

Abstract
Erythropoietic protoporphyria (EPP) is an inherited disorder of heme biosynthesis that results from a partial deficiency of ferrochelatase (FECH). Recently, we have shown that the inheritance of the common hypomorphic IVS3-48C allele trans to a deleterious mutation reduces FECH activity to below a critical threshold and accounts for the photosensitivity seen in patients. Rare cases of autosomal recessive inheritance have been reported. We studied a cohort of 173 white French EPP families and a group of 360 unrelated healthy subjects from four ethnic groups. The prevalences of the recessive and dominant autosomal forms of EPP are 4% (95% confidence interval 1–8) and 95% (95% confidence interval 91–99), respectively. In 97.9% of dominant cases, an IVS3-48C allele is coinherited with the deleterious mutation. The frequency of the IVS3-48C allele differs widely in the Japanese (43%), southeast Asian (31%), white French (11%), North African (2.7%), and black West African (<1%) populations. These differences can be related to the prevalence of EPP in these populations and could account for the absence of EPP in black subjects. The phylogenic origin of the IVS3-48C haplotypes strongly suggests that the IVS3-48C allele arose from a single recent mutational event. Estimation of the age of the IVS3-48C allele from haplotype data in white and Asian populations yields an estimated age three to four times younger in the Japanese than in the white population, and this difference may be attributable either to differing demographic histories or to positive selection for the IVS3-48C allele in the Asian population. Finally, by calculating the KA/KS ratio in humans and chimpanzees, we show that the FECH protein sequence is subject to strong negative pressure. Overall, EPP looks like a Mendelian disorder, in which the prevalence of overt disease depends mainly on the frequency of a single common single-nucleotide polymorphism resulting from a unique mutational event that occurred 60,000 years ago.
Complex phenotype/genotype relationships in simple Mendelian disorders have often been attributed to environmental factors or to modifier genes that modulate the clinical expression of a gene defect at a major locus. An alternative hypothesis is that, in some dominantly inherited disorders, clinical manifestations are not simply a matter of haploinsufficiency and that some additional deficiency may be necessary for the phenotype to be expressed (Delaunay 2002; Emison et al. 2005). In these disorders, slight differences in the expression of the normal allele in trans to the mutated allele may have a major impact on the clinical expression of the disease by reducing gene expression below a critical threshold. The penetrance of a dominant mutation may thus be modulated by functional polymorphisms occurring at the same locus.
Erythropoietic protoporphyria (EPP) (MIM 177000) is an inherited disorder due to a partial deficiency of ferrochelatase (FECH) (EC 4.99.1.1.), the final enzyme in the heme biosynthesis pathway (Anderson et al. 2001). FECH is an inner membrane mitochondrial enzyme that catalyzes the insertion of ferrous iron into protoporphyrin IX to form heme. FECH deficiency in bone marrow erythroid cells is responsible for the overproduction and accumulation of protoporphyrin IX in erythrocytes, which leads to a secondary accumulation of protoporphyrin in the plasma, skin, bile, and feces (Anderson et al. 2001). The most common clinical manifestation is lifelong acute photosensitivity of the skin in response to exposure to the sun, which develops early in childhood. Although EPP is generally a benign condition, hepatic complications, such as cholelithiasis or, in rare cases (∼2%), cirrhosis and even rapidly fatal liver disease, may occur (Bloomer 1988; Doss and Frank 1989; Todd 1994; Meerman 2000). EPP cases have been reported in Europe, the United States, and Japan. So far, to our knowledge, no case of EPP has been reported in a black African subject.
More than 85 mutations in the FECH gene, including missense, nonsense, splicing, deletions, and insertions, have been identified in EPP families (fig. 1A) (Rüfenacht et al. 1998; Yotsumoto et al. 2001; Chen et al. 2002; Yasui et al. 2002; Wiman et al. 2003; Onaga et al. 2004; Whatley et al. 2004; Human Gene Mutation Database). EPP is characterized by a high allele heterogeneity, although some founder mutations have been reported in Swiss patients (Rüfenacht et al. 1998). Most individuals who are heterozygous for these mutations are asymptomatic, despite having FECH activity levels that are only half the normal value (Nordmann and Deybach 1990). For protoporphyrin to accumulate sufficiently to cause photosensitivity, FECH activity has to fall below a critical threshold of ∼35% of the normal level (Bonkowsky et al. 1975; Rossi et al. 1988; Nordmann and Deybach 1990).
Figure 1.

A, Exon/intron organization of the human FECH gene and reported mutations responsible for EPP. Previously unreported mutations are shown in bold. B, Differences between human and chimpanzee cDNA. Bold vertical lines denote exons, and unbroken lines denote introns. c = cDNA Sequence (human [GenBank accession number NM000140]; chimpanzee [GenBank accession number DQ149645]). p = Protein sequence (GenBank accession number NP000131). g = FECH Gene sequence (GenBank accession number AJ250235).
On the basis of enzymatic assays and molecular analysis in EPP families, two modes of inheritance have been proposed. In rare cases, the disorder is transmitted as an autosomal recessive trait, with both parents transmitting a molecular defect to the affected children, who typically have a residual lymphocyte FECH activity of <10% of normal (Deybach et al. 1986; Norris et al. 1990; Lamoril et al. 1991; Sarkany et al. 1994; Goerz et al. 1996; Whatley et al. 2004). So far, to our knowledge, only seven cases of autosomal recessive transmission have been documented (Lamoril et al. 1991; Sarkany et al. 1994; Poh-Fitzpatrick et al. 2002; Whatley et al. 2004). In most cases, the inheritance of EPP is described as an autosomal dominant disorder with incomplete penetrance. We have recently demonstrated that patients with the dominant form usually share a hypomorphic allele that is common in the general population in trans to a rare loss-of-function allele (Went and Klasen 1984; Gouya et al. 1996, 1999, 2002). A common intronic SNP, IVS3-48C, is responsible for the low expression of the hypomorphic allele by modulating the use of a constitutive cryptic acceptor splice site; the aberrantly spliced mRNA is degraded by a nonsense-mediated decay mechanism, leading to a lower steady-state level of FECH mRNA. In overt cases, this low steady-state level of normal mRNA results in an additional FECH enzyme deficiency, which is necessary for protoporphyrin overproduction and photosensitivity to occur (Gouya et al. 2002).
In this article, we report our study of two large cohorts of white French (WF) EPP families and a control group of 360 unrelated healthy subjects from four different ethnic groups. We first established the relative prevalence of the autosomal recessive versus the dominant forms and, then, in the dominant form, we identified the different mechanisms that underlie the incomplete penetrance of FECH mutations. We then studied the IVS3-48T/C polymorphism in different populations, investigating the relationship between IVS3-48C allele prevalence and overt EPP occurrence. Finally, we focused our attention on the phylogenic origin of the IVS3-48C allele and estimated when the IVS3-48C allele first appeared.
