

Abstract
Objective
We tested the hypothesis that hypoxia inhibits currents through L-type Ca2+ channels and inhibits norepinephrine-induced rises in intracellular Ca2+ in cremasteric arteriolar muscle cells, thus accounting for the inhibitory effect of hypoxia on norepinephrine-induced contraction of these cells.
Methods
Single smooth muscle cells were enzymatically isolated from second-order and third-order arterioles from hamster cremaster muscles. The effects of hypoxia (partial pressure of oxygen: 10–15 mm Hg) were examined on Ba2+ (10 mM) currents through L-type Ca2+ channels by use of the perforated patch clamp technique. Also, the effect of hypoxia on norepinephrine-induced calcium changes was studied using Fura 2 microfluorimetry.
Results
Hypoxia inhibited the norepinephrine-induced (10 μM) contraction of single arteriolar muscle cells by 32.9 ± 5.6% (mean ± SE, n = 4). However, hypoxia had no significant effect on whole-cell currents through L-type Ca2+ channels: the peak current densities measured at +20 mV were −3.83 ± 0.40 pA/pF before hypoxia and −3.97 ± 0.36 pA/pF during hypoxia (n = 15; p > 0.05). In addition, hypoxia did not inhibit Ca2+ transients in arteriolar muscle cells elicited by 10 μM norepinephrine. Instead, hypoxia increased basal Ca2+ (13.8 ± 3.2%) and augmented peak Ca2+ levels (29.4 ± 7.3%) and steady-state Ca2+ levels (15.2 ± 5.4%) elicited by 10 μM norepinephrine (n = 21; p < 0.05).
Conclusions
These data indicate that hypoxia inhibits norepinephrine-induced contraction of single cremasteric arteriolar muscle cells by a mechanism that involves neither L-type Ca2+ channels nor norepinephrine-induced Ca2+ mobilization. Instead, our findings suggest that hypoxia must inhibit norepinephrine-induced contraction by affecting a component of the signaling pathway that lies downstream from the increases in Ca2+ produced by this neurotransmitter.
Keywords: arterioles, calcium ions, contraction, hypoxia, ion channels, microcirculation, oxygen, vascular smooth muscle
INTRODUCTION
Arterioles in the microcirculation are exquisitely sensitive to changes in the partial pressure of oxygen (PO2) in their environment. Decreases in PO2 cause arteriolar dilation (7,8,16,20), whereas increases in oxygen tension cause arteriolar constriction (2,3,10,11,14). However, the mechanism by which changes in PO2 produce changes in arteriolar tone remain unclear.
Recently, Jackson (12) showed that hypoxia inhibits norepinephrine-induced contraction of single arteriolar muscle cells. This effect of reduced PO2 was not related to membrane hyperpolarization or activation of outward potassium currents. However, the inhibitory effect of hypoxia could be reversed by agents or conditions that depolarized the arteriolar muscle cells. Therefore, it was hypothesized that reduced PO2 acted on, or through, a process that was voltage-sensitive.
Hypoxia inhibits currents through L-type calcium channels (CaL channels) in some vascular cells (4–6). Norepinephrine-induced contraction of single arteriolar muscle cells depends on Ca2+ influx through CaL channels (13). Thus, these channels could be the voltage-dependent target of action of hypoxia in arteriolar muscle cells, as previously proposed (12). Therefore, the purpose of the present study was to determine whether hypoxia inhibits norepinephrine-induced contraction of arteriolar muscle cells by inhibiting the function of CaL channels.
MATERIALS AND METHODS
Animal and Tissue Preparation
All animal use was approved by the Institutional Animal Care and Use Committee at Western Michigan University. Hamsters were killed by an overdose of pentobarbital (>150 mg/kg intraperitoneally) or by asphyxiation with CO2. Single arteriolar muscle cells from the cremaster muscles of male golden Syrian hamsters (weight: 80–160 g; Charles River Laboratories, Wilmington, MA) were isolated enzymatically, as previously described (13). Briefly, the cremaster muscles were removed, rinsed, and placed in a zero Ca2+ physiological salt solution (PSS) at 4 °C with the following composition: NaCl 140 mM; KCl 5 mM; MgCl2 1 mM; 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 10 mM; glucose 10 mM; pH 7.4 adjusted with NaOH; 295–300 mOsm. Cremaster muscles were transferred to a cooled (4 °C), water-jacketed dissection chamber (Radnoti Glass, Monrovia, CA) containing dissociation solution (DS; PSS containing 0.1 mM Ca2+, 10 μM diltiazem, 10 μM sodium nitroprusside, and 1% albumin). Cremaster muscles were pinned out flat with insect pins on pads made of Sylgard (Dow Corning, Midland, MI) that were placed in the bottom of the dissection chamber. Second-order and third-order arterioles were hand-dissected out of the muscle, cut into lengths of ~800 μm, and incubated in DS containing 26 U/mL papain and 1 mg/mL dithioerythritol at 37 °C for 35 minutes. The arteriolar segments then were incubated in 1 mL of DS containing 1.95 U/mL collagenase, 1 mg/mL trypsin inhibitor, and 75 U/mL elastase at 37 °C for 17–22 minutes. The vessel segments were washed in DS without enzymes, and single arteriolar muscle cells were dispersed by gentle trituration of the arteriolar segments with an Eppendorf-style pipettor. The cell isolate was placed in a 1.5-mL siliconized-polypropylene microcentrifuge tube and stored at room temperature for up to 4 hours. Aliquots of cells from the isolate then were placed in a chamber mounted on the stage of an Eclipse TE300 inverted microscope (Nikon, Tokyo, Japan), with the additional details for each specific set of experiments listed below. During all experiments, flow was maintained through the chamber by gravity from 60 mL reservoirs at a rate of 1–3 mL/minute with bath solutions specific for each protocol (see below). All experiments were performed at room temperature (20–25 °C).
