increases in O2− levels in the absence of estrogen over this brief time period (fluorescence intensity: con, 629.5 ± 28; L-NMMA, 691.7 ± 23; n=6; p > 0.12; Figure 3A). In the absence of L-NMMA, 100nM estrogen did not affect fluorescence intensity (10-30 min; data not shown). In contrast, in the presence of 100μM L-NMMA estrogen increased O2− -mediated fluorescence significantly after only 10 minutes (fluorescence intensity: L-NMMA, 688.5 ± 61; L-NMMA ± E2, 887.6 ± 68; n=6; p < 0.001; Figure 3B), with estrogen-stimulated O2− production reaching a plateau after 20-30 minutes (20 min: 985.6 ± 74; 30 min: 1048 ± 83; n=6). There was no significant increase in O2− fluorescence intensity after 20 minutes (p >0.05; n=6). These fluorescence studies on single coronary myocytes are entirely consistent with our functional studies on intact coronary arteries, and strongly support our hypothesis that estrogen stimulates O2− production in coronary myocytes to contract coronary arteries.
Figure 3.
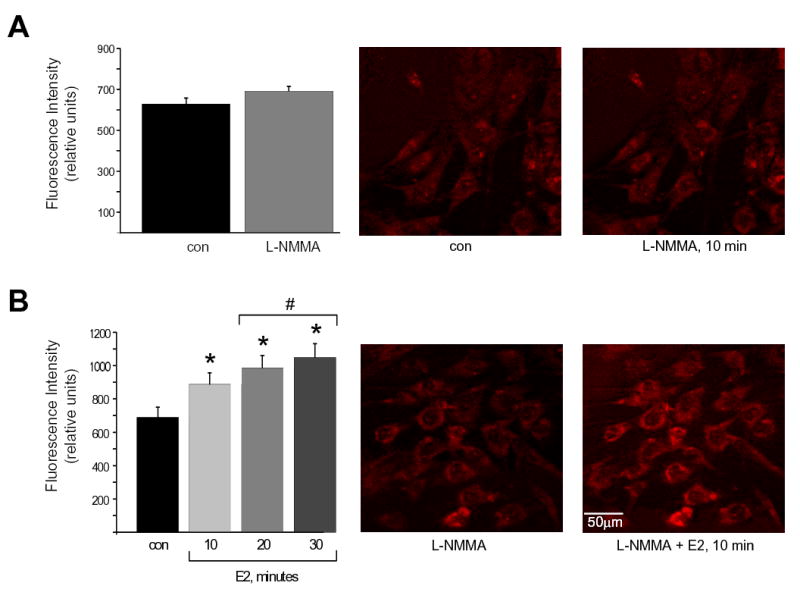
Estrogen stimulates generation of superoxide in NOS-uncoupled, coronary artery myocytes. (A) Cellular fluorescence images of human coronary artery myocytes loaded with dihydroethidium (DHE, 2μM; 45 min) to indicate O2− generation. In these control experiments fluorescence measurements were taken before and 10 min after treatment with 10μM L-NMMA (middle and right panels, respectively). Average fluorescence intensity from 6 experiments ± S.E. is illustrated in the bar graph (left panel). There was no change in fluorescence intensity. (B) Estrogen-stimulated DHE fluorescence of human coronary artery myocytes. Myocytes were loaded with DHE as above, and treated with 10μM L-NMMA to uncouple NOS activity (middle panel). Treating these same cells with 100nM 17β-estradiol (E2; 10-30 min) enhanced O2− -induced fluorescence. The average effect of estrogen in 6 experiments ± S.E. is illustrated in the bar graph (left panel). *indicates a significant increase in fluorescence in estrogen-treated cells (p < 0.001). #indicates a further significant increase in fluorescence at 20-30 min exposure to estrogen compared to the 10-min exposure time (p<0.05). There was no significant change in fluorescence intensity between 20 and 30 min (n=6).