Subjects, Material, and Methods
Study Subjects
We investigated a first cohort of 241 subjects from 113 WF EPP families recruited at the Centre Français des Porphyries between 1990 and 2004 (fig. 2). The 241 subjects consisted of 113 overt patients with a typical history of skin photosensitivity and a high level of free protoporphyrin in their erythrocytes (32,443 ± 21,243 [mean ± SD] nM; normal <1,900 nM) plus 128 clinically unaffected relatives. We assayed peripheral lymphocyte FECH enzyme activity in 95 of the 113 overt patients. Family studies (nuclear or extended families) allowed us to perform an IVS3-48T/C segregation analysis in 53 cases.
Figure 2.

Schematic representation of the clinical, enzymatic, and molecular data of the 95 EPP families for which both FECH enzyme activity and gene defect were available. Wt = Wild-type allele; M =FECH-Mutated allele.
The second cohort consisted of 60 nuclear (n=25) or extended (n=35) French EPP families, who were recruited at the Centre Français des Porphyries between 1975 and 2000 and had well-documented clinical histories and FECH enzyme activity levels, which allowed us to assess the correlation between the IVS3-48C allele frequency and the prevalence of overt EPP. These families were independent of the 113 described above, and their EPP diagnosis was based on clinical data and the measurement of lymphocyte FECH activity (overt EPP patients, 1.5 ± 0.39 [mean ± SD] nM) and erythrocyte protoporphyrin levels (overt EPP patients, 36,840 ± 44,468 [mean ± SD] nM). No DNA was available from these subjects. Available for study were 361 subjects consisting of 60 probands, 152 relatives whose lymphocyte FECH activity was at least 50% below normal, and 149 relatives whose lymphocyte FECH activity was in the normal range. The 301 relatives were distributed across 100 different generations.
Three hundred sixty subjects unrelated to EPP patients were studied: 80 WF; 75 North Africans (NA) originating from Algeria, Morocco, or Tunisia; 115 southeast Asians (SEA) originating from Vietnam, Cambodia, or Shanghai, China; and 100 black West Africans (WA) originating from Cameroon, the Ivory Coast, or Nigeria.
These procedures involving human subjects were performed in accordance with the 1983 revision of the Declaration of Helsinki, and the study was approved by the Hospital Ethics Committee of Hôpital Ambroise Paré, in Paris.
Characterization of the Specific FECH Mutation
Genomic DNA was extracted from peripheral blood leukocytes with the use of a QIAamp DNA purification kit (Qiagen). All coding regions, the intron/exon junctions with 200–300 bp flanking sequence, 1.3 kb of 5′ noncoding region, and the two polyadenylation signal regions were PCR amplified and subjected to direct sequencing using fluorescent ddNTPs (BigDye) and an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Primers and conditions are available from the corresponding author on request. The presence or absence of mutations was confirmed by the sequencing of both strands. We also sequenced DNA samples from three chimpanzees (Pan troglodytes), using the same set of primers (GenBank accession number DQ149645).
Intragenic Polymorphism Genotyping
We analyzed the 12 intragenic SNPs distributed over the FECH gene: ss46563186, ss46563187, ss46563188, and rs17063905 are located in the promoter region; rs2269219 is in intron 1; ss46563189 is in intron 2; IVS3-48T/C (rs2272783) is in intron 3; ss46563190 is in intron 4; ss46563191 is in intron 5; ss46563192 and ss46563193 are in intron 9; and rs8339 is in the noncooling region of exon 11. Human FECH genomic DNA and cDNA are numbered in accordance with the reference sequences, with GenBank accession numbers AJ 250235 and NM000140, respectively, with the A of the ATG initiation codon shown as “+1.” The symbols used to designate genes in this article follow the guidelines of the HUGO Gene Nomenclature Committee (Povey et al. 2001). After PCR amplification of the appropriate fragments, PCR products were digested with HinfI, HaeII, DdeI, AluI, Cac8I, NlaIII, TseI, BstNI, BsrI, BsmAI, and TaqIα restriction enzymes. SNP genotyping of rs8339 was performed using denaturing gradient gel electrophoresis, as described elsewhere (Gouya et al. 1999). All primers and conditions are available on request. IVS3-48T/C data for the Japanese population were provided by Nakamura Yusuke (Human Genome Center, Institute of Medical Science, University of Tokyo) and are available in the dbSNP.
Haplotype Analysis
Haplotypes were estimated from unphased genotypes by use of the expectation maximization (EM) algorithm included in the software package ARLEQUIN ver 2.000 (Schneider et al. 1997). Phylogenetic analysis of the individual haplotypes used the minimal spanning network included in ARLEQUIN.
Lymphocyte FECH Enzyme Assay and Erythrocyte Protoporphyrin Measurements
FECH activity was determined by fluorometric measurement of zinc-mesoporphyrin formation after incubation for 60 min at 37°C, as established by Li et al. (1987), but with some modifications. For routine assays, a peripheral lymphocyte homogenate was prepared in 50 mM Tris-HCl (pH 7.6) and 20% glycerol, and the protein concentration was measured using the Bradford method (Bio-Rad reagents). The reaction consisted of a 5-min preincubation at 37°C, with 200 μl of lymphocyte homogenate, 200 μl of incubation buffer (250 mM Tris-HCl [pH 7.6], 1% [v/v] Triton X-100, and 1.75 mM palmitic acid), and 40 μl of 0.5-mM mesoporphyrin (final concentration 43 μM). Then, 20 μl of 1-mM zinc acetate solution was added (final concentration 43 μM), and the incubation continued for a further 60 min. A blank was prepared without the cell homogenate. The reaction was stopped by the addition of a dimethyl sulfoxide/methanol mixture (30:70 [v/v]). After it was centrifuged, the supernatant was transferred to a fluorometry cell, and the fluorescence was measured at 580 nm, with an excitation wavelength of 410 nm. The enzymatic activity was expressed as nanomoles of zinc-mesoporphyrin formed per hour per milligram of protein at 37°C (normal value = 4.83 ± 0.91 [mean ± SD] nmol mesozinc/h/mg). The erythrocyte protoporphyrin was determined by standard methods (Deacon and Elder 2001).
Statistical Analysis
The Student t test or the Mann-Whitney test was used to compare group means of continuous data. Standard deviations were calculated for group means. The genotype distribution of the 12 polymorphisms in the WF, WA, and SEA populations and for the IVS3-48T/C SNP in the NA population satisfied the Hardy-Weinberg equilibrium (χ2 test, P>.5).
Estimate of the Age and Growth Rate of the IVS3-48C Mutation
We performed a joint estimate of these quantities, using the method developed by Austerlitz et al. (2003). This method provides a joint estimate of the age of the mutation—that is, the time elapsed since the appearance of the common ancestor of the mutation carriers in the population and the growth rate of the number of copies since this appearance. This common ancestor can be either a new mutant or a migrant who brought the gene into the population or the only individual who left offspring carrying this mutation in the present population because of a bottleneck. Four different populations were studied: the 80 WF subjects, the 92 Chinese from the SEA population, the 50 parents from 25 fully genotyped EPP families, and the 44 Japanese subjects from the International HapMap Project.