Contraction Assays
The contractility of single arteriolar muscle cells in response to norepinephrine was assessed as previously described (13) with minor modifications. Small coverslips (1 cm2) were placed in the bottom of the chamber to allow the transfer of sets of cells into and out of the chamber. The coverslips were coated with a cell adhesive (Cell-Tak; Becton Dickinson, Franklin Lakes, NJ) to facilitate the adherence of cells to the coverslips. An aliquot (100 μl) of the cell isolate was placed on the coverslip, and cells were allowed to adhere for 10 minutes. After the cells adhered, the chamber was perfused with PSS containing 2 mM Ca2+ for 10 minutes. The contraction of single smooth muscle cells in response to micropipette-applied norepinephrine (10 μM) then was determined either during control conditions or during hypoxia (see below), as described previously (13). Briefly, a micropipette filled with norepinephrine was positioned adjacent to a cell with a micro-manipulator. Norepinephrine containing PSS was ejected from the pipette by pressurization with a water manometer. The micropipette was moved within the field of view by use of a micromanipulator. The micropipette and the field of view were both moved against the direction of chamber flow. This prevented cells that were not directly exposed to norepinephrine from contracting. At least 30 cells were tested from each aliquot, and cells were counted as contracting if they shortened >30% within 15 seconds after norepinephrine exposure. After determining the number of cells contracting during either the control condition or hypoxia, a second aliquot of cells was placed on a second Cell-Tak-coated cover-slip in the chamber, and the norepinephrine-dependent contractility then was determined for the remaining oxygen tension (only one condition was tested in each aliquot). Thus, if contractility under control conditions was studied in the first aliquot of cells, the contractility under hypoxia was determined in the second aliquot of cells. The order of the treatments was randomized to obviate time-dependent differences in reactivity, and only one oxygen tension was tested for each aliquot. Data were normalized to the fraction of cells that contracted in response to norepinephrine (i.e., the number that contracted divided by the total number of cells tested) under control conditions and were expressed as the percentage of the control, as described previously (13). Bath solutions were bubbled vigorously with room air (control PO2- 159 mm Hg) or with 100% N2 (hypoxia PO2: 10–15 mm Hg measured in the chamber), as previously described (12). Oxygen tensions were measured with a miniature Clark-type oxygen electrode (model # MI-730; Microelectrodes, Inc., Bedford, NH) at the beginning and end of our experiments. Oxygen tensions did not differ between these two time points, nor did they differ from values measured in previous experiments (12).
Patch Clamp Methods
An aliquot (approximately 100 μl) of cell-containing solution was placed in a 1-mL chamber mounted on the stage of an Eclipse TE300 (Nikon) inverted microscope. Cells were allowed to adhere to the bottom of the chamber for approximately 10 minutes. After a 10-minute wash period with 2 mM Ca2+-containing PSS, heat-polished borosilicate patch clamp pipettes (tip resistances: 2–5 MΩ when filled with pipette solution; see below) were placed on the membranes of single arteriolar muscle cells with a hydraulic micromanipulator. Micropipette tips were filled with the following solution: CsCl 143 mM; MgCl2 1 mM; ethylene glycolbis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) 0.5 mM; HEPES 10 mM; pH 7.2 with CsOH. The micropipettes were back-filled with the same solution also containing 50–240 μg/mL amphotericin B. High-resistance seals of >10 GΩ were obtained, and the perforated patch configuration was achieved using amphotericin B (to obviate cell dialysis) while observing increases in capacitative current. After obtaining stable seals, the chamber was perfused with a bath solution containing: tetraethylammonium chloride (TEACl) 10 mM; 4-aminopyridine 10 mM; NaCl 125 mM; BaCl2 10 mM; MgCl2 1 mM; HEPES 10 mM; and glucose 10 mM; pH 7.4 with CsOH. Ba2+ was used as the charge carrier to amplify currents through CaL channels and to obviate Ca2+-dependent run-down of the currents (18).
