

Abstract
Objective
The mechanism by which elevated extracellular potassium ion concentration ([K+]o) causes dilation of skeletal muscle arterioles was evaluated.
Methods
Arterioles (n = 111) were hand-dissected from hamster cremaster muscles, cannulated with glass micropipettes and pressurized to 80 cm H2O for in vitro study. The vessels were superfused with physiological salt solution containing 5 mM KCl, which could be rapidly switched to test solutions containing elevated [K+]o and/or inhibitors. The authors measured arteriolar diameter with a computer-based diameter tracking system, vascular smooth muscle cell membrane potential with sharp micropipettes filled with 200 mM KCl, and changes in intracellular Ca2+ concentration ([Ca2+]i) with Fura 2. Membrane currents and potentials also were measured in enzymatically isolated arteriolar muscle cells using patch clamp techniques. The role played by inward rectifier K+ (KIR) channels was tested using Ba2+ as an inhibitor. Ouabain and substitution of extracellular Na+ with Li+ were used to examine the function of the Na+/K+ ATPase.
Results
Elevation of [K+]o from 5 mM up to 20 mM caused transient dilation of isolated arterioles (27 ± 1 μm peak dilation when [K+]o was elevated from 5 to 20 mM, n = 105, p < .05). This dilation was preceded by transient membrane hyperpolarization (10 ± 1 mV, n = 23, p < .05) and by a fall in [Ca2+]i as indexed by a decrease in the Fura 2 fluorescence ratio of 22 ± 5% (n = 4, p < .05). Ba2+ (50 or 100 μM) attenuated the peak dilation (40 ± 8% inhibition, n = 22) and hyperpolarization (31 ± 12% inhibition, n = 7, p < .05) and decreased the duration of responses by 37 ± 11% (n = 20, p < 0.05). Both ouabain (1 mM or 100 μM) and replacement of Na+ with Li+ essentially abolished both the hyperpolarization and vasodilation.
Conclusions
Elevated [K+]o causes transient vasodilation of skeletal muscle arterioles that appears to be an intrinsic property of the arterioles. The results suggest that K+-induced dilation involves activation of both the Na+/K+ ATPase and KIR channels, leading to membrane hyperpolarization, a fall in [Ca2+]i, and culminating in vasodilation. The Na+/K+ ATPase appears to play the major role and is largely responsible for the transient nature of the response to elevated [K+]o, whereas KIR channels primarily affect the duration and kinetics of the response.
Keywords: arterioles, ATP-sensitive potassium channels, barium ions, calcium ions, Fura 2, inward-rectifier potassium channels, membrane potential, Na+, K+ ATPase, ouabain, potassium channels, potassium ions, skeletal muscle, vasodilation
INTRODUCTION
Skeletal muscle contraction leads to large increases in blood flow to the active muscle through dilation of resistance arteries and arterioles (32). However, the mechanisms underlying this active or functional hyperemia remain poorly understood. Potassium ions released from active skeletal muscle have been implicated in this functional hyperemia for over 60 years (10,34). Numerous studies have shown that elevated extracellular concentration of K+ ([K+]o) can cause vasodilation and that the K+ released from skeletal muscle fibers during the repolarization phase of muscle action potentials can accumulate in the interstitial space at concentrations sufficient to cause vasodilation (10,18,30,34). Nonetheless, the cellular mechanisms by which the vascular smooth muscle cells in the walls of skeletal muscle arterioles relax to cause vasodilation in response to elevated [K+]o remain unclear. Early studies suggested that elevated [K+]o caused vasodilation by activation of the electrogenic Na+/K+ ATPase and transient membrane hyperpolarization (10,16,35). Recent in vivo studies of rat cremaster muscle microcirculation support this hypothesis (20). In contrast, Loeb et al. (19) suggested that K+-induced dilation in the same tissue involves activation of inward rectifier K+ [KIR] channels. This latter hypothesis is supported by a number of recent in vitro studies, performed in small cerebral (17,27,28,39) and coronary (17,31) arteries. The purpose of the present study was to (1) determine the effects of elevated [K+]o on the diameter of cannulated arterioles isolated from hamster cremaster muscles as an in vitro model system to study the mechanism of action of elevated [K+]o on skeletal muscle arterioles and (2) evaluate the roles played by the Na+/K+ ATPase and KIR channels in dilation of skeletal muscle arterioles in response to elevated [K+]o.
MATERIALS AND METHODS
Animals and Tissue Preparation
All animal use was approved by the Institutional Animal Care and Use Committee at Western Michigan University and was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council (USA). Male golden Syrian hamsters (80–150 g, Charles River Laboratories, Wilmington, MA) were euthanized with sodium pentobarbital (>150 mg/kg body weight, ip) or by CO2 asphyxiation and their right testicles and surrounding cremaster muscles were removed and washed in cold Ca2+-free physiological salt solution (PSS: 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, pH 7.4). The tissue was then placed in a water-jacketed dissection chamber maintained at 4°C, filled with Ca2+-free PSS containing 0.1% bovine serum albumin (BSA), sodium nitroprusside (10 μM), and diltiazem (10 μM). The low Ca2+ concentration and the vasodilators helped to prevent vasospasm that may occur during dissection. The cremaster muscle then was dissected from the testicle and pinned to a Sylgard 184 (Dow Corning Corp., Midland, MI) pad on the bottom of the chamber. Second-order arterioles, 1–2 mm long and devoid of side branches, then were isolated by hand dissection with the aid of a stereomicroscope. The vessel segments were excised and transferred to a custom-built lexan cannulation chamber (0.7 mL) using a Wiretrol pipettor (50–100 μL; Drummond Scientific, Broomall, PA) pretreated with 10% BSA to reduce adhesion of the isolated vessels to the glass barrel of the pipettor. The cannulation chamber was filled with Ca2+-free PSS containing 0.1% BSA, diltiazem (10 μM) and sodium nitroprusside (10 μM) at room temperature and its bottom was formed from a No. 1 coverslip.