The next series of experiments demonstrated that the biochemical environment of NOS is a critical factor in determining the physiological response of coronary arteries to estrogen. Further in vitro contractile studies demonstrated that in the presence of only L-NMMA, 300nM estrogen contracted coronary arteries by an average of 214.2 ± 136% (n=5; Figure 4A). In contrast, when arteries were pretreated with both 100μM L-NMMA and 1mM L-arginine (to increase NO availability and attenuate NOS-dependent O2− production), 300nM estrogen now relaxed these non-precontracted coronary arteries by an average of 172.4 ± 48% (n=6; p<0.02 compared to the L-NMMA-only group). In these same vessels, subsequent addition of exogenous O2− (i.e., 10μM pyrogallol, a O2− donor) reversed the relaxation effect of estrogen and contracted arteries by an average of 141.5 ± 53% (n=6; Figure 4A). These findings support a critical role for NOS activity in mediating effects of estrogen on coronary arteries. Identification of the Type 1 (n)NOS isoform as a potential estrogen target molecule in coronary arteries was demonstrated by immunoblot studies which detected expression of only the nNOS isoform in coronary artery smooth muscle from either pigs or humans (Figure 4B; n=4). In contrast, both nNOS and eNOS were detected in homogenates (i.e., muscle + endothelium) of porcine coronary arteries (data not shown). Therefore, it is most likely the nNOS isoform that mediates the dual effect of estrogen on coronary smooth muscle.
Figure 4.
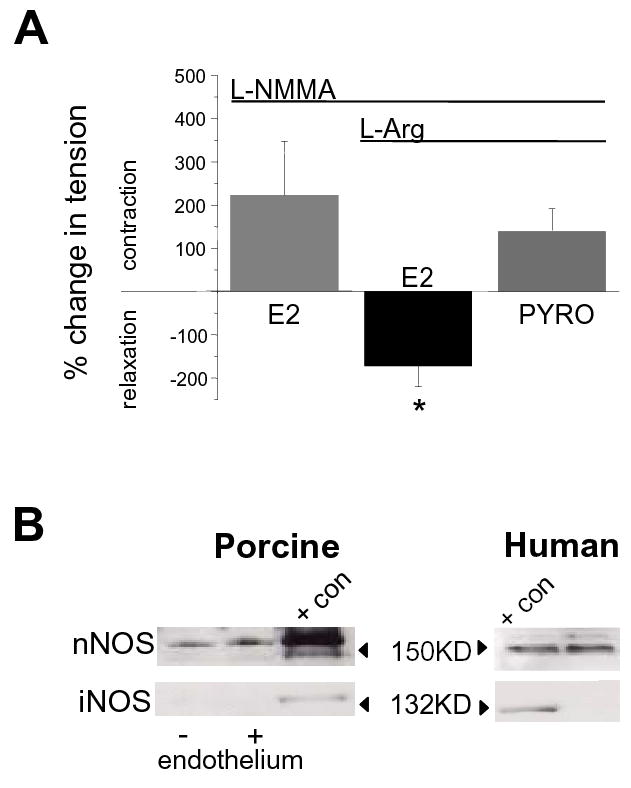
Type 1 (n)NOS is a likely target of estrogen action in coronary artery smooth muscle. (A) Average effect of estrogen (E2, 300nM; ± S.E.) on isometric tension of endothelium-denuded porcine coronary arteries. Estrogen contracted arteries pretreated (20-30 min) with 100μM L-NMMA (n=5). In contrast, estrogen relaxed arteries pretreated with both 100μM L-NMMA and 1mM L-arginine (n=6). In these same arteries addition of 10μM pyrogallol (PYRO), a O2− donor, reversed the relaxation effect of estrogen and induced a further contraction (n=6). Period of drug exposure is indicated by the lines above the graph. (B) Typical immunoblots of NOS isoform expression detected in porcine coronary artery homogenate and human coronary myocytes. Presence of endothelium is as indicated for porcine arteries. Lanes containing exogenous purified protein controls are indicated as “+ con”. In endothelium-denuded vessels or single myocytes nNOS was the only isoform detected.