Results
Mutation Analysis
We conducted a FECH molecular study on 113 overt EPP patients available between 1990 and 2004 from the Centre Français des Porphyries. Twenty-five of these EPP patients had already been studied and reported but were re-evaluated here for the IVS3-48T/C polymorphism (Lamoril et al. 1991; Rüfenacht et al. 1998; Gouya et al. 1996, 1999, 2002), and 88 new cases were investigated. The 88 new patients were subjected to the direct sequencing of all coding regions, the intron/exon junctions, the 1.3-kb upstream promoter region, and the two polyadenylation signal regions. In 18 overt patients (15.9%), we were not able to identify any FECH gene defect, even though the clinical manifestations, the erythrocyte protoporphyrin level, and the FECH enzyme activity closely matched EPP. The possibility that the entire gene had been deleted was excluded by the evidence that they were heterozygous for at least two intragenic SNPs. Of the 95 patients in whom gene defects were identified, 90 were heterozygous for a FECH mutation and 5 displayed compound heterozygosity. Twenty-seven previously unreported mutations were identified (fig. 1A and table 1). These were highly heterogeneous and usually family specific. The predominant type of mutations were missense mutations in the coding regions (36.4%), followed by intronic mutations predicting anomalous RNA splicing (24.2%), small deletions and insertions (21.2%), and nonsense mutations (15.2%). Most of the mutations produced null alleles. This distribution is similar to previously published EPP mutation patterns (Rüfenacht et al. 1998; Schneider-Yin et al. 2000).
Table 1.
EPP Patients with Compound Heterozygotism for Two Rare FECH Alleles
| Patient | FECHActivitya | ErythrocyteProtoporphyrinb | Allele 1 | Allele 2 |
| P1 | 0.2 | 50,000 | S222G, IVS3-48C | D274N, IVS3-48T |
| P2 | 0.5 | 50,000 | P192T, IVS3-48T | D283G, IVS3-48T |
| P3 | 0.5 | 60,000 | G55C, IVS3-48T | M267I, IVS3-48T |
| P4 | 0.3 | 26,000 | D274N, IVS3-48T | L384S, IVS3-48T |
| P5 | 1.4 | 4,800 | IVS8-2A→G, IVS3-48T | Y191H, IVS3-48T |
In nanomoles of mesozinc/hour/milligram of protein at 37 °C, measured in peripheral lymphocytes (normal value [mean ± SD] = 4.83 ± 0.91 ).
In nanomoles/liter (N<1,900).
FECH Enzyme Assay
In the 18 overt patients in whom mutation testing failed to identify any gene defect, enzyme activity was 60%–70% below normal, and this enzyme deficiency was always shown to be transmitted through two to three generations, which allowed us to detect asymptomatic carriers in family studies. To evaluate the accuracy of lymphocyte FECH activity assay in EPP diagnosis, we also compared the FECH enzymatic data to the molecular data in the 95 cases for which both sets of data were available. In these families, lymphocyte FECH activity was measured in 223 subjects: 95 probands, 61 relatives carrying a FECH gene defect, and 67 mutation-free subjects (fig. 2). The 156 individuals carrying at least one FECH gene defect displayed 50%–90% lower FECH activity than the normal control group (fig. 3). The lowest FECH activities were observed in patients with two molecular defects (figs. 2 and 3; 0.375 ± 0.15 [mean ± SD] nmol mesozinc/h/mg of protein at 37°C), and asymptomatic carriers showed higher activity (2.42±0.41 nmol mesozinc/h/mg) than overt patients (1.45±0.32 nmol mesozinc/h/mg, P<10-4). In addition, when the 67 relatives without the FECH mutation were classified according to their IVS3-48T/C genotype, we found that the subjects who were IVS3-48T/T homozygous (5.03±0.66 nmol mesozinc/h/mg) had higher FECH activity than those who were IVS3-48T/C heterozygous (4.20±0.60 nmol mesozinc/h/mg, P<10-4).
Figure 3.

Peripheral lymphocyte FECH activity (nanomoles of mesozinc/hour/milligram of protein at 37°C) in six different groups are represented as box plots showing the median, the quartiles, and the 90th and the 10th percentiles. Additionally, the mean ± SD values are as follows: control group, 4.83 ± 0.91; IVS3-48T/T healthy relative group, 5.03 ± 0.66; IVS3-48T/C healthy relative group, 4.20 ± 0.60; asymptomatic carrier group, 2.42 ± 0.41; symptomatic patient group with the dominant form, 1.45 ± 0.32; and symptomatic patient group with the recessive form, 0.375 ± 0.15.
EPP Inheritance
Four EPP families displayed clear recessive inheritance (fig. 2). These patients presented with <10% residual lymphocyte FECH activity, and this was associated with two FECH gene defects (fig. 2 and table 1). The FECH gene segregation study shows that each of the parents of these patients presents one mutant allele associated with an ∼50% decrease in FECH activity (data not shown). The four patients (P1, P2, P3, and P4) (table 1) were found to be compound heterozygotes for missense mutations. Two mutations (P192T and D383G) had previously been reported in dominant EPP, and a novel one, D274N, was found twice (in P1 and P4) in this study.
On the basis of molecular and enzymatic data, 90 EPP families displayed a clear autosomal dominant form, with a single molecular FECH defect associated with 30%–40% residual FECH activity (fig. 2).
Unexpectedly, we identified compound heterozygosity in one patient (P5) with 30% residual FECH activity (fig. 2 and table 1). Overt patient P5 had two trans mutations: an IVS8-2A→G on one allele, disrupting the consensus acceptor splice site of intron 8, and a Y191H substitution on the other. Both of these mutations are unique to this family. The expression in the prokaryotic system of the missense Y191H mutation shows 72% residual FECH activity when compared with the normal human cDNA construct (Rüfenacht et al. 1998). With the assumption that the IVS8-2A→G mutation is responsible for the exon 8 skipping, resulting in complete loss of function, and that the Y191H substitution decreases the allelic FECH activity by 28%, in a diploid system the global residual FECH activity should be ∼35%. This is, indeed, in the same range as the activity found in this patient’s P5 lymphocytes (table 1). Both parents are IVS3-48T homozygous. In agreement with molecular data, the parent transmitting the IVS8-2A→G mutation displays a 50% decrease in FECH activity, and the parent carrying the Y191H mutation has a FECH activity in the normal range (4.6 nmol mesozinc/h/mg of protein at 37°C) compared with the control group (4.83 ± 0.91 [mean ± SD] nmol mesozinc/h/mg) (fig. 3).
Altogether, 90 patients (90/95: 95% [95% CI 91–99]) presented with autosomal dominant EPP, and 4 (4/95: 4% [95% CI 1–8]) presented with autosomal recessive EPP. The classification of patient P5 is controversial and depends on whether one considers the mutation data or the enzyme data (fig. 2).