Arteriole Cannulation
Arterioles were cannulated using glass micropipettes with tip outer diameters of 30–50 μm, filled with PSS containing CaCl2 (1.8 mM). Micropipettes were formed from glass tubing (borosilicate capillary tubing, Warner Instruments, Hamden, CT) using a pipette-puller and a microforge. The ends of the pipettes were fire polished and bent to a 45° angle such that when positioned in the chamber, the last 3–5 mm of the pipettes were parallel to the bottom of the chamber. The pipettes were mounted in commercially available pipette holders with side ports (Warner Instruments, Hamden, CT) that allowed pressurization of the pipette lumens. These holders were mounted on small, 3-axis micromanipulators (MT-XYZ, Newport, Irvine, CA) so that the tips of the pipettes could be positioned in the cannulation chamber in 3 dimensions. With the aid of a stereomicroscope, one end of an arteriole was gently pulled onto a pipette tip and secured with 2 strands of 11–0 nylon monofilament (Ashaway Line and Twine, Ashaway, RI), using sharpened #5 Dumont forceps. The pipette lumen was pressurized to 8 cm H2O to flush blood from the vessel lumen. The other end of the arteriole then was cannulated and tied in place with a second pair of monofilament knots.
The cannulation chamber was transferred to the stage of an inverted microscope when only diameter or diameter and intracellular Ca2+ were measured, or to the stage of a Leica DMLFS/A upright microscope equipped with a Gibraltar fixed stage (Burleigh Instruments, Fishers, NY) and a long working distance, electrically isolated, water immersion objective when diameter and membrane potential were measured. Arterioles were pressurized to 80 cm H2O with no flow through their lumens and superfused with room-temperature PSS, and the distance between the 2 pipette tips was adjusted to approximate the in vivo length of the arterioles. The vessels were tested for leaks by closing the stopcock between the vessel and the fluid column and monitoring diameter for several minutes. If a diameter change was noted (indicating a leak in the vessel), the experiment was terminated and a new vessel selected. If no leaks were detected, then the pressure was reduced to 20 cm H2O and the bath was slowly warmed to 34°C with a continuous flow (2 mL/min) of 1.8 mM CaCl2-containing PSS. The solution flowed by gravity from a heated bath through heated tubing into the chamber, and bath temperature was monitored by a temperature probe placed near the bath outflow port. Heated solution continually flowed in one end of the chamber and was aspirated from the other end by suction.
Vessels were allowed to reach physiological temperature and equilibrate under no-luminal-flow conditions for 30–60 min at 20 cm H2O. The pressure was then increased to 70–80 cm H2O and the arterioles allowed to stabilize for an additional 15–30 min. Vessels used in this study all displayed substantial myogenic tone and myogenic reactivity as assessed by the reduction in diameter initiated by the increased lumenal pressure and by their dilation in response to elevated extracellular potassium and other dilators (see Results for more information). Cannulated vessels remained physiologically viable for at least 4–5 h under these conditions. At the end of all experiments, arterioles were superfused with Ca2+-free PSS at room temperature so that vessel maximal diameter could be determined.
In a subset of vessels, we damaged/removed endothelial cells by perfusion of the arterioles with an air bubble as has been used by others (8). After recording control responses, 0.5 mL of air was injected into the tubing connected to the upstream pipette. The stopcock occluding the outflow from the downstream pipette was then opened, allowing the air bubble to perfuse the lumen of the arteriole. After passage of the air bubble, the vessels were allowed to regain tone before being restimulated with 20 mM [K+]o. If tone was not reestablished, the vessels were briefly exposed to 10 μM norepinephrine. After washout of the norepinephrine, if tone was not reestablished, the experiment was terminated. Efficacy of endothelial cell removal/damage was verified by loss of dilation induced by acetylcholine (see Results).
Measurement of Diameter
Arterioles were transilluminated and visualized using either 20× or 40× long working distance objectives. Images of arterioles were captured by video cameras and the outputs were displayed using Diamtrak software (T.O. Neild, Flinders University, Adelaide, Australia) (24) and associated video capture cards on Windows compatible computers. The Diamtrak systems allowed measurement of vessel diameter at >10 Hz and output continuous analog signals proportional to diameter with an accuracy of ±1–2 μm. The analog signals from the Diamtrak systems were then recorded on computer-based data acquisition systems for later analysis and display.