The pipette voltage was controlled and the current was measured with an Axopatch 200A amplifier controlled by a computer running pClamp 7 or pClamp 8 software (Axon Instruments, Forest City, CA). Signals were passed through a four-pole Bessel filter with a cutoff frequency of 1 KHz, digitized at 5 KHz, and stored on a computer hard drive for subsequent analysis. Whole-cell Ba2+ currents were normalized to cell capacitance to account for differences in cell size or changes in membrane area. Cell capacitance was estimated by integration of the capacitive transient generated by 10 mV hyperpolarizing pulses from the holding potential after electronic cancellation of pipette-patch capacitance. The mean cell capacitance was 18.0 ± 0.7 pF, and the mean access resistance was 10.0 ± 0.4 MΩ (n = 31).
Two different voltage-clamp protocols were used to assess the effects of hypoxia on CaL channels. In the first protocol, to determine the effect of hypoxia on the activation of CaL channels, cells were held at −70 mV. The membrane potential was stepped for 200 milliseconds from −90 to +60 mV (in increments of 10 mV), and the peak CaL channel currents were measured. In a second voltage-clamp protocol that was designed to study the steady-state inactivation of CaL channels, cells were held at −80 mV and were subjected to a conditioning pulse of 1000 milliseconds in duration (starting at −90 mV and increasing in increments of 10 mV up to +60 mV) to inactivate an increasing portion of the CaL channels. After the conditioning pulse, the membrane potential was stepped back to −80 mV for 20 milliseconds to deactivate any noninactivated channels before applying the test potential of +20 mV for 200 milliseconds. Peak CaL channel currents then were measured at the test potential of +20 mV (this test potential yielded maximal currents in the activation protocol; see RESULTS). In each cell, one of the voltage-clamp protocols was performed under control conditions (room air) and was repeated after 10 minutes of hypoxia. Ba2+ currents also were measured in these cells after recovery from hypoxia and did not differ from currents obtained during control conditions (data not shown).
Inactivation data were displayed as I/Imax, where Imax is the average maximum current amplitude elicited during the test pulse to +20 mV after conditioning potentials that caused no inactivation (i.e., potentials more negative than −40 mV). A Boltzmann distribution curve was fit to the data using the following equation: I/Imax = (1/[1 + exp(V0.5 − V)/k]) + C, where V0.5 is the conditioning potential resulting in 50% inactivation, V is the conditioning potential, k is the slope factor, and C is the noninactivating component.
Measurement of Intracellular Calcium
Aliquots of cells (100 μl) were placed onto Cell-Tak-treated coverslips that were placed in the bottom of the chamber. After allowing the cells to settle and adhere to the coverslips, the cells were loaded with 1 μM Fura 2-(acetyloxy) methyl ester (AM) (in 2 mM CaCl2 PSS with 0.05% dimethyl sulfoxide and 1% bovine serum albumin) for 30 minutes and then were washed for 30 minutes with 2 mM Ca2+-containing PSS.
Fura 2 fluorescence from single cells was measured using a Ratiomaster microscope-based photometry system equipped with a microscope photometer and a DeltaRam high speed multi-illuminator and shutter system (Photon Technologies, Inc., Lawrenceville, NJ). For fluorescence measurements, emission at 510 nm was sampled at 20 Hz for the excitation wavelengths of 340 and 380 nm.
After the subtraction of background fluorescence, the ratio of fluorescence emission for 340/380 nm illumination ([F340 − Background340]/[F380 − Background380] = R) was calculated and used as an index of [Ca2+]i. This was done rather than a calculation of actual [Ca2+]i because of inherent uncertainties in the conversion of the ratios to Ca2+ concentration using an in vitro calibration. [Ca2+]i was estimated for display purposes using the equation [Ca2+]i = Kd*β;* (R − Rmin)/(Rmax − R), where Rmin and Rmax are the minimum and maximum R values, and μ is the ratio of emission intensities at 380 nm in zero Ca2+/saturating Ca2+ as determined by in situ calibration. A Kd value of 235 nM, as determined by in vitro calibration of our system, was used in these calculations.
To test the hypothesis that hypoxia prevents the norepinephrine-induced contraction of single arteriolar muscle cells by inhibiting Ca2+ increases, cells were stimulated with micropipette-applied norepinephrine (10 μM) under control conditions and after 10 minutes of hypoxia, and then were stimulated a third time after cells were re-exposed to room air (recovery). Thus, the effects of all three conditions (i.e., control, hypoxia, and recovery from hypoxia) on norepinephrine-induced contractility were tested in each cell. Preliminary data indicated that the repeated application of norepinephrine under control conditions yielded similar Ca2+ transients. On some experimental days, up to four cells were examined for each isolate.