A final series of experiments was conducted to shed some light on the molecular basis of estrogen-induced coronary artery contraction. The mechanism(s) converting the expected estrogen relaxation response into a O2−-mediated contractile effect is (are) unknown, but would most likely involve an increase in activator calcium in smooth muscle. To test this hypothesis we placed coronary arteries in a nominally calcium-free solution, uncoupled NOS activity, and then obtained a complete concentration-response relationship for estrogen-induced contraction. In the absence of extracellular calcium estrogen, at any concentration, had no effect on vessel tension (Figure 5A; n=4), indicating that the contractile response to estrogen did not involve significant release of calcium from intracellular stores. In contrast, when extracellular calcium was restored to normal (2mM) levels, vessels contracted immediately with an average development of 168.4 ± 37 mg tension/mg tissue (n=4). As expected, subsequent addition of 10mM tiron to scavenge O2− reduced calcium-induced contraction significantly (37.0 ± 9.0%; n=4; p = 0.002; data not shown). These findings demonstrate that estrogen-induced, O2−-mediated coronary artery contraction is dependent upon influx of extracellular calcium. This mechanism was further substantiated by observing that estrogen-induced contraction (in normal extracellular calcium) was completely attenuated by 1μM nifedipine, an inhibitor of L-type calcium channels (n=3; Figure 5B), suggesting that estrogen/O2− opens calcium channels in the plasma membrane of NOS-uncoupled coronary artery myocytes. In addition, the contractile effect of estrogen was prevented by indomethacin, an inhibitor of prostaglandin synthesis. As summarized in Figure 5B, pretreating coronary arteries with 10μM indomethacin completely blocked the contractile response to estrogen (n=3). These findings suggest that estrogen-generated O2− does not act directly on membrane ion channels, but instead stimulates production of a vasoconstrictor prostaglandin.
Figure 5.
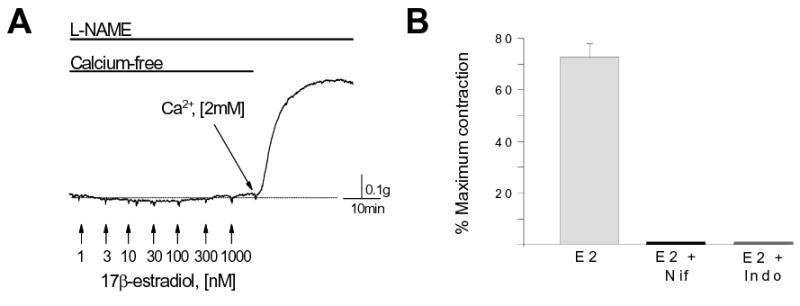
Estrogen-induced coronary artery contraction involves calcium influx. (A) Arteries were incubated in a nominally calcium-free solution with 100μM L-NAME to uncouple NOS activity (30 min). 17β-estradiol (1-1000nM) had no effect on contractile activity. Restoration of extracellular [Ca2+] to 2mM evoked an immediate contraction in the presence of estrogen. (B) Average contractile response to 30nM 17β-estradiol (E2; 100μM N-PLA pretreatment; n=10). This contractile effect of estrogen was prevented by pretreating arteries with either 1μM nifedipine (Nif; 30min) or 10μM indomethacin (Indo; 30min). Each bar represents the mean contractile response ± S.E.