Measurement of Membrane Potential
Membrane potential of smooth muscle cells in the walls of a subset of arterioles was measured using sharp glass microelectrodes. Electrodes were pulled on a Flaming-Brown-style pipette puller (P-97, Sutter Instruments) from borosilicate glass tubing containing a glass filament (1 mm O.D., World Precision Instruments, Sarasota, FL). The pipettes were filled with 200 mM KCl and had tip resistances of 200–300 MΩ. They were connected to the head-stage of a microelectrode amplifier (Warner Instruments) by a silver wire inserted into the back of the pipette via a Lucite pipette holder. The head-stage and electrode were mounted on a 3-way hydraulic manipulator that, in turn, was mounted on a course mechanical micromanipulator. The reference electrode was a Ag-AgCl pellet placed in the outflow portion of the chamber. The vessels were isolated from the toxic effects of the reference electrode (14) by the unidirectional flow pattern in the chamber. The output of the microelectrode amplifier was recorded simultaneously with diameter signals with the use of a MacLab (ADInstruments, Colorado Springs, CO) data acquisition system. Successful impalements were signified by a rapid development of a negative potential that was stable for at least 15 s and returned to within a few millivolts of baseline upon withdrawal of the electrode from the cell. The baseline potential before impalement and after withdrawal were averaged and used to determine membrane potential.
When membrane potential was measured, arterioles were stretched over a small rectangle of Sylgard glued to the bottom of the chamber to stabilize the vessels and to facilitate cell impalement. To verify that we recorded from smooth muscle cells in some experiments, the tips of pipettes were filled with 200 mM KCl containing 10 mM Alexa Fluor 488. This highly fluorescent dye served as an excellent marker of cell impalement and appeared to label only single smooth muscle cells as others have reported using different dyes (7,38). We observed that if we approached the arterioles with the electrode oriented along their long axis, near the edge of the vessel, and were gentle in our insertion, that essentially all impalements were smooth muscle cells. We found it very difficult to impale endothelial cells from this approach unless we inserted the electrode near the midline of the vessel and drove the electrode through the wall of the arteriole and hit the endothelial cells on the opposite side. In a typical experiment, membrane potential was measured in 2–4 cells in the absence and presence of inhibitors and then the results of these multiple impalements were averaged and used as a single value for any given experiment.
Measurement of Changes in Intracellular Calcium
After arterioles were cannulated, warmed, pressurized, and equilibrated, the smooth muscle cell layer was preferentially loaded (23) with 5 μM Fura 2 AM (Molecular Probes, Eugene, OR) in 0.5% dimethyl sulfoxide (DMSO) and 0.1% BSA in PSS for 60 min. After the incubation period, vessels were washed for 30 min to allow for deesterification of the Fura 2 AM.
Fura 2 fluorescence from the smooth muscle cell layer was measured using a Ratiomaster microscope-based photometry system, which included a TE300 inverted microscope (Nikon, Tokyo, Japan), a D-104 photometer, and a DeltaRam high-speed multi-illuminator and shutter system controlled by FeliX 1.42 software (Photon Technologies, Lawrenceville, NJ) essentially as described previously (3). For fluorescence measurements, emission at 510 nm was sampled at 20 Hz for the excitation wavelengths of 340 and 380 nm.
Arteriolar background fluorescence was measured before vessels were loaded with Fura 2 AM. Background fluorescence was subtracted from fluorescence measurements of Fura 2 loaded arterioles and the ratio of fluorescence emission for 340/380 nm illumination [(F340 − Background340)/(F380 − Background380) = R] was calculated and used as an index of [Ca2+]i (9). We expressed our Fura 2 measurements as ratios and did not convert them to actual [Ca2+]i because of inherent uncertainties in conversion of the ratios to Ca2+ concentration using an in vitro calibration (36).
In all experiments, the effect of 20 mM KCl was tested before loading the arteriolar smooth muscle with Fura 2 AM. We found no differences in baseline tone or responses to 20 mM KCl, suggesting that loading the smooth muscle with Fura 2 did not alter tone or responsiveness to 20 mM KCl (data not shown).
Isolation of Single Arteriolar Muscle Cells
Single arteriolar muscle cells were isolated enzymatically as previously described (15). Second-order arterioles were hand-dissected out of the muscle as described above, cut into lengths of ~800 μm, and incubated in dissociation solution (DS, Ca2+-free PSS containing 0.1% BSA, 10 μM diltiazem, 10 μM sodium nitroprusside) containing 26 U/mL papain and 1 mg/mL dithioerythritol at 37°C for 35 min. The arteriolar segments then were incubated in 1 mL of DS containing 1.95 U/mL collagenase, 1 mg/mL trypsin inhibitor, and 75 U/mL elastase at 37°C for 17–22 min. The vessel segments were washed in DS without enzymes and single arteriolar muscle cells were dispersed by gentle trituration of the arteriolar segments with a 1000-μL Eppendorf-style pipette. The cell isolate was placed in a 1.5-mL siliconized-polypropylene microcentrifuge tube and stored at room temperature for up to 4 h. Aliquots of cells from the isolate were placed in a chamber mounted on the stage of a Nikon Eclipse TE300 inverted microscope with additional detail for each specific set of experiments listed below. During all experiments, flow was maintained through the chamber by gravity from 60-mL reservoirs at a rate of 1–3 mL/min with bath solutions specific for each protocol (see below). All experiments were performed at room temperature (20–25°C).