Prevalence of the FECH IVS3-48C Allele in Patients with Overt EPP and in Asymptomatic Carriers
Haplotyping studies of the 113 overt EPP patients and of the 61 asymptomatic carriers showed that none of the 61 asymptomatic carriers had the IVS3-48C polymorphism. In contrast, 93 of 95 overt patients with a dominant form of EPP did have an IVS3-48C allele. Three of the four EPP patients with recessive inheritance are IVS3-48T homozygous, and one is IVS3-48T/C heterozygous (fig. 2). Fifty-three nuclear EPP families were available for a segregation study of the FECH alleles. This demonstrated that the vast majority of overt patients present an IVS3-48C polymorphism in trans to the mutant FECH gene. No IVS3-48C allele was found in the P5 family and in two families with dominant EPP (FECH gene mutations: IVS9-2A→G and C411G). We were unable to identify any additional mutations in the two remaining families, but, in one patient, clinical manifestations of the disease appeared belatedly at the age of 30 years, just after the patient experienced partial hepatic resection for multiple hepatic adenomas attributed to prolonged exposure to contraceptive pills (fig. 2).
Prevalence of IVS3-48C Polymorphism in WF, NA, WA, and SEA Populations
We determined the genotype of the IVS3-48T/C polymorphism of 80 WF, 75 NA, 115 SEA, and 100 WA subjects. Table 2 shows that there are major differences between the four populations studied, with regard to their IVS3-48T/C polymorphism frequency. We identified a north-south gradient from 11.3% in the WF population to 2.7% in the NA population and to <1% in WA subjects. In the SEA population, the IVS3-48C allele occurs at a high frequency (31%).
Table 2.
FECH IVS3-48T/C Polymorphism in WF, NA, WA, SEA, and Japanese Populations
| No. of subjects with genotypea |
No. (%) of alleles |
|||||
| Population | No. ofsubjects | C/C | C/T | T/T | C | T |
| WF | 80 | 0 | 18 | 62 | 18 (11.3) | 142 |
| NA | 75 | 0 | 4 | 71 | 4 (2.7) | 146 |
| WA | 100 | 0 | 1 | 99 | 1 (<1) | 199 |
| SEA | 115 | 16 | 39 | 60 | 71 (31.0) | 159 |
| Japanb | 549 | NA | NA | NA | 475 (43.3) | 623 |
NA = Not available.
Correlation between IVS3-48C Allele Frequency and Overt EPP Prevalence
To confirm that the penetrance of EPP mutations in the WF population was, indeed, determined by the frequency of the IVS3-48C hypomorphic allele, we studied a second set of EPP families (n=60; see “Study Subjects” subsection). Using multigenerational pedigrees, we estimated the number of cases in which one or several EPP patients were found in generations different from that of the probands. When two or more siblings were affected, only one was considered in the model. This led us to count nine occurrences in 100 generations. With the assumption that the coinheritance of a FECH gene defect and a low-expression IVS3-48C FECH allele is required to have overt clinical manifestations of the disease, the theoretical number of occurrences with at least one overt patient in 100 generations in EPP families should be 100×11%=11. This theoretical value is close to the nine subjects with overt EPP patients found in our set of 60 EPP families (binomial law, P=.11).
IVS3-48C FECH Alleles: Haplotypic and Phylogenic Analyses
Twelve polymorphisms were used in the SEA, WF, and WA populations to construct haplotypes estimated by the EM algorithm. All SNPs were found to comply with the Hardy-Weinberg equilibrium. Twenty-two IVS3-48C haplotypes were assigned from unphased genotype data, but most of them had a frequency of <1% (data not shown) and were not taken into consideration. The five most common haplotypes (HC1–HC5) are shown in table 3. Haplotypes HC1, HC2, and HC3 and haplotypes HC1, HC2, HC4, and HC5 account for most of the IVS3-48C haplotypes found in the SEA and WF populations, respectively (table 3). Haplotypes HC1 and HC2 occur in both the SEA and WF populations but at different frequencies. Haplotype HC1 is, by far, the most frequent in the SEA population (23% of all haplotypes and 71% of IVS3-48C haplotypes) but occurs at a much lower frequency in the WF population (1.2% of all haplotypes and 12% of IVS3-48C haplotypes) and in the only WA subject to display a IVS3-48C polymorphism. The HC1 haplotype differs from the closest African haplotype (root haplotype [RH] in table 3) at three sites, two of them being monomorphic in the WA population (ss46563187 and IVS3-48T/C [table 3]). Haplotype HC2 occurs in SEA with a 5% frequency of IVS3-48C haplotypes (1.7% of all SEA haplotypes) and 13% in WF (1.3% of all WF haplotypes). Haplotypes HC3, HC4, and HC5 are population specific. We then performed a phylogenic analysis of the IVS3-48C haplotypes. We found that most of the 22 IVS3-48C haplotypes, and especially the five most common IVS3-48C haplotypes, are closely related to each other. Haplotype HC1 represents the consensus haplotype and was progressively shown to be connected to haplotypes HC2 to HC5, either by single-site difference (using the minimum spanning network algorithm implemented in ARLEQUIN ver. 2.000) or by recombination events between IVS3-48C and IVS3-48T haplotypes. In the most parsimonious model, one single-site mutation and three recombinations between IVS3-48C and IVS3-48T haplotypes explain the HC1 to HC5 phylogeny, as proposed in fig. 4. IVS3-48C haplotypes may originate from a very frequent (9%) WA RH (table 3 and fig. 4).
Table 3.
Identification and Frequency of IVS3-48C Haplotypes in WF, SEA, and WA Populations
| Snpa |
Haplotype Frequency |
||||||||||||||
| Haplotype | ss46563186 (5′NC) | ss46563187 (5′NC) | ss46563188 (5′NC) | rs17063905 (5′NC) | rs2269219 (I1) | ss46563189 (I2) | rs2272783 (I3) | ss46563190 (I4) | ss46563191(I5) | ss46563192 (I9) | ss46563193 (I9) | ssrs8339 (E11, 3′NC) | WF | SEA | WA |
|
IVS3-48C FECHb/All FECHc: |
|||||||||||||||
| HC1 | C | A | G | G | T | A | C | A | T | C | G | T | 12/1.2 | 71/23 | 100/<1 |
| HC2 | C | A | G | G | T | A | C | C | T | C | G | C | 12/1.2 | 5/1.7 | 0 |
| HC3 | C | A | G | G | T | A | C | A | G | C | G | T | 0 | 6.8/2.1 | 0 |
| HC4 | C | G | G | G | T | A | C | A | G | C | G | C | 13/1.3 | 0 | 0 |
| HC5 | C | G | G | G | T | A | C | A | G | C | G | T | 12/1.2 |
0 |
0 |
| RH HaplotypeFrequencyd: |
|||||||||||||||
| RH | C | G | G | G | T | A | T | A | G | C | G | T | 1.2 | <1 | 9 |
Accession numbers are according to dbSNP. NC = Noncooling region. I = Intron. E = Exon.
Proportion (%) of HC1 to HC5 haplotypes among IVS3-48C FECH haplotypes.
Proportion (%) of HC1 to HC5 haplotypes among all FECH haplotypes.
Proportion (%) of haplotypes among all FECH haplotypes.
Figure 4.