DISCUSSION
Estrogen-stimulated NO production from endothelium and within the vascular wall contributes to beneficial effects of estrogen on the cardiovascular system, as reflected by earlier clinical studies demonstrating that estrogen reduces overall cardiovascular risk by approximately 50% (13, 23, 31). In contrast, the more recent WHI trials have reported deleterious effects of postmenopausal estrogen replacement on cardiovascular function (e.g., a nearly 30% increase in coronary heart disease) (30). The reasons for these discrepancies are not apparent. Interestingly, however, WHI conclusions are based solely on findings from older (age range: 50-79; mean age: 63.3) postmenopausal women. One aspect of aging that the WHI did not take into account is increasing evidence that as a woman ages there is a decline in factors critical for maintaining NOS in the “coupled” (i.e., NO-producing) state. NOS possesses both oxygenase and reductase domains with binding sites for several cofactors, and the disposition of the microenvironment around NOS will determine its primary product. For example, NOS can catalyze an “uncoupled” NADPH oxidation to generate superoxide rather than NO (1), and O2− has been suggested to play a role in age-related increased coronary vascular resistance due to its oxidative potential and ability to further reduce NO bioavailability. (10)
There is evidence that the aging process could be associated with increased uncoupling of NOS activity. For example, aging reduces the levels of L-arginine and tetrahydrobiopterin (BH4), both of which are cofactors required to sustain NO production from NOS activity. Serum L-arginine levels and NO production decrease with age (27, 28), which is associated with enhanced oxidative stress on the cardiovascular system (40). Moreover, recent studies indicate that arginase activity is upregulated in the aging vasculature, thus lowering the availability of L-arginine for NOS activity in either endothelium or VSM (4). Concomitant with age-dependent loss of L-arginine, there is also evidence that BH4 synthesis declines with age (7). BH4, which is essential for generating NO and inhibiting O2− production from nNOS (25), is decreased by nearly 30-fold by diet-induced hyperlipidemia (35). Loss of BH4 would increase the tendency to uncouple NOS activity as atherosclerosis progresses to further predispose arteries to oxidative damage. Therefore, it appears increasingly likely that as humans age and atherosclerosis progresses there is reduced NO bioavailability and enhanced oxidative stress. Our findings suggest that a NOS stimulator, such as estrogen given in HRT, would very likely accelerate the normal age-related decline in cardiovascular function, possibly via enhanced generation of ROS. Not coincidentally, among the three NOS isoforms it is nNOS that has the greatest propensity to generate O2− as uncoupling increases (1, 14). This fact, together with our findings that nNOS is an estrogen target within the vascular wall, may form the basis of an explanation as to why the WHI observed mainly harmful effects of estrogen in postmenopausal women: HRT-stimulated O2− production.
There are several mechanisms whereby estrogen could increase O2− production in VSM, such as by stimulating NADPH oxidase, xanthine oxidase, mitochondrial oxidase, and/or NOS. For example, NADPH oxidase is expressed in VSM and appears to be an important source of O2− production (18, 34); however, estrogen inhibits NADPH oxidase activity in other cell types (32) and decreases NADPH expression in endothelial cells (36). In the present study the response of coronary arteries to estrogen was critically dependent upon L-arginine balance; i.e., in the presence L-arginine, estrogen relaxed arteries, but addition of inhibitory L-arginine analogs converted estrogen into a coronary vasoconstrictor (see Figures 1 and 4A). These findings point strongly to NOS as the source of estrogen-induced production of either O2− or NO in endothelium-damaged arteries. While the present findings certainly cannot rule out other potential sources of estrogen-stimulated O2− production, potential contribution of O2− from NOS-independent sources would seem to play a minor role, if any, in mediating the vascular effects of estrogen in coronary arteries. L-arginine, it appears, is a sine qua non of estrogen-induced coronary relaxation. It seems doubtful that L-arginine would also play such a significant role in preventing O2− generation from NADPH oxidase or xanthine oxidase in the vascular wall. In contrast, L-arginine is highly effective in blunting estrogen-stimulated O2−-mediated contraction of coronary arteries. Therefore, we conclude that the most likely molecular target mediating both estrogen-induced relaxation and contraction of endothelium-denuded coronary arteries is NOS expressed in VSM.