Patch Clamp Methods
Methods are similar to those previously used in this laboratory (12,15). Heat-polished, borosilicate patch pipettes (tip resistances = 2–5 MΩ when filled with pipette solution, see below) were placed on the membranes of single arteriolar cells with the aid of an hydraulic micromanipulator. Application of negative pressure to the back of the pipettes yielded high-resistance seals (>10 GΩ ) and the whole-cell configuration was obtained using amphotericin B (240 μg/mL) to perforate the cell-attached patch (29). Access was observed by monitoring increases in capacitive current induced by 10 mV hyperpolarizing pulses from a holding potential of −60 mV. The pipette voltage (or current) was controlled and current (or pipette potential) was measured with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA). The amplifier was controlled and signals were acquired by a computer equipped with a Digidata 1200a DMA interface and running p-Clamp or Axoscope software (Axon Instruments, Foster City, CA). Signals were passed through a 4-pole Bessel filter with a cutoff frequency of 1 kHz, digitized at 5 kHz, and stored on the computer’s hard disk for later analysis. All whole-cell currents were normalized to cell capacitance estimated by integration of the capacitive transient generated by 10 mV hyperpolarizing pulses (filtered at 5 kHz, sampled at 25 kHz) after electronic cancellation of pipette-patch capacitance. The pipette solution contained 100 mM K-aspartate, 43 mM KCl, 1 mM MgCl2, 10 mM HEPES, and 0.5 mM EGTA, pH 7.0, using NaOH.
Currents through KIR channels were recorded using 500 ms ramp protocols, where cells were stepped from a holding potential of −60 to −100 mV and then increased in a linear fashion from −100 to 0 mV over a 500 ms interval. Alternatively, step protocols were employed where cells were held at −60 mV and then stepped, in 10-mV increments, to test potentials from −90 to 0 mV for 400 ms. Average currents recorded during the last 100 ms were used for analysis. Similar results were obtained with both methods. Typically, 3 protocols were performed under each experimental condition, and the results averaged for analysis and display.
Materials
Except as noted, all drugs and chemicals were obtained from Sigma (St. Louis, MO). Elastase was obtained from Calbiochem (San Diego, CA) and BSA was obtained from USB (Amersham, Cleveland, OH). Solutions with elevated [K+] were made by equimolar substitution of KCl for NaCl. The osmolarity of all solutions was adjusted to 295 mOsm using sucrose as the osmolyte, as needed. Cromakalim and glibenclamide were dissolved in DMSO at a concentration of 10 mM and then diluted in PSS to their final concentrations. Ouabain was dissolved directly in warm PSS at the concentration used.
Statistics
Data are reported as means ± standard errors and n values represent the number of arterioles studied. Statistical significance was determined using Student’s t tests (2 groups) or analysis of variance (more than 2 groups) as appropriate with post hoc analysis performed using the Student–Newman–Keuls test (26). All comparisons were performed at the 95% confidence level.
RESULTS
Isolated Hamster Cremasteric Arterioles
A total of 111 arterioles were studied. Their maximum diameters were 106 ± 2 μm and all developed substantial resting tone when pressurized: resting diameters were 59 ± 1 μm, 56 ± 1% of their maximal diameters (p < .05 compared to maximum diameters). This tone appeared to be largely dependent on Ca2+ influx through L-type, voltage-gated Ca2+ channels as it was eliminated by exposure of the vessels to the Ca2+ channel blocker diltiazem (10 μM): in 6 arterioles with resting diameters of 71 ± 5 μm, diltiazem (10 μM) dilated the vessels to 106 ± 7 μm, a diameter that was not significantly different from that observed in the absence of external Ca2+ (113 ± 6 μm, p > .05).
Elevated [K+]o Dilates Isolated Cremasteric Arterioles
Increasing superfusate [K+] from 5 mM to 8, 12.5, or 20 mM caused concentration-dependent dilation of cannulated arterioles (Figure 1). Elevation of [K+]o from 5 to 20 mM produced the maximal and most consistent response. Therefore, all subsequent experiments were performed using this concentration.
Figure 1.
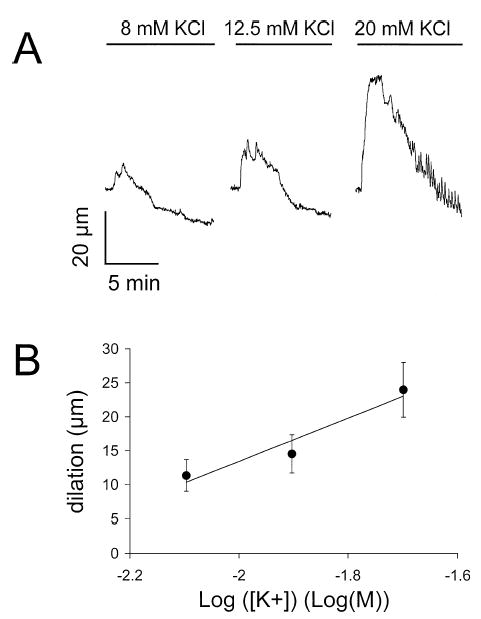
Elevated [K+]o causes concentration-dependent, transient dilation of skeletal muscle arterioles. (A) Digitized diameter records for a cannulated arteriole when [K+]o was elevated from 5 mM to 8, 12.5, or 20 mM as indicated. (B) Summary of the results from 11 experiments (mean resting diameter = 57 ± 5 μm, mean maximum diameter = 105 ± 5 μm) and the mean ± SE dilation induced by elevation of [K+]o from 5 mM to 8, 12.5, or 20 mM. Regression analysis showed a significant linear relationship (slope = 32 ± 11 μm/log(M), intercept = 77 ± 21, < .05, n = 33 arterioles), as indicated by the solid line.