Materials
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) with the following exceptions: elastase was purchased from Calbiochem (La Jolla, CA); bovine serum albumin was purchased from USB (Cleveland, OH); and Fura 2-AM was purchased from Molecular Probes (Eugene, OR). Amphotericin B was dissolved in dimethylsulfoxide, and nifedipine was dissolved in 100% ethanol. All solutions were prepared using 18 MΩ reagent-grade water.
Statistical Analyses
The data are presented as the mean ± SE. Data were analyzed by paired Student’s t tests or analysis of variance where appropriate. If a treatment effect was found by analysis of variance, the means were compared by Student-Newman-Keuls post hoc analysis. Statistical comparisons were performed at the 95% confidence level. For the inactivation data, a Boltzmann distribution curve was fit using KaleidaGraph software (version 3.5; Synergy Software, Reading, PA).
RESULTS
Hypoxia Inhibits Norepinephrine-Induced Contraction
Hypoxia significantly inhibited the contraction of arteriolar muscle cells in response to 10 μM norepinephrine. In contraction assays (four isolates; a total of 412 cells were observed with 205 cells under control conditions and 207 during hypoxia), 83.3 ± 2.7% of the cells contracted in response to norepinephrine under control conditions, and this value was reduced to 56.1 ± 6.2% of cells during hypoxia (p < 0.05). Thus, hypoxia reduced the contractility of single arteriolar muscle cells to 67.0 ± 5.6% of the control (p < 0.05) (Fig. 1).
Figure 1.
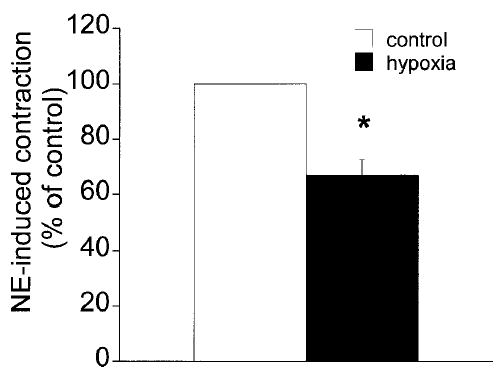
Norepinephrine-induced contraction of arteriolar muscle cells is inhibited by hypoxia. The data are given as the mean ± SE. The fraction of cells, expressed as a percentage of the control fraction, that contracted in response to micropipette-applied norepinephrine (10 μM; n = 4). * denotes significant difference from the control value (p < 0.05).
Hypoxia Does Not Affect CaL Currents in Arteriolar Muscle Cells
Barium currents elicited by stepping from a holding potential of −70 mV to test potentials from −90 mV to +60 mV are shown in Fig. 2. The currents activated at −40 mV and were maximal at +20 mV. Superfusion of the cells with dihyropyridine, nifedipine (1 μM) substantially (>90% inhibition) and reversibly inhibited the currents (Fig. 2A). Figure 2B shows a barium current trace before and during nifedipine application on depolarization of a cell to +20 mV. The voltage-dependent characteristics of these currents, along with the block by nifedipine, confirm that currents induced by step depolarizations were carried through CaL channels. No currents were seen during more negative test potentials (i.e., those expected to elicit T-type currents), either in the absence or presence of nifedipine, indicating that T-type calcium channels were absent in these cells (15).
Figure 2.
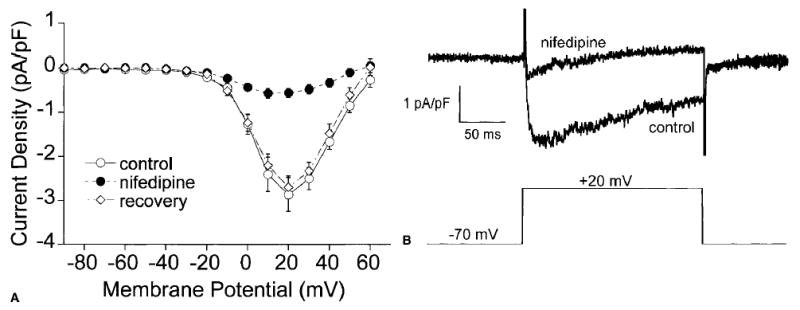
Nifedipine inhibits CaL currents in arteriolar muscle cells. The data are given as the mean ± SE. (A) The mean current-value (I-V) relationship (n = 8) for CaL channel currents measured at the peak of test pulses in the absence (○), presence (•), and after washout of 1 μM nifedipine (⋄). (B) Raw current traces (top) from a single smooth muscle cell in the presence and absence of nifedipine during a depolarization from −70 mV to +20 mV (bottom).