Phylogeny of the IVS3-48C haplotypes presenting the more parsimonious combination of single-site mutations and recombination events. Each haplotype is represented by a circle, the area of which represents the relative frequency of that haplotype amongst the overall IVS3-48C haplotypes. Each circle is subdivided to show the proportion of the individual haplotype frequency in each of the WF, SEA, and WA population groups. The solid lines connect haplotypes by single-site difference with precision of the position directly on the line. Dashed lines denote recombination events. The root haplotype (RH) is where the IVS3-48C mutation probably occurs. With a frequency of 9%, RH is the second most frequent WA FECH haplotype. The haplotype names are the same as in table 3.
Estimation of the Date of the Appearance of the IVS3-48C Mutation
We calculated the date of the most recent common ancestor of IVS3-48C haplotypes, using four different data sets. With two of them, those of the WF population and the WF EPP families, the age of the allele was estimated to be ∼40,000–60,000 years. In contrast, the estimate calculated from the Japanese and the Chinese data set was only ∼15,000 years, which was associated with a higher rate of population growth (table 4).
Table 4.
Estimated Growth Rate and Age of the IVS3-48C Allele (with 95% CIs) in Three Different Populations and in WF EPP families
| Population | 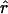 |
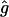 |
Age(years)a |
| Japanese | 1.027 (1.024–1.033) | 529.0 (471.2–636.5) | 13,225 (11,780–15,912) |
| Chinese | 1.019 (1.017–1.024) | 714.2 (634.9–849) | 17,855 (15,872–21,225) |
| EPP Families | 1.007 (1.006–1.009) | 1,655.4 (1,431.7–2,055.3) | 41,385 (35,792–51,382) |
| WF | 1.004 (1.004–1.006) | 2,431.0 (2,089.6–3,044.6) | 60,775 (52,240–76,115) |
Assuming that one generation = 25 years.
KA/KS Study
To estimate the level of constraint of the FECH gene, we aligned the orthologous human and chimpanzee (Pan troglodytes) FECH sequences. Compared with the human consensus sequence, the chimpanzees show five nonsynonymous mutations restricted to exon 2 (in codons 24, 31, 39, and 45) that code for the signal peptide targeting FECH in the mitochondria and three synonymous mutations in exon 3, 8, and 9 (in codons 100, 273, and 336) that code for the mature part of the protein (fig. 1B). Considering only the coding part for the mature protein, we observed a ratio of zero amino acid–altering mutations (nonsynonymous, KA when normalized to the number of relevant sites) to three silent mutations (synonymous, KS).
Discussion
Extensive, ongoing research into low penetrance in simple Mendelian disorders is now beginning to bear fruit, with important implications for the understanding of the etiology of disease and for the development of new prevention and treatment strategies. Environmental factors and modifier genes are often invoked as modulators of the clinical expression of a gene defect at a major locus. In EPP, for protoporphyrin to accumulate sufficiently to cause clinical symptoms, a reduction in FECH activity below a critical threshold of ∼35% is required. To account for this reduction, several mechanisms have been suggested, including the coinheritance of either two trans FECH gene defects or of one FECH defect and a common hypomorphic FECH allele (Gouya et al. 2002). To determine the respective relevance of these mechanisms to the clinical expression of EPP, we have conducted a large retrospective and prospective mutation and enzyme study in EPP patients. In accordance with the generally accepted autosomal dominant inheritance, most of the patients have a single mutation restricted to one allele. We achieved an overall detection rate of 83% (95 of 113 patients), which matches rates reported in previous studies: Rüfenacht et al. (1998), using DGGE screening in 29 Swiss and French EPP families (90%) and Whatley et al. (2004), analyzing 14 British EPP patients (80%). In 2 of 18 cases with an unidentified mutation, a segregation study of 12 intragenic polymorphisms covering the entire FECH gene reveals a large intragenic deletion that is currently being characterized (data not shown). It remains to be determined whether the other 16 negative cases simply correspond to a lack of sensitivity of the method, to the location of mutations in unexplored parts of the gene, or to major gene rearrangement. In any case, in these families, the FECH assay is a valuable tool for detecting asymptomatic carriers, since it appears to provide a highly specific and sensitive diagnosis of EPP. Indeed, in our study, the assays of FECH activity identified all EPP carriers of a FECH gene defect whether they were symptomatic or not (n=156), and there was no overlap between healthy relatives and asymptomatic patients. When both molecular and enzyme data were used to analyze EPP families, the FECH assay unambiguously differentiated between the healthy subjects (i.e., those without a mutant allele) and symptomatic or asymptomatic carriers of a mutant FECH allele. In addition, the FECH assay is also sensitive enough to discriminate between IVS3-48T/T and IVS3-48T/C groups in normal subjects (fig. 3). Furthermore, autosomal recessive EPP patients, although clinically indistinguishable from those with the dominant form, could easily be distinguished from them by this assay, since they had residual FECH activity of <10%.
In animals, recessive inheritance is the rule, with cases reported in bovines (Ruth et al. 1977), mice (Tutois et al. 1991), and zebrafish (Childs et al. 2000). In our cohort, 4% of patients with overt EPP presented a recessive form of the disease, a frequency similar to that reported by Whatley et al. (2004) in the United Kingdom (3%). In the seven families with recessive EPP previously reported, the mutations were mainly missense mutations (Lamoril et al. 1991; Sarkany et al. 1994; Poh-Fitzpatrick et al. 2002; Whatley et al. 2004), and, in the three new patients in our study, the mutations found were always missense mutations, whereas, in dominant EPP, missenses account for only 34.6% of all mutations. Functional studies of missense mutations have shown that those found in compound heterozygous subjects have detectable residual activity, in contrast with those found in dominant EPP, which are much lower or undetectable (Rüfenacht et al. 1998; Whatley et al. 2004). Taken together with the low frequency of “null alleles” in recessive EPP, these data highlight the absolute need to maintain a minimum threshold level of heme biosynthesis in cells. The extremely low FECH activity measured in the recessive forms could be a major risk factor for hepatic effects. Severe liver disease occurs in 2% of dominant forms (Bloomer 1988; Todd 1994; Meerman 2000) but in 30% of the 10 known EPP patients with recessive inheritance. This shows how important it is to detect patients with recessive inheritance. Nevertheless, so far, three of our recessive EPP patients with the lowest FECH activity (P1, P2, and P4) are still free of severe hepatic injury, suggesting that other genetic or environmental factors may also contribute to EPP liver complications.
As a general detection strategy of the recessive forms of EPP, Whatley et al. (2004) proposed first to genotype patients for the IVS3-48T/C polymorphism and to select IVS3-48T homozygous patients for a complete mutational analysis. Using this strategy, we would have missed one of our four patients with recessive inheritance (P1 in table 1). If applied in Japan, this strategy would miss >60% of recessive cases, since the IVS3-48C allele frequency is 42%. The best screening method remains either the systematic molecular study of the entire FECH gene, with a risk of missing one mutation in 15% of the cases, or the lymphocyte FECH assay.