In most vessels studied, the dilation induced by elevated [K+]o was transient: the vessels dilated and then returned, often in a sawtooth fashion, to, or below, their original diameters despite maintained elevation of [K+]o (Figure 1A). For all vessels studied, exposure to 20 mM K+ dilated arterioles from 58 ± 1 μm to 86 ± 2 μm (n = 105, p < .05, maximum diameter = 106 ± 2 μm). Of the 105 vessels in which 20 mM K+-induced dilation was examined, 87 (83%) displayed transient responses. In the remaining 18 vessels studied (17%), the K+-induced dilation appeared to be well maintained for tens of minutes. The difference in responses did not correlate with the size of vessel studied, but did appear to correlate with different animals. In several instances, 2 vessels isolated from the same animal appeared to have similar responses (transient or sustained).
Elevated [K+]o Hyperpolarizes Isolated Cremasteric Arterioles
In a subset of experiments we measured the membrane potential of smooth muscle cells in these cannulated arterioles. We verified that we were recording from vascular muscle cells by using Alexa Fluor 488 to label impaled cells in some experiments (Figure 2). As shown in Figure 3, elevation of [K+]o from 5 to 20 mM resulted in membrane hyperpolarization that preceded dilation of the arterioles. Typically, the membrane potential would slowly begin to recover, culminating in spike-like potentials that preceded, and correlated well with the sawtooth pattern of diameter recovery in those vessels that displayed transient responses (Figure 3). We found it very difficult to keep electrodes in smooth muscle cells for the entire course of the response. However, we consistently were able to record the initial hyperpolarization since this usually plateaued before much change in diameter. Because of this problem, as well as the variability of the time of occurrence and the pattern of action potentials, summary data were computed only for the nadir of the initial hyperpolarization. However, as will be subsequently shown, this correlated well with the initial peak dilations that we observed. In 23 arterioles, resting membrane potential was −28 ± 1 mV and hyperpolarized to −38 ± 1 mV (p < .05) when [K+]o was raised from 5 to 20 mM. This hyperpolarization preceded dilation by 6.3 ± 0.4 s, after which the vessel diameters increased from 58 ± 3 μm to 79 ± 4 μm (p < .05) (see Figure 3). Membrane potential was recorded in 6 vessels that displayed relatively sustained K+-induced dilation. In 5 of these arterioles, elevated [K+]o caused hyperpolarization that was followed by a low frequency of membrane potential spikes that were associated with small oscillations in diameter. In the remaining vessel, the membrane simply remained hyperpolarized in the presence of elevated [K+]o.
Figure 2.
Alexa Fluor 488-labeled arteriolar muscle cell. Shown is a combined transmitted light and epifluorescent image of an isolated arteriole that was pinned to the bottom of the chamber and from which membrane potential of arteriolar muscle cells was recorded. The image was captured with a CCD camera and a Scion image capture card controlled by a Macintosh G-4 computer running NIH Image software. Similar results were obtained in cannulated, pressurized arterioles (data not shown). Bar = 50 μm.
Figure 3.
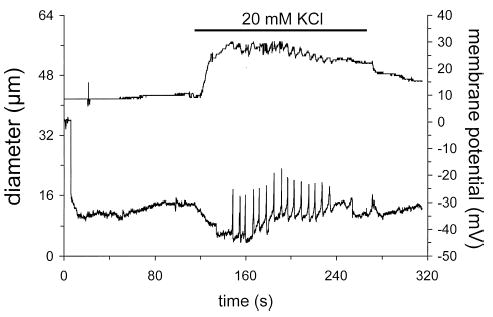
Elevated [K+]o causes dilation and hyperpolarization of skeletal muscle arterioles. Shown is a digitized, simultaneous recording of arteriolar diameter (top trace) and membrane potential (bottom trace) of an arteriolar muscle cell in a cannulated, pressurized cremasteric arteriole. When [K+]o was elevated from 5 mM to 20 mM (as indicated by the solid bar above the diameter trace), the arteriolar muscle cell hyperpolarized by about 10 mV and this change in membrane potential preceded arteriolar dilation. At the peak of the hyperpolarization, action potential-like spikes in membrane potential began. These membrane potential spikes preceded and correlated well with step-like decreases in arteriolar diameter. Upon return to 5 mM K+, both diameter and membrane potential recovered.
Elevated [K+]o Alters Intracellular Calcium in Isolated Cremasteric Arterioles
We assessed the effects of elevated [K+]o on the [Ca2+]i of arteriolar muscle cells in 4 cannulated, pressurized arterioles using the Ca2+-sensitive dye, Fura 2 AM. We found that elevation of [K+]o from 5 to 20 mM induced an initial 22 ± 5% decrease (p < .05) in the Fura 2 fluorescence ratio and a 32 ± 7 μm dilation of the arterioles (resting diameter = 72 ± 4 μm, peak diameter in 20 mM K+ = 103 ± 10 μm, p < .05, maximum diameter = 109 ± 11 μm). The decreases in the Fura 2 ratio preceded the dilations by 3.7 ± 0.4 s (n = 4). Continued exposure to 20 mM KCl induced oscillations in the Fura 2 ratio and arteriolar diameter in 3 of 4 experiments (Figure 4A). Similar to the initial dilation, these diameter oscillations were always preceded by oscillations in the Fura 2 ratio by approximately 4 s, consistent with the oscillations in diameter being caused by changes in smooth muscle cell [Ca2+]i (Figure 4B). In the remaining arteriole, elevation of [K+]o resulted in a fall in the Fura 2 ratio that remained below baseline while the K+ concentration was elevated.