Hypoxia did not alter the activation of Ba2+ currents through CaL channels in any manner. Current amplitudes were similar at all voltages before and during superfusion of the cells with hypoxic solutions (Fig. 3A). Peak current densities at +20 mV were −3.83 ± 0.40 pA/pF before hypoxia and −3.97 ± 0.36 pA/pF during hypoxia (n = 15; p > 0.05). Neither did hypoxia affect the steady-state inactivation of CaL channels in single arteriolar muscle cells (Fig. 3B). Hypoxia had no effect on the currents measured, or on the current–voltage relationship in any of the cells tested. Under control conditions, the test potential resulting in 50% inactivation (V0.5) was −4.6 ± 1.4 mV, a value that was not significantly different from the value of −2.9 ± 1.9 mV that was observed in the same cells during hypoxia (n = 8; p > 0.05).
Figure 3.
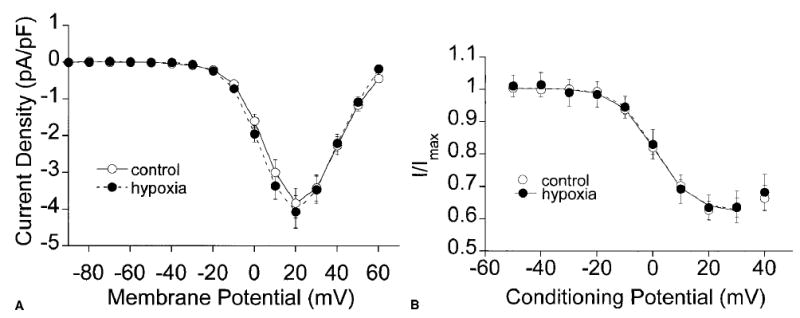
Hypoxia does not inhibit currents through CaL channels in arteriolar muscle cells. All data are given as the mean ± SE. (A) Mean current–voltage relationship (n = 15) of CaL channel currents during the activation protocol before (○;) and during hypoxia (•). (B) Normalized inactivation (n = 8) of CaL in arteriolar smooth muscle cells before (○) and during hypoxia (•).
Hypoxia Does Not Inhibit Norepinephrine-Induced Calcium Changes in Arteriolar Muscle Cells
A representative trace of norepinephrine-induced changes in Fura 2 fluorescence during control conditions is shown in Fig. 4. Similar to the responses of other smooth muscles (25,26), the norepinephrine-induced calcium transients in arteriolar smooth muscle cells could be described by the following three components: a baseline, followed by a peak on application of norepinephrine, and a plateau that was maintained until norepinephrine removal. The Fura 2 ratio returned to baseline values on norepinephrine removal.
Figure 4.
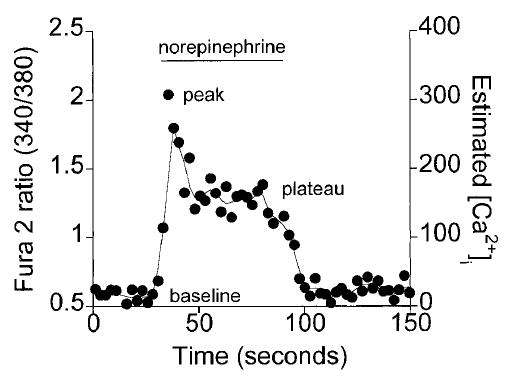
Norepinephrine-induced Ca2+ transient in an arteriolar muscle cell, as indicated by a representative Fura 2 ratio trace. The Fura 2 ratio changes in response to 60 seconds of norepinephrine were characterized by a pre-norepinephrine baseline, a peak on norepinephrine application, followed by a maintained plateau until norepinephrine removal.
Hypoxia did not inhibit norepinephrine-induced changes in the Fura 2 ratio in arteriolar muscle cells (Fig. 5). Instead, exposure to hypoxia slightly augmented the norepinephrine-induced changes in the ratio. The mean control Fura 2 ratio values for the baseline, peak, and plateau during norepinephrine exposure were 0.65 ± 0.03, 2.22 ± 0.13, 1.21 ± 0.06, respectively (21 cells), whereas during hypoxia the values for the baseline, peak, and plateau were 0.73 ± 0.02, 2.83 ± 0.21, and 1.38 ± 0.07, respectively (p < 0.05). On returning to control conditions (recovery), the mean baseline ratio (0.68 ± 0.02) and the mean norepinephrine-induced plateau (1.24 ± 0.07) returned to values that were not significantly different from the control value. However, the norepinephrine-induced peak remained elevated (2.73 ± 0.22; p < 0.05 versus control). It should be noted that in all of the cells tested, hypoxia either augmented (16 cells) or did not change (5 cells) the norepinephrine-induced Fura 2 ratios. In none of the cells tested did hypoxia inhibit the norepinephrine-induced changes in the Fura 2 ratio.