It remains unclear as to which NOS isoform could mediate the acute effects of estrogen on VSM. Interestingly, we found N-PLA to be highly effective in converting the “normal” relaxation effect of estrogen into vasoconstriction. Because N-PLA exhibits high (3000-fold) selectivity for nNOS over iNOS and a 150-fold greater selectivity over eNOS (43), our studies implicate Type 1 (n)NOS as mediating estrogen effects in CASMC. The fact that our studies were performed in either endothelium-denuded vessels or myocytes in culture cast further doubt upon eNOS as an estrogen target molecule in our experiments. Furthermore, immunoblot studies detected significant expression of only nNOS protein in coronary artery smooth muscle from either porcine or human vessels (whereas eNOS was detected only in endothelium-intact arteries). Therefore, consistent findings from this combination of functional and molecular approaches strongly suggest that it is the nNOS isoform that functions as novel estrogen target protein in both porcine and human coronary smooth muscle. Previous studies have demonstrated expression of nNOS in VSM (5, 6, 29), and it is nNOS activity in coronary smooth muscle that maintains flow-induced vasodilation in eNOS-KO mice (17).
Our findings pose the intriguing possibility that a single molecule, nNOS, can mediate either estrogen-induced coronary artery dilation (via NO) or constriction (via O2−). Fluorescence studies support this hypothesis, as we previously reported estrogen-stimulated NOS activity in HCASMC under normal (i.e., “NOS-coupled”) conditions (39), and now report estrogen-stimulated O2− generation in NOS-uncoupled HCASMC (Figure 3B). Therefore, it appears very likely that nNOS expressed in coronary artery smooth muscle influences vascular reactivity, and we speculate that this endothelium-independent mechanism could be especially important in older women with increasingly damaged or dysfunctional endothelium so often seen in aging, atherosclerosis, and/or diabetes (12, 41). In addition, estrogen should also modulate vascular reactivity in men. Aromatase is expressed in VSM (16), thus providing a local mechanism for de novo synthesis of estrogen in the vasculature of both males and females. Our findings now provide a novel molecular mechanism whereby intravascular synthesis of estrogen could impact cardiovascular function in both women and men to produce either physiological or pathophysiological effects.
Estrogen stimulates nNOS activity in hippocampal neurons (15), and early studies suggested beneficial effects of estrogen on brain function; however, WHI findings suggest potentially deleterious consequences of the steroid on global cognitive function in older women (26). As in the vasculature, it is unclear how estrogen could produce such opposite effects in the brain. Interestingly, Chen et al. (7) have reported a dramatic (81.5%) loss of GTP-cyclohydrolase-I immunoreactivity in brains of aged vs. young humans and other primates. This enzyme is rate-limiting for the synthesis of BH4, and loss of this co-factor should accelerate NOS uncoupling in the brain and other tissues leading to greater oxidative stress. This model is remarkably consistent with our present findings of dual and opposite effects of estrogen in coronary arteries. Therefore, we propose that how estrogen influences the critical balance of NO vs. O2− might have important implications on normal cellular function in a variety of systems, and explain, at least in part, the controversies concerning the complicated physiology and pharmacology of estrogens, HRT, and aging. Our findings also raise the very intriguing question: If estrogen is indeed a stimulator of oxidative stress in older women, should traditional concepts of menopause need to be reconsidered? In other words, could age-related loss of ovarian function actually be an adaptive mechanism to reduce estrogen-stimulated pathologies due to elevated ROS production as NOS becomes increasingly uncoupled with age?
The fact that estrogen cannot contract arteries in the absence of extracellular calcium or in the presence of nifedipine (with normal extracellular calcium) indicates a dependency upon influx of calcium through dihydropyridine-sensitive calcium channels. In contrast, previous findings suggest that estrogen inhibits calcium channel activity in neurons and A7r5 vascular cells (19, 42). The present findings, however, indicate a stimulatory effect of estrogen on calcium channels in “NOS-uncoupled” vessels, thereby leading to contraction. Moreover, it is most likely not O2− that opens calcium channels directly; rather, our results with indomethacin suggest that estrogen/O2− generates production of a contractile prostaglandin which then induces contraction. Interestingly, we had demonstrated previously that indomethacin also inhibits the contractile effect of another oxidant, H2O2, which also can either contract or relax coronary arteries (2). Because O2− is readily converted to H2O2 via SOD, it is possible that the observed contractile effect of estrogen might involve local production of H2O2 (or another oxidant species) in coronary artery smooth muscle; however, further studies are needed to identify other ROS and prostaglandins that might play a role in estrogen-induced vascular contraction.