Figure 4.
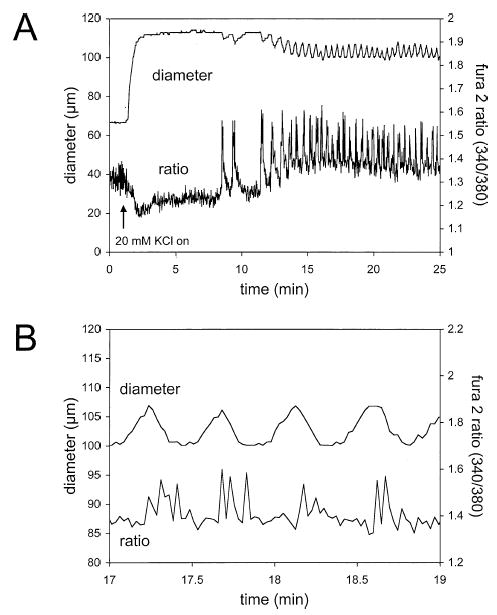
Elevated [K+]o causes dilation and decreases in intracellular Ca2+ of skeletal muscle arterioles. (A) Digitized diameter and Fura 2 fluorescence ratio traces in response to elevation of [K+]o from 5 to 20 mM as indicated by the arrow. Upon exposure to elevated K+, the Fura 2 ratio decreased, indicative of a fall in intracellular Ca2+. This change in Ca2+ occurred prior to the change in diameter (see (B)). Transient oscillations in the Fura 2 ratio were then observed that preceded but correlated well with oscillations in diameter. (B) An expanded segment of the record shown in (A) to better demonstrate the temporal relationship between the Ca2+ signal and diameter.
K+-Induced Dilation of Cremasteric Arterioles Does Not Require Endothelium
Endothelial cells can influence smooth muscle cells in a number of ways. Therefore, we assessed the effects of removal of endothelial cells on K+-induced dilation. We found it very difficult to adequately remove endothelial cells from the arterioles while leaving the smooth muscle viable. However, we were able to collect data on 5 vessels. We found that perfusion of these cannulated arterioles with an air bubble had no significant effect on resting diameter: prior to air bubble perfusion diameter was 53 ± 2 μm, dilating to 81 ± 6 μm in response to elevated K+, while after air bubble perfusion resting diameter was 65 ± 2 μm, dilating to 80 ± 2 μm in response to 20 mM K+ (max diameter = 99 ± 2 μm). A 2 × 2 factorial analysis of variance indicated no significant effect of air bubble perfusion on resting diameter (p > .05), and the vessels significantly dilated (p < .05) to similar diameters (p > .05) upon exposure to 20 mM K+. In contrast, arteriolar dilation induced by a maximally effective concentration of acetylcholine (5–10 μM) was completely eliminated, indicating complete loss of a functional endothelium: prior to air bubble perfusion, arterioles dilated by 38 ± 3 μm (n = 5, p < .05), after air bubble perfusion the arteriolar diameter change averaged 0.7 ± 0.6 μm (n = 5, p > .05).
Barium Blunts K+-Induced Dilation of Cannulated Arterioles
Superfusion of arterioles with 50 μM Ba2+, which has been demonstrated to abolish K+-induced hyperpolarization and dilation in other systems (17,27,28,31,39), had no significant effect on resting diameter: control diameter = 55 ± 5 μm, diameter in 50 μM BaCl2 = 53 ± 5 μm (n = 8, p > .05). However, this KIR channel blocker inhibited K+-induced dilation by 50%: elevation of [K+]o from 5 to 20 mM resulted in a 24 ± 4 μm dilation under control conditions that was reduced to 12 ± 4 μm during superfusion with 50 μM BaCl2 (n = 8, p < .05). This inhibition appeared to be selective because dilation induced by acetylcholine (5 μM) was unaffected by Ba2+ exposure: prior to 50 μM Ba2+, arterioles dilated 46 ± 8 μm when exposed to 5 μM acetylcholine; during superfusion with 50 μM Ba2+, vessels dilated 46 ± 9 μm (n = 4, p > .05).
Increasing the concentration of BaCl2 to 100 μM produced no further inhibition of K+-induced dilation (Figure 5), suggesting that these concentrations of Ba2+ were maximally effective. In the steady state, 100 μM BaCl2 slightly depolarized arterioles from −30 ± 1 mV to −26 ± 1 mV (n = 7, p < .05) and caused a small decrease in resting arteriolar diameter: prior to Ba2+ exposure resting diameter was 59 ± 4 μm; during superfusion with 100 μM Ba2+, diameter was 51 ± 5 μm (n = 14, p > .05, max diameter = 109 ± 5 μm). Similar to its effect on diameter responses, 100 μM Ba2+ also blunted the 20 mM K+-induced hyperpolarization (Figure 5).