Figure 5.
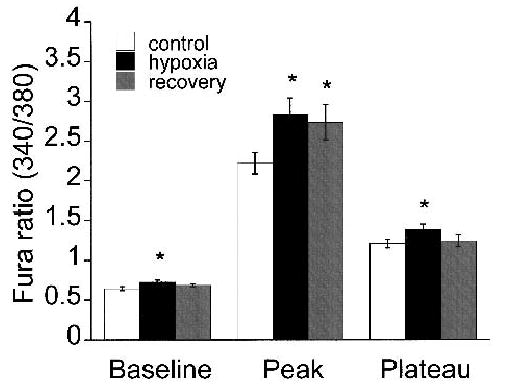
Hypoxia does not inhibit norepinephrine-induced Ca2+ in arteriolar muscle cells. All data are given as the mean ± SE. Summary (21 cells) of the baseline, peak, and plateau Fura 2 ratios from arteriolar muscle cells in response to norepinephrine before (white bars), during (black bars), and after recovery (gray bars) from hypoxia. * denotes a significant difference from control value (p < 0.05).
It has been reported that hypoxia may lead to an increase in the Fura 2 ratio by eliminating O2-induced quenching of Fura 2 fluorescence (23). To test whether changes in PO2 may alter the Fura 2 ratio independent from changes in intracellular Ca2+, single arteriolar muscle cells were loaded with Fura 2-AM (see METHODS) and permeabilized with 10 μM ionomycin, and the extracellular Ca2+ concentration was set at 500 nM (to fix [Ca2+]i at the same level). Fura 2 fluorescence remained unaltered while changing from room air to hypoxia and then back to room air (data not shown). These data suggest that changes in oxygen tension did not directly affect Fura 2 fluorescence. Thus, norepinephrine-induced changes in the Fura 2 ratio were due to changes in [Ca2+]i and likely were not due to a direct effect of the changes in oxygen tension.
DISCUSSION
This study demonstrates that hypoxia, although inhibiting the contractility of arteriolar muscle cells, did not inhibit currents through CaL channels, nor their voltage-dependent activation or inactivation. Hypoxia also did not inhibit [Ca2+]i changes elicited by norepinephrine. Thus, the inhibitory effects of hypoxia on norepinephrine-induced contraction must occur at steps downstream from norepinephrine-induced Ca2+ mobilization.
Wu et al. (27) reported that rat cremasteric arteriolar muscle cells display Ba2+ currents that seemed to be carried through CaL channels. Our findings confirm and extend these observations. We found that Ba2+ currents in hamster cremasteric arteriolar muscle cells were inhibited by nifedipine and were voltage-dependent, activating at −40 mV with peak amplitudes at +20 mV. These characteristics are consistent with the expression of high-voltage-activated, dihydropyridine-sensitive CaL channels (15). In addition, we report here for the first time the steady-state voltage-dependent inactivation of CaL channels in hamster cremasteric arteriolar muscle cells. Inactivation began at −20 mV, with half-maximal inactivation occurring at −4.6 ± 1.4 mV. These data are similar to those seen in other studies of vascular smooth muscle when 10 mM barium was used as the charge carrier (21,22).
CaL channel current amplitudes, and voltage-dependent activation and inactivation were unaffected by hypoxia. These data are in direct opposition to those from several studies in which hypoxia was found to reversibly inhibit CaL channels in smooth muscle from conduit arteries (5,6) and in cardiac myocytes (4). However, our data are in agreement with the findings of Tateishi and Faber (24), who found that hypoxia did not reverse constrictions induced by the direct activation of CaL channels with KCl or the CaL channel agonist SDZ-202-791 in rat cremaster arterioles. It may be that the CaL channels expressed in arteriolar muscle cells differ somehow from those expressed in larger vessels or elsewhere in the cardiovascular system. Nonetheless, our data suggest that the inhibitory effect of hypoxia on norepinephrine-induced contraction in arteriolar muscle cells does not involve the hypoxia-induced inhibition of currents through CaL channels.
The application of norepinephrine to single arteriolar smooth muscle cells induced calcium changes that were similar to those seen in other smooth muscle cells (25,26). Hypoxia did not inhibit these Ca2+ transients in any of the cells tested. Instead, we found that hypoxia increased resting Ca2+ levels and augmented norepinephrine-induced increases in intracellular Ca2+ in most of the cells. These observations suggest that the inhibition of norepinephrine-induced contraction produced by hypoxia must occur downstream from norepinephrine-induced mobilization of Ca2+ and suggest that hypoxia decreased the Ca2+ sensitivity of the arteriolar muscle cells.