In summary, we propose that estrogen, per se, is neither “good” nor “bad” in terms of producing physiological effects. Rather, an important action of estrogen is to stimulate NOS activity. It is the disposition of NOS (either coupled or uncoupled) that will then determine which product predominates (NO or O2−, respectively), and therefore, whether the overall effect of estrogen will be beneficial or harmful. Evidence from both clinical and basic research studies demonstrates that aging is associated with depletion of cofactors essential to maintain NOS in the coupled state. We hypothesize, therefore, that NOS activity (particularly that of nNOS) would become increasingly uncoupled as women and men age, thereby increasing the predisposition to oxidative damage in the cardiovascular and other systems. Accordingly, HRT would tend to be harmful to older, postmenopausal women, yet would tend to produce primarily salutary effects in younger women (as is often observed with oral contraceptive regimens).
Acknowledgments
This study was supported by grants from the National Institutes of Health (NHLBI: HL64779 and HL07389, REW; HL-68026, SAB) and the American Heart Association (REW: 9950179N; DF: 0330196N). We wish to thank Ms. Karen Powell and Ms. Nancy Godin for their technical assistance, and Ms. Louise Meadows for her expertise in performing arterial tension studies. We also recognize the kind cooperation of Thomson Meat Packing, Lanier’s Meat Packing, and Happy Valley Meat Processing companies.
References
- 1.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barlow RS, White RE. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. Am J Physiol. 1998;275:H1283–1289. doi: 10.1152/ajpheart.1998.275.4.H1283. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett SR, Bennett PR, Campa JS, Dennes WJ, Slater DM, Mann GE, Poston L, Poston R. Expression of nitric oxide synthase isoforms in pregnant human myometrium. J Physiol 521 Pt. 1999;3:705–716. doi: 10.1111/j.1469-7793.1999.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- 5.Boulanger CM, Heymes C, Benessiano J, Geske RS, Levy BI, Vanhoutte PM. Neuronal nitric oxide synthase is expressed in rat vascular smooth muscle cells: activation by angiotensin II in hypertension. Circ Res. 1998;83:1271–1278. doi: 10.1161/01.res.83.12.1271. [DOI] [PubMed] [Google Scholar]
- 6.Brophy CM, Knoepp L, Xin J, Pollock JS. Functional expression of NOS 1 in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2000;278:H991–997. doi: 10.1152/ajpheart.2000.278.3.H991. [DOI] [PubMed] [Google Scholar]
- 7.Chen EY, Kallwitz E, Leff SE, Cochran EJ, Mufson EJ, Kordower JH, Mandel RJ. Age-related decreases in GTP-cyclohydrolase-I immunoreactive neurons in the monkey and human substantia nigra. J Comp Neurol. 2000;426:534–548. [PubMed] [Google Scholar]
- 8.Chester AH, Jiang C, Borland JA, Yacoub MH, Collins P. Oestrogen relaxes human epicardial coronary arteries through non-endothelium-dependent mechanisms. Coron Artery Dis. 1995;6:417–422. doi: 10.1097/00019501-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Conklin BS, Fu W, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir decreases endothelium-dependent vasorelaxation and increases superoxide in porcine arteries. Cardiovasc Res. 2004;63:168–175. doi: 10.1016/j.cardiores.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 10.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- 11.Darkow DJ, Lu L, White RE. Estrogen relaxation of coronary artery smooth muscle is mediated by nitric oxide and cGMP. Am J Physiol. 1997;272:H2765–2773. doi: 10.1152/ajpheart.1997.272.6.H2765. [DOI] [PubMed] [Google Scholar]