Dominant inheritance occurred in 95% of cases, and the IVS3-48C low-expression mechanism was irrelevant only in 2 of the 90 patients (>2%). In one case, the clinical manifestation of the disease may have been precipitated by an hepatic resection for adenoma that reduced the liver’s protoporphyrin clearance capacity. This patient might otherwise have remained asymptomatic. It will be of interest to follow her EPP symptomatology after hepatic regeneration. These two mechanisms leading to overt EPP should be added to other rare mechanisms reported elsewhere that reduce FECH activity by >50%, such as acquired FECH gene mutations (Aplin et al. 2001) or epigenetic control of FECH expression (Onaga et al. 2004). As another physiopathological mechanism, the possibility of dominant negative mutations has been suggested on the basis of an EPP animal model (Magness et al. 2002) or in vitro FECH expression studies (Najahi-Missaoui and Dailey 2005; Ohgari et al. 2005) but remains to be stated in human EPP families.
In patient P5, the strict respect of the Mendelian rules supports his classification in the recessive form of EPP (i.e., two mutations). Nevertheless, as described in the “Results” section, the parent transmitting the Y191H mutation shows a lymphocyte FECH activity in the normal range. If we consider the Y191H as deleterious, this subject would be, in our experience, the first carrier of a FECH gene defect with a normal residual FECH activity. In fig. 3, we can observe that the mean FECH activity between the IVS3-48T/T healthy relatives and the IVS3-48T/C healthy relatives differ significantly (Mann-Whitney test, P<.0001) although there is a large overlap between the two groups. This indicates that, at the individual level, a subject heterozygous for a wild-type FECH allele and a hypomorphic FECH allele (IVS3-48C allele or a mild missense mutation like the Y191H mutant) is not distinguishable for FECH activity from a subject carrying two wild-type FECH alleles.
The presence of the hypomorphic IVS3-48C common allele trans to a mutated FECH allele appears to be generally required for clinical expression of EPP (>97% of the patients with autosomal dominant disease). Recent studies in Japan, North America, Sweden, Israel, and the United Kingdom all provide independent confirmation of the preeminence of this original mode of inheritance (Yasui et al. 2002; Risheg et al. 2003; Schoenfeld et al. 2003; Wiman et al. 2003; Whatley et al. 2004). We could also assess the role of the IVS3-48C allele low-expression mechanism by analyzing the correlation between clinical EPP prevalence and IVS3-48C allele frequency in different populations. We observed a north-south and east-west gradient range from <1% in WA subjects to 31% in SEA subjects and 43% in Japan (Human Genome Center, Institute of Medical Science, University of Tokyo). In a cohort of 60 EPP patients independent of the 113 families above, we have shown that the 11% IVS3-48C allele frequency observed in France can account for EPP penetrance and correlates to EPP prevalence. Kondo et al. (2004) recently published extensive epidemiological data on EPP in Japan. They showed that 51% of Japanese EPP cases occurred in families with some other history of EPP, compared with only 15% (χ2=28; P<10-4) of French EPP cases, suggesting that the penetrance of FECH mutations is higher in Japan. This could be due to the IVS3-48C allele frequency of 43% in Japan, which is four times higher than that in France. Nevertheless, further studies in Japan are need to confirm the hypothesis. Moreover, to our knowledge, EPP has never been demonstrated in black sub-Saharan Africans. Although melanodermy probably protects against photosensitivity, other cutaneous porphyria affect some black patients in South Africa (P. Meisner and R. Hift, South African Porphyria Center, personal communication). These findings support the proposal that the low prevalence of EPP and the absence of the IVS3-48C allele in the WA population may be linked, a notion reinforced by the appparent absence of EPP cases in the black African population living in France (data not shown). It also suggests that diet, lifestyle, and other nongenetic factors probably have no impact on EPP penetrance. Altogether, we report convergent evidence that overt EPP prevalence is dependent on IVS3-48C allele frequency.
IVS3-48C allele frequency in the WA population is very low, if not zero. The single HC1 haplotype found in a Nigerian subject probably results from a recent admixture with Eurasian genes, as two alleles are restricted to non-African populations. The five most common IVS3-48C haplotypes can easily be connected, sharing a restricted haplotype and the IVS3-48C allele that appears on a common RH African haplotype that differs from HC5 only by the IVS3-48C allele. These observations strongly suggest that IVS3-48C alleles originate from a founder effect that happened after the population had moved out of Africa. Moreover, the study of a dinucleotide repeat (ATn) in intron 2 shows that, in 20 unrelated white EPP subjects, the IVS3-48C allele was invariably associated with the A9 allele on the same chromosome, whereas control white subjects are highly polymorphic (n=60; nine different alleles, data not shown). This lack of IVS3-48C ATn variability suggests rapid expansion after a bottleneck (Zietkiewicz et al. 2003). In SEA, the IVS3-48C allele frequency is associated with a different IVS3-48C haplotype distribution pattern (fig. 4), which suggests independent expansions that could correspond to the northern and southern routes of migration referred to in the literature (Lahr and Foley 1998; Kivisild et al. 1999). All these observations are compatible with the historically documented Upper Paleolithic expansions dated 30,000–60,000 years ago. We then estimated the time of introduction of the IVS3-48C allele more precisely on the basis of the decay of allele association through time (Labuda et al. 1997; Austerlitz et al. 2003). By use of the WF or the WF EPP families’ data sets, the estimates were ∼40,000–60,000 years (assuming 25 years per generation), in accordance with our precedent estimation. Interestingly, estimates based on populations with high IVS3-48C allele frequency, like the Japanese and the Chinese populations, seemed to give a more recent occurrence of ∼13,000–17,000 years. If we assume that the IVS3-48C mutation occurred only once, this discrepancy can be attributed to either a particular pattern of growth in the SEA population or to the positive selection of IVS3-48C haplotypes.
Finally, we estimated the neutrality of sequence differences found between human and chimpanzee FECH genes, by calculating the ratio of nonsynonymous to synonymous mutations. Restricting our calculations to the coding region for the mature part of the protein, we found KA/KS=0, indicating strongly negative Darwinian selection of the mutant altering the function of the protein. It remains to be seen whether the rapid expansion across Asia of IVS3-48C haplotypes, despite a context of a strongly negative impact on FECH function, should be attributed to genetic drift alone or to positive selection.
In conclusion, on the basis of molecular and enzyme data, the conventional Mendelian rules of recessive and dominant inheritance of a genetic trait appear to be too restrictive in EPP. Several mechanisms appear to contribute to bringing the residual FECH activity below the critical threshold. In >90% of cases, overt EPP results from the cosegregation of a severe mutation trans to the hypomorphic IVS3-48C allele, and the high prevalence in Europe, the United States, and Asia of the IVS3-48C allele results in a pseudodominant inheritance of EPP. The phylogenic origin of this common single SNP and its geographic distribution mainly contribute to the clinical expression of EPP. It remains to be seen whether demographic events or selection explains the rapid expansion of IVS3-48C haplotypes across Asia. Finally, as far as we are aware, EPP may be the only rare monogenic trait for which the prevalence of overt disease is dependent upon the frequency of a common variant, IVS3-48T/C SNP, resulting from a unique mutational accident that occurred 60,000 years ago.