There are a number of studies indicating that hypoxia can desensitize vascular smooth muscle to agonist-induced increases in [Ca2+]i (i.e., the Ca2+ response is not changed, but the agonist-induced contraction is inhibited). In rat cerebral and small mesenteric arteries, hypoxia reduced arginine vasopressin-induced force, but [Ca2+]i changes were essentially unchanged (1). In porcine coronary arteries, hypoxia inhibited force but not increases in [Ca2+]i in response to KCl or the thromboxane analog U46619 (19). Hypoxia caused a significant dilation in pressurized cerebral arteries without decreasing [Ca2+]i (9), suggesting that, in addition to agonist-induced tone, pressure-induced vascular tone may also be inhibited by hypoxia without a reduction in [Ca2+]i. In other nonvascular smooth muscle, hypoxia also inhibits agonist-induced force production without inhibiting [Ca2+]i changes (17).
Taken together, these data suggest that hypoxia can inhibit force in smooth muscle cells independent of decreases in intracellular calcium. Our finding that norepinephrine-induced [Ca2+]i changes in arteriolar muscle cells were not reduced despite a reduction in norepinephrine-dependent contractility is consistent with the above findings. The mechanisms responsible for the hypoxia-induced change in Ca2+ sensitivity remain to be established.
In conclusion, hypoxia inhibits norepinephrine-induced contraction of smooth muscle cells isolated from skeletal muscle arterioles. However, this effect cannot be attributed to a reduction in Ca2+ mobilization because hypoxia did not inhibit CaL channel currents, nor did hypoxia inhibit norepinephrine-induced Ca2+ transients. These findings suggest that hypoxia may inhibit the contraction of arteriolar muscle cells by exerting effects downstream from the increases in Ca2+ such that the cells become desensitized to [Ca2+]i.
Acknowledgments
The authors are grateful for the superb technical assistance of Sue Kovats.
Footnotes
Supported by Public Health Service grant HL 32469 to Dr. Jackson and American Heart Association Postdoctoral Fellowship 0120607Z to Dr. Cohen.
References
- 1.Aalkjaer C, Lombard JH. Effect of hypoxia on force, intracellular pH and Ca2+ concentration in rat cerebral and mesenteric small arteries. J Physiol. 1995;482:409–419. doi: 10.1113/jphysiol.1995.sp020528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bongard O, Bounameaux H, Fagrell B. Effects of oxygen inhalation on skin microcirculation in patients with peripheral arterial occlusive disease. Circulation. 1992;86:878–886. doi: 10.1161/01.cir.86.3.878. [DOI] [PubMed] [Google Scholar]
- 3.Bredle DL, Bradley WE, Chapler CK, Cain SM. Muscle perfusion and oxygenation during local hyperoxia. J Appl Physiol. 1988;65:2057–2062. doi: 10.1152/jappl.1988.65.5.2057. [DOI] [PubMed] [Google Scholar]
- 4.Fearon IM, Palmer AC, Balmforth AJ, Ball SG, Varadi G, Peers C. Modulation of recombinant human cardiac L-type Ca2+ channel α1C subunits by redox agents and hypoxia. J Physiol. 1999;514:629–637. doi: 10.1111/j.1469-7793.1999.629ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franco-Obregon A, Lopez-Barneo J. Low PO2 inhibits calcium channel activity in arterial smooth muscle cells. Am J Physiol. 1996;271:H2290–H2299. doi: 10.1152/ajpheart.1996.271.6.H2290. [DOI] [PubMed] [Google Scholar]
- 6.Franco-Obregon A, Urena J, Lopez-Barneo J. Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc Natl Acad Sci U S A. 1995;92:4715–4719. doi: 10.1073/pnas.92.10.4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frisbee JC. Impaired dilation of skeletal muscle microvessels to reduced oxygen tension in diabetic obese Zucker rats. Am J Physiol. 2001;281:H1568–H1574. doi: 10.1152/ajpheart.2001.281.4.H1568. [DOI] [PubMed] [Google Scholar]
- 8.Frisbee JC, Roman RJ, Krishna UM, Falck JR, Lombard JH. Relative contributions of cyclooxygenase- and cytochrome P450 ω-hydroxylase-dependent pathways to hypoxic dilation of skeletal muscle resistance arteries. J Vasc Res. 2001;38:305–314. doi: 10.1159/000051061. [DOI] [PubMed] [Google Scholar]
- 9.Gebremedhin D, Bonnet P, Greene AS, England SK, Rusch NJ, Lombard JH, Harder DR. Hypoxia increases the activity of Ca2+-sensitive K+ channels in cat cerebral arterial muscle cell membranes. Pflugers Arch. 1994;428:621–630. doi: 10.1007/BF00374586. [DOI] [PubMed] [Google Scholar]