- 12.Egashira K, Inou T, Hirooka Y, Kai H, Sugimachi M, Suzuki S, Kuga T, Urabe Y, Takeshita A. Effects of age on endothelium-dependent vasodilation of resistance coronary artery by acetylcholine in humans. Circulation. 1993;88:77–81. doi: 10.1161/01.cir.88.1.77. [DOI] [PubMed] [Google Scholar]
- 13.Fletcher SW, Colditz GA. Failure of estrogen plus progestin therapy for prevention. Jama. 2002;288:366–368. doi: 10.1001/jama.288.3.366. [DOI] [PubMed] [Google Scholar]
- 14.Gorren AC, Mayer B. The versatile and complex enzymology of nitric oxide synthase. Biochemistry (Mosc) 1998;63:734–743. [PubMed] [Google Scholar]
- 15.Grohe C, Kann S, Fink L, Djoufack PC, Paehr M, van Eickels M, Vetter H, Meyer R, Fink KB. 17beta-estradiol regulates nNOS and eNOS activity in the hippocampus. Neuroreport. 2004;15:89–93. doi: 10.1097/00001756-200401190-00018. [DOI] [PubMed] [Google Scholar]
- 16.Harada N, Sasano H, Murakami H, Ohkuma T, Nagura H, Takagi Y. Localized expression of aromatase in human vascular tissues. Circ Res. 1999;84:1285–1291. doi: 10.1161/01.res.84.11.1285. [DOI] [PubMed] [Google Scholar]
- 17.Huang A, Sun D, Shesely EG, Levee EM, Koller A, Kaley G. Neuronal NOS-dependent dilation to flow in coronary arteries of male eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2002;282:H429–436. doi: 10.1152/ajpheart.00501.2001. [DOI] [PubMed] [Google Scholar]
- 18.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 19.Lee DY, Chai YG, Lee EB, Kim KW, Nah SY, Oh TH, Rhim H. 17Beta-estradiol inhibits high-voltage-activated calcium channel currents in rat sensory neurons via a non-genomic mechanism. Life Sci. 2002;70:2047–2059. doi: 10.1016/s0024-3205(01)01534-x. [DOI] [PubMed] [Google Scholar]
- 20.McNeil PL, Miyake K, Vogel SS. The endomembrane requirement for cell surface repair. Proc Natl Acad Sci U S A. 2003;100:4592–4597. doi: 10.1073/pnas.0736739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 22.Miller FJ, Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 23.Mosca L, Manson JE, Sutherland SE, Langer RD, Manolio T, Barrett-Connor E. Cardiovascular disease in women: a statement for healthcare professionals from the American Heart Association. Writing Group. Circulation. 1997;96:2468–2482. doi: 10.1161/01.cir.96.7.2468. [DOI] [PubMed] [Google Scholar]
- 24.Mugge A, Riedel M, Barton M, Kuhn M, Lichtlen PR. Endothelium independent relaxation of human coronary arteries by 17 beta-oestradiol in vitro. Cardiovasc Res. 1993;27:1939–1942. doi: 10.1093/cvr/27.11.1939. [DOI] [PubMed] [Google Scholar]
- 25.Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J Biol Chem. 1999;274:9573–9580. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 26.Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. Jama. 2003;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- 27.Reckelhoff JF, Kellum JA, Blanchard EJ, Bacon EE, Wesley AJ, Kruckeberg WC. Changes in nitric oxide precursor, L-arginine, and metabolites, nitrate and nitrite, with aging. Life Sci. 1994;55:1895–1902. doi: 10.1016/0024-3205(94)00521-4. [DOI] [PubMed] [Google Scholar]
- 28.Reckelhoff JF, Kellum JA, Jr, Racusen LC, Hildebrandt DA. Long-term dietary supplementation with L-arginine prevents age-related reduction in renal function. Am J Physiol. 1997;272:R1768–1774. doi: 10.1152/ajpregu.1997.272.6.R1768. [DOI] [PubMed] [Google Scholar]