Acknowledgments
This research was supported by GIS-Maladie Rares 2005 Research Network on Rare Disorders. We thank S. Sassa, S. Taketani, P. Meisner, and R. Hift, for providing additional information about the clinical expression of EPP in their respective countries; R. Aquaron and M. Traoré, for providing samples from North African and West African countries; and Guillaume Lecointre, for chimpanzee DNA samples. The French control group was drawn from the Centre d’Etude du Polymorphisme Humain (CEPH, Paris). This study was supported by a grant from GIS-Institut des maladies rares, INSERM-AFM-Ministère de la Recherché.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- ARLEQUIN, http://anthropologie.unige.ch/arlequin/
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for chimpanzee FECH sequence [DQ149645], human FECH cDNA [NM000140], human FECH gene [AJ250235], and human FECH protein [NP000131]
- Human Gene Mutation Database (HGMD), http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html/
- Human Genome Center, Institute of Medical Science, University of Tokyo, http://www.hgc.ims.u-tokyo.ac.jp/
- International HapMap Project, http://www.hapmap.org/index.html.en/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for EPP) [PubMed]
References
- Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited disease. 8th ed. McGraw-Hill, New York, pp 2961–3062 [Google Scholar]
- Aplin C, Whatley SD, Thompson P, Hoy T, Fisher P, Singer C, Lovell CR, Elder GH (2001) Late-onset erythropoietic porphyria caused by a chromosome 18q deletion in erythroid cells. J Invest Dermatol 117:1647–1649 10.1046/j.0022-202x.2001.01560.x [DOI] [PubMed] [Google Scholar]
- Austerlitz F, Kalaydjieva L, Heyer E (2003) Detecting population growth, selection and inherited fertility from haplotypic data in humans. Genetics 165:1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomer JR (1988) The liver in protoporphyria. Hepatology 8:402–407 [DOI] [PubMed] [Google Scholar]
- Bonkowsky HL, Bloomer JR, Ebert PS, Mahoney MJ (1975) Heme synthetase deficiency in human protoporphyria: demonstration of the defect in liver and cultured skin fibroblasts. J Clin Invest 56:1139–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FP, Risheg H, Liu Y, Bloomer J (2002) Ferrochelatase gene mutations in erythropoietic protoporphyria: focus on liver disease. Cell Mol Biol (Noisy-le-grand) 48:83–89 [PubMed] [Google Scholar]
- Childs S, Weinstein BM, Mohideen MA, Donohue S, Bonkovsky H, Fishman MC (2000) Zebrafish dracula encodes ferrochelatase and its mutation provides a model for erythropoietic protoporphyria. Curr Biol 10:1001–1004 10.1016/S0960-9822(00)00653-9 [DOI] [PubMed] [Google Scholar]
- Deacon AC, Elder GH (2001) ACP best practice no 165: front line tests for the investigation of suspected porphyria. J Clin Pathol 54:500–507 10.1136/jcp.54.7.500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay J (2002) Molecular basis of red cell membrane disorders. Acta Haematol 108:210–218 10.1159/000065657 [DOI] [PubMed] [Google Scholar]
- Deybach JC, Da Silva V, Pasquier Y, Nordmann Y (1986) Ferrochelatase in human erythropoietic protoporphyria: the first case of a homozygous form of the enzyme deficiency. In: Nordmann Y (ed) Porphyrines and porphyries. John Libbey, Paris, pp 163–173 [Google Scholar]
- Doss MO, Frank M (1989) Hepatobiliary implications and complications in protoporphyria, a 20-year study. Clin Biochem 22:223–229 10.1016/S0009-9120(89)80081-5 [DOI] [PubMed] [Google Scholar]
- Emison ES, McCallion AS, Kashuk CS, Bush RT, Grice E, Lin S, Portnoy ME, Cutler DJ, Green ED, Chakravarti A (2005) A common sex-dependent mutation in a RET enhancer underlies Hirschsprung disease risk. Nature 434:857–863 10.1038/nature03467 [DOI] [PubMed] [Google Scholar]
- Goerz G, Bunselmeyer S, Bolsen K, Schurer NY (1996) Ferrochelatase activities in patients with erythropoietic protoporphyria and their families. Br J Dermatol 134:880–885 10.1046/j.1365-2133.1996.121856.x [DOI] [PubMed] [Google Scholar]
- Gouya L, Deybach JC, Lamoril J, Da Silva V, Beaumont C, Grandchamp B, Nordmann Y (1996) Modulation of the phenotype in dominant erythropoietic protoporphyria by a low expression of the normal ferrochelatase allele. Am J Hum Genet 58:292–299 [PMC free article] [PubMed] [Google Scholar]
- Gouya L, Puy H, Lamoril J, Da Silva V, Grandchamp B, Nordmann Y, Deybach JC (1999) Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood 93:2105–2110 [PubMed] [Google Scholar]
- Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, Da Silva V, Grandchamp B, Deybach JC (2002) The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet 30:27–28 10.1038/ng809 [DOI] [PubMed] [Google Scholar]
- Kivisild T, Bamshad MJ, Kaldma K, Metspalu M, Metspalu E, Reidla M, Laos S, Parik J, Watkins WS, Dixon ME, Papiha SS, Mastana SS, Mir MR, Ferak V, Villems R (1999) Deep common ancestry of indian and western-Eurasian mitochondrial DNA lineages. Curr Biol 9:1331–1334 10.1016/S0960-9822(00)80057-3 [DOI] [PubMed] [Google Scholar]
- Kondo M, Yano Y, Shirataka M, Urata G, Sassa S (2004) Porphyrias in Japan: compilation of all cases reported through 2002. Int J Hematol 79:448–456 10.1532/IJH97.03127 [DOI] [PubMed] [Google Scholar]
- Labuda D, Zietkiewicz F, Labuda M (1997) The genetic clock and the age of the founder effect in growing populations: a lesson from the French Canadians and Ashkenazim. Am J Hum Genet 61:768–771 [PMC free article] [PubMed] [Google Scholar]
- Lahr MM, Foley RA (1998) Towards a theory of modern human origins: geography, demography, and diversity in recent human evolution. Am J Phys Anthropol Suppl 27:137–176 [DOI] [PubMed] [Google Scholar]
- Lamoril J, Boulechfar S, de Verneuil H, Grandchamp B, Nordmann Y, Deybach JC (1991) Human erythropoietic protoporphyria: two point mutations in the ferrochelatase gene. Biochem Biophys Res Commun 181:594–599 10.1016/0006-291X(91)91231-Z [DOI] [PubMed] [Google Scholar]
- Li FM, Lim CK, Peters TJ (1987) An HPLC assay for rat liver ferrochelatase activity. Biomed Chromatogr 2:164–168 10.1002/bmc.1130020408 [DOI] [PubMed] [Google Scholar]