Figure 5.
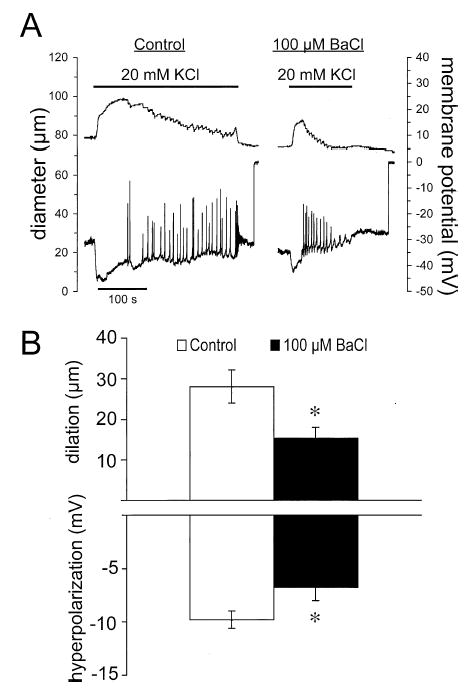
Barium attenuates K+-induced dilation and hyperpolarization. (A) Digitized typical records of diameter (top traces) and membrane potential (bottom traces) before (control) and during superfusion with 100 μM Ba2+ in response to elevation of [K+]o from 5 to 20 mM. Barium decreased the amplitude and the duration of both the diameter and membrane potential responses. (B) Summary data for the peak diameter and membrane potential responses. Data are means ± SE (n = 7), *significantly different from control response, p < .05.
Barium ions (50 or 100 μM) also substantially reduced the duration of the dilator response to elevated [K+]o, as can be seen in Figure 5A. To quantify this effect, we estimated the time from exposure of the arterioles to 20 mM K+ to the time of 50% recovery of baseline diameter after the peak dilation (t½). In 20 paired experiments in which this measure could be accurately quantified, we found that exposure to Ba2+ (50 or 100 μM) decreased the t½ to 63 ± 11% of the control value of 319 ± 65 s (p < .05).
Barium ions can also block ATP-sensitive K+ (KATP) channels (28). To exclude a role for these channels in the effects of elevated [K+]o we assessed the effects of glibenclamide, a relatively selective KATP channel inhibitor (28). Glibenclamide had no significant effect on resting diameter in this series of experiments: control diameter was 53 ± 4 μm and the diameter in the presence of glibenclamide was 55 ± 6 μm (n = 6, p > .05). Glibenclamide (1 μM) severely reduced dilations induced by the KATP channel opener cromakalim (10 μM (28)) from 51 ± 7 μm to 10 ± 3 μm (n = 6, p < .05). However, in contrast, glibenclamide (1 μM) had no significant effect on dilations induced by 20 mM K+: prior to exposure to glibenclamide, arterioles dilated 25 ± 3 μm, whereas during glibenclamide superfusion, arterioles dilated 21 ± 4 μm (n = 6, p > .05).
K+-Induced Currents and Membrane Potential in Single Arteriolar Muscle Cells
The effects of elevated [K+]o on currents and membrane potential in single, enzymatically isolated arteriolar muscle cells were also examined. We found that increasing extracellular K+ from 5 to 20 mM K+ activated an inwardly rectifying current that could be completely inhibited by 200 μM Ba2+ (Figure 6) in whole-cell voltage clamp experiments. However, there did not appear to be any Ba2+-sensitive outward component to this current, as required for elevated K+ to cause membrane hyperpolarization. Consistent with this observation, we found that increasing [K+]o from 5 to 10 or 20 mM caused only depolarization of these cells in current clamp experiments (Figure 7). Cremasteric arteriolar muscle cells were capable of hyperpolarization as 10 μM cromakalim increased membrane potential from −36 ± 2 mV to −62 ± 2 mV (n = 4, p < .05).
Figure 6.
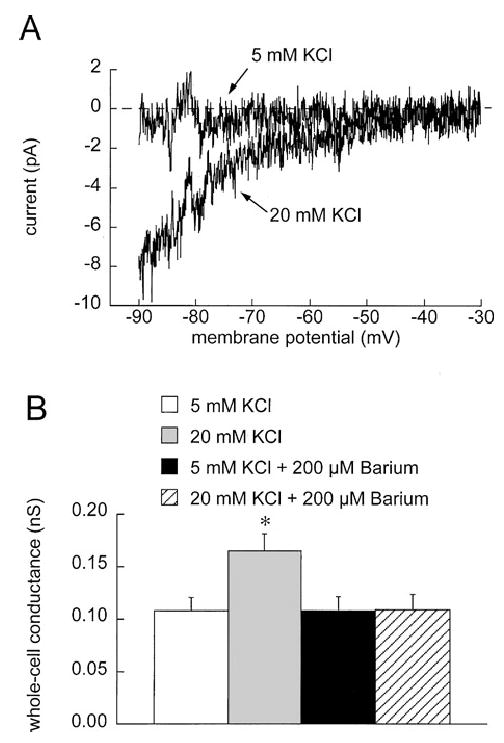
Elevated [K+]o activates Ba2+-sensitive currents in cremasteric arteriolar muscle cells. (A) Ba2+-sensitive currents, present when cells were exposed to 5 mM K+, or 20 mM K+ were obtained by subtracting currents obtained during 500-ms ramp protocols in the absence and presence of 200 μM Ba2+. This high concentration of Ba2+ was used to insure complete inhibition of K+-activated currents. 20 mM K+ activated a Ba2+-sensitive, inwardly rectifying current. Note that no outward component of this current was observed. To summarize and quantify this K+-activated current, we performed regression analysis on the current-voltage data from −90 to −40 mV to obtain slope conductances. (B) In 6 cells, 20 mM K+ significantly (*p < .05 compared to 5 mM K+) increased the slope conductance, and this increase could be abolished by 200 μM Ba2+.