- 10.Jackson WF. Arteriolar oxygen reactivity is inhibited by leukotriene antagonists. Am J Physiol. 1989;257:H1565–H1572. doi: 10.1152/ajpheart.1989.257.5.H1565. [DOI] [PubMed] [Google Scholar]
- 11.Jackson WF. Arteriolar oxygen reactivity: Where is the sensor? Am J Physiol. 1987;253:H1120–H1126. doi: 10.1152/ajpheart.1987.253.5.H1120. [DOI] [PubMed] [Google Scholar]
- 12.Jackson WF. Hypoxia does not activate ATP-sensitive K+ channels in arteriolar muscle cells. Microcirculation. 2000;7:137–145. [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson WF, Huebner JM, Rusch NJ. Enzymatic isolation and characterization of single vascular smooth muscle cells from cremasteric arterioles. Microcirculation. 1997;4:35–50. doi: 10.3109/10739689709148316. [DOI] [PubMed] [Google Scholar]
- 14.Lombard JH, Kunert MP, Roman RJ, Falck JR, Harder DR, Jackson WF. Cytochrome P-450 ω;-hydroxylase senses O2 in hamster muscle, but not cheek pouch epithelium, microcirculation. Am J Physiol. 1999;276:H503–H508. doi: 10.1152/ajpheart.1999.276.2.H503. [DOI] [PubMed] [Google Scholar]
- 15.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 16.Messina EJ, Sun D, Koller A, Wolin MS, Kaley G. Role of endothelium-derived prostaglandins in hypoxia-elicited arteriolar dilation in rat skeletal muscle. Circ Res. 1992;71:790–796. doi: 10.1161/01.res.71.4.790. [DOI] [PubMed] [Google Scholar]
- 17.Obara K, Bowman PS, Ishida Y, Paul RJ. Effects of hypoxia on [Ca2+]i, pHi and myosin light chain phosphorylation in guinea-pig taenia caeci. J Physiol. 1997;503:427–433. doi: 10.1111/j.1469-7793.1997.427bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuhmann K, Romanin C, Baumgartner W, Groschner K. Intracellular Ca2+ inhibits smooth muscle L-type Ca2+ channels by activation of protein phosphatase type 2B and by direct interaction with the channel. J Gen Physiol. 1997;110:503–513. doi: 10.1085/jgp.110.5.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu S, Bowman PS, Thorne G, III, Paul RJ. Effects of hypoxia on isometric force, intracellular Ca2+, pH, and energetics in porcine coronary artery. Circ Res. 2000;86:862–870. doi: 10.1161/01.res.86.8.862. [DOI] [PubMed] [Google Scholar]
- 20.Skinner MR, Marshall JM. Studies on the roles of ATP, adenosine and nitric oxide in mediating muscle vasodilatation induced in the rat by acute systemic hypoxia. J Physiol. 1996;495:553–560. doi: 10.1113/jphysiol.1996.sp021615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smirnov SV, Aaronson PI. Ca2+ currents in single myocytes from human mesenteric arteries: Evidence for a physiological role of L-type channels. J Physiol. 1992;457:455–475. doi: 10.1113/jphysiol.1992.sp019387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smirnov SV, Knock GA, Belevych AE, Aaronson PI. Mechanism of effect of extracellular pH on L-type Ca2+ channel currents in human mesenteric arterial cells. Am J Physiol. 2000;279:H76–H85. doi: 10.1152/ajpheart.2000.279.1.H76. [DOI] [PubMed] [Google Scholar]
- 23.Stevens T, Fouty B, Cornfield D, Rodman DM. Reduced PO2 alters the behavior of Fura-2 and Indo-1 in bovine pulmonary artery endothelial cells. Cell Calcium. 1994;16:404–412. doi: 10.1016/0143-4160(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 24.Tateishi J, Faber JE. Inhibition of arteriole α2- but not α1-adrenoceptor constriction by acidosis and hypoxia in vitro. Am J Physiol. 1995;268:H2068–H2076. doi: 10.1152/ajpheart.1995.268.5.H2068. [DOI] [PubMed] [Google Scholar]
- 25.Tognarini DP, Moulds RF. Intracellular Ca2+ and contractile responses to α1-adrenoceptor subtype activation in rat aortic vascular smooth muscle. Eur J Pharmacol. 1997;322:31–36. doi: 10.1016/s0014-2999(96)00978-8. [DOI] [PubMed] [Google Scholar]
- 26.Vandeputte C, Giummelly P, Atkinson J, Delagrange P, Scalbert E, Capdeville-Atkinson C. Melatonin potentiates NE-induced vasoconstriction without augmenting cytosolic calcium concentration. Am J Physiol. 2001;280:H420–H425. doi: 10.1152/ajpheart.2001.280.1.H420. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by αvβ3 and α5β1 integrin ligands. J Cell Biol. 1998;143:241–252. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]