- Magness ST, Maeda N, Brenner DA (2002) An exon 10 deletion in the mouse ferrochelatase gene has a dominant-negative effect and causes mild protoporphyria. Blood 100:1470–1477 10.1182/blood-2001-12-0283 [DOI] [PubMed] [Google Scholar]
- Meerman L (2000) Erythropoietic protoporphyria: an overview with emphasis on the liver. Scand J Gastroenterol Suppl (232):79–85 [PubMed] [Google Scholar]
- Najahi-Missaoui W, Dailey HA (2005) Production and characterization of erythropoietic protoporphyric heterodimeric ferrochelatases. Blood 106:1098–1104 10.1182/blood-2004-12-4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordmann Y, Deybach JC (1990) Human hereditary porphyrias. In: Dailey HA (ed) Biosynthesis of heme and chlorophylls. McGraw-Hill, New York, pp 491–542 [Google Scholar]
- Norris PG, Nunn AV, Hawk JL, Cox TM (1990) Genetic heterogeneity in erythropoietic protoporphyria: a study of the enzymatic defect in nine affected families. J Invest Dermatol 95:260–263 10.1111/1523-1747.ep12484876 [DOI] [PubMed] [Google Scholar]
- Ohgari Y, Sawamoto M, Yamamoto M, Kohno H, Taketani S (2005) Ferrochelatase consisting of wild-type and mutated subunits from patients with a dominant-inherited disease, erythropoietic protoporphyria, is an active but unstable dimer. Hum Mol Genet 14:327–334 10.1093/hmg/ddi029 [DOI] [PubMed] [Google Scholar]
- Onaga Y, Ido A, Uto H, Hasuike S, Kusumoto K, Moriuchi A, Numata M, Nagata K, Hori T, Hayashi K, Tsubouchi H (2004) Hypermethylation of the wild-type ferrochelatase allele is closely associated with severe liver complication in a family with erythropoietic protoporphyria. Biochem Biophys Res Commun 321:851–858 10.1016/j.bbrc.2004.06.178 [DOI] [PubMed] [Google Scholar]
- Poh-Fitzpatrick MB, Wang X, Anderson KE, Bloomer JR, Bolwell B, Lichtin AE (2002) Erythropoietic protoporphyria: altered phenotype after bone marrow transplantation for myelogenous leukemia in a patient heteroallelic for ferrochelatase gene mutations. J Am Acad Dermatol 46:861–866 10.1067/mjd.2002.120460 [DOI] [PubMed] [Google Scholar]
- Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H (2001) The HUGO Gene Nomenclature Committee (HGNC). Hum Genet 109:678–680 10.1007/s00439-001-0615-0 [DOI] [PubMed] [Google Scholar]
- Risheg H, Chen FP, Bloomer JR (2003) Genotypic determinants of phenotype in North American patients with erythropoietic protoporphyria. Mol Genet Metab 80:196–206 10.1016/j.ymgme.2003.07.001 [DOI] [PubMed] [Google Scholar]
- Rossi E, Costin KA, Garcia-Webb P (1988) Ferrochelatase activity in human lymphocytes, as quantified by a new high-performance liquid-chromatographic method. Clin Chem 34:2481–2485 [PubMed] [Google Scholar]
- Rüfenacht UB, Gouya L, Schneider-Yin X, Puy H, Schafer BW, Aquaron R, Nordmann Y, Minder EI, Deybach JC (1998) Systematic analysis of molecular defects in the ferrochelatase gene from patients with erythropoietic protoporphyria. Am J Hum Genet 62:1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruth GR, Schwartz S, Stephenson B (1977) Bovine protoporphyria: the first nonhuman model of this hereditary photosensitizing disease. Science 198:199–201 [DOI] [PubMed] [Google Scholar]
- Sarkany RP, Alexander GJ, Cox TM (1994) Recessive inheritance of erythropoietic protoporphyria with liver failure. Lancet 344:958–959 10.1016/S0140-6736(94)92314-0 [DOI] [PubMed] [Google Scholar]
- Schneider S, Kueffer J-M, Roessli D, Excoffier L (1997) Arlequin version 1.1: a software for population genetic data analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva [Google Scholar]
- Schneider-Yin X, Gouya L, Meier-Weinand A, Deybach JC, Minder EI (2000) New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care. Eur J Pediatr 159:719–725 10.1007/s004310000494 [DOI] [PubMed] [Google Scholar]
- Schoenfeld N, Mamet R, Minder EI, Schneider-Yin X (2003) A “null allele” mutation is responsible for erythropoietic protoporphyria in an Israeli patient who underwent liver transplantation: relationships among biochemical, clinical, and genetic parameters. Blood Cells Mol Dis 30:298–301 10.1016/S1079-9796(03)00040-8 [DOI] [PubMed] [Google Scholar]
- Todd DJ (1994) Erythropoietic protoporphyria. Br J Dermatol 131:751–766 [DOI] [PubMed] [Google Scholar]
- Tutois S, Montagutelli X, Da Silva V, Jouault H, Rouyer-Fessard P, Leroy-Viard K, Guenet JL, Nordmann Y, Beuzard Y, Deybach JC (1991) Erythropoietic protoporphyria in the house mouse: a recessive inherited ferrochelatase deficiency with anemia, photosensitivity, and liver disease. J Clin Invest 88:1730–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Went LN, Klasen EC (1984) Genetic aspects of erythropoietic protoporphyria. Ann Hum Genet 48:105–117 [DOI] [PubMed] [Google Scholar]
- Whatley SD, Mason NG, Khan M, Zamiri M, Badminton MN, Missaoui WN, Dailey TA, Dailey HA, Douglas WS, Wainwright NJ, Elder GH (2004) Autosomal recessive erythropoietic protoporphyria in the United Kingdom: prevalence and relationship to liver disease. J Med Genet 41:e105 10.1136/jmg.2003.016121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiman A, Floderus Y, Harper P (2003) Novel mutations and phenotypic effect of the splice site modulator IVS3-48C in nine Swedish families with erythropoietic protoporphyria. J Hum Genet 48:70–76 10.1007/s100380300009 [DOI] [PubMed] [Google Scholar]
- Yasui Y, Muranaka S, Tahara T, Shimizu R, Watanabe S, Horie Y, Nanba E, Uezato H, Takamiyagi A, Taketani S, Akagi R (2002) A new ferrochelatase mutation combined with low expression alleles in a Japanese patient with erythropoietic protoporphyria. Clin Sci (Lond) 102:501–506 [PubMed] [Google Scholar]
- Yotsumoto S, Shimada S, Terasaki K, Taketani S, Kobayashi K, Saheki T, Kanzaki T (2001) A novel A(-4)-to-G acceptor splice site mutation leads to three bases insertion in ferrochelatase mRNA in a patient with erythropoietic protoporphyria. J Invest Dermatol 117:159–161 10.1046/j.0022-202x.2001.01365.x [DOI] [PubMed] [Google Scholar]
- Zietkiewicz E, Yotova V, Gehl D, Wambach T, Arrieta I, Batzer M, Cole DE, Hechtman P, Kaplan F, Modiano D, Moisan JP, Michalski R, Labuda D (2003) Haplotypes in the dystrophin DNA segment point to a mosaic origin of modern human diversity. Am J Hum Genet 73:994–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]