- 29.Rosenfeld CR, Chen C, Roy T, Liu X. Estrogen selectively up-regulates eNOS and nNOS in reproductive arteries by transcriptional mechanisms. J Soc Gynecol Investig. 2003;10:205–215. doi: 10.1016/s1071-5576(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 30.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. Jama. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 31.Stampfer MJ, Colditz GA. Estrogen replacement therapy and coronary heart disease: a quantitative assessment of the epidemiologic evidence. Prev Med. 1991;20:47–63. doi: 10.1016/0091-7435(91)90006-p. [DOI] [PubMed] [Google Scholar]
- 32.Sumi D, Hayashi T, Matsui-Hirai H, Jacobs AT, Ignarro LJ, Iguchi A. 17beta-estradiol inhibits NADPH oxidase activity through the regulation of p47phox mRNA and protein expression in THP-1 cells. Biochim Biophys Acta. 2003;1640:113–118. doi: 10.1016/s0167-4889(03)00026-0. [DOI] [PubMed] [Google Scholar]
- 33.Tostes RC, Nigro D, Fortes ZB, Carvalho MH. Effects of estrogen on the vascular system. Braz J Med Biol Res. 2003;36:1143–1158. doi: 10.1590/s0100-879x2003000900002. [DOI] [PubMed] [Google Scholar]
- 34.Ungvari Z, Csiszar A, Edwards JG, Kaminski PM, Wolin MS, Kaley G, Koller A. Increased superoxide production in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003;23:418–424. doi: 10.1161/01.ATV.0000061735.85377.40. [DOI] [PubMed] [Google Scholar]
- 35.Vasquez-Vivar J, Duquaine D, Whitsett J, Kalyanaraman B, Rajagopalan S. Altered tetrahydrobiopterin metabolism in atherosclerosis: implications for use of oxidized tetrahydrobiopterin analogues and thiol antioxidants. Arterioscler Thromb Vasc Biol. 2002;22:1655–1661. doi: 10.1161/01.atv.0000029122.79665.d9. [DOI] [PubMed] [Google Scholar]
- 36.Wagner AH, Schroeter MR, Hecker M. 17beta-estradiol inhibition of NADPH oxidase expression in human endothelial cells. Faseb J. 2001;15:2121–2130. doi: 10.1096/fj.01-0123com. [DOI] [PubMed] [Google Scholar]
- 37.White RE. Estrogen and vascular function. Vascular Pharmacoloy. 2002;38:73–80. doi: 10.1016/s0306-3623(02)00129-5. [DOI] [PubMed] [Google Scholar]
- 38.White RE, Darkow DJ, Lang JL. Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP-dependent mechanism. Circ Res. 1995;77:936–942. doi: 10.1161/01.res.77.5.936. [DOI] [PubMed] [Google Scholar]
- 39.White RE, Han G, Maunz M, Dimitropoulou C, El-Mowafy AM, Barlow RS, Catravas JD, Snead C, Carrier GO, Zhu S, Yu X. Endothelium-independent effect of estrogen on Ca2+-activated K+ channels in human coronary artery smooth muscle cells. Cardiovasc Res. 2002;53:650–661. doi: 10.1016/s0008-6363(01)00428-x. [DOI] [PubMed] [Google Scholar]
- 40.Yu BP, Chung HY. Oxidative stress and vascular aging. Diabetes Res Clin Pract. 2001;54 (Suppl 2):S73–80. doi: 10.1016/s0168-8227(01)00338-2. [DOI] [PubMed] [Google Scholar]
- 41.Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. 1993;92:652–662. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, Ram JL, Standley PR, Sowers JR. 17 beta-Estradiol attenuates voltage-dependent Ca2+ currents in A7r5 vascular smooth muscle cell line. Am J Physiol. 1994;266:C975–980. doi: 10.1152/ajpcell.1994.266.4.C975. [DOI] [PubMed] [Google Scholar]
- 43.Zhang HQ, Fast W, Marletta MA, Martasek P, Silverman RB. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-L-arginine. J Med Chem. 1997;40:3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]