Figure 7.
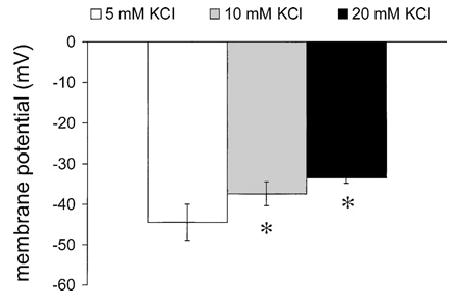
Elevated [K+]o does not hyperpolarize single cremasteric arteriolar muscle cells. Data are mean ± SE (n = 6) membrane potentials recorded in current clamp using the perforated patch technique, in 5, 10, and 20 mM K+. Elevated [K+]o was observed only to depolarize the cells. *Significantly different from potential in 5 mM K+, p < .05.
Subsequently, we also studied membrane potential responses in unpressurized arterioles that were simply pinned to the bottom of the recording chamber. We found that in this situation, elevation of [K+]o from 5 to 20 mM caused only a very small, transient hyperpolarization (−2.2 ± 0.7 mV; n = 9, p < .05) from a resting potential of −45 ± 4 mV that was significantly smaller than the hyperpolarization observed in pressurized arterioles ( p < .05). This small response was always followed by a steady depolarization of 13 ± 2 mV in contrast to the spike activity that was observed in pressurized vessels.
Ouabain Inhibits K+-Induced Dilation
To assess the role played by the Na+/K+-ATPase in K+-induced dilation, we examined the effects of ouabain on responses elicited by 20 mM K+. Superfusion of cannulated arterioles with a high concentration of ouabain (1 mM) (as has been used by others (20)) caused a transient constriction and membrane depolarization that subsided within 20 min (data not shown). In the steady state, this cardiac glycoside had no significant effect on resting diameter or membrane potential. Prior to exposure to 1 mM ouabain, diameters were 60 ± 3 μm, whereas after 20 min exposure to 1 mM ouabain they were 57 ± 3 μm (n = 18, p > .05; max. diameter = 108 ± 4μm). Similarly, membrane potentials under control conditions were −28 ± 2 mV, and were −27 ± 2 mV after 20 min exposure to 1 mM ouabain (n = 4, p > .05). However, ouabain (1 mM) abolished the dilation and hyperpolarization in most experiments (Figure 8), often reversing them to constriction and depolarization, respectively (Figure 8A).
Figure 8.
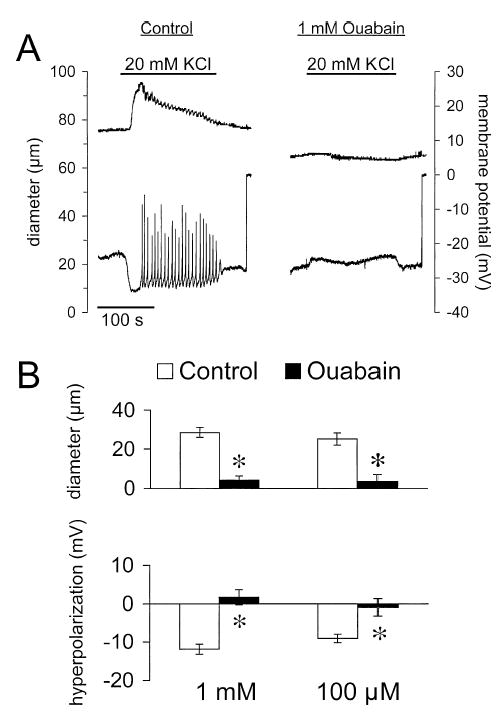
Ouabain inhibits K+-induced dilation and hyperpolarization. (A) Digitized typical records of diameter (top trace) and membrane potential (bottom trace) from a cannulated arteriole in response to elevation of [K+]o from 5 to 20 mM (solid bars over diameter trace) in the absence (control) and presence of 1 mM ouabain. In the presence of ouabain, dilation and hyperpolarization were reversed to constriction and depolarization. (B) Summary of the peak responses in the absence (control) and presence of either 1 or 100 μM ouabain. Data are means ± SE (n = 18 for diameter and n = 4 for membrane potential for 1 mM ouabain; n = 19 for diameter and n = 6 for membrane potential for 100 μM ouabain). *Significantly different from control and not significantly different from zero, p < .05.
Similar effects were observed with a 10-fold lower concentration of ouabain. As with the higher concentration, ouabain (100 μM) caused a transient constriction and membrane depolarization that subsided after 10–20 min (data not shown). In the steady state, this concentration of ouabain had no significant effect on membrane potential: control membrane potential was −28 ± 1 mV, whereas membrane potential during superfusion with 100 μM ouabain was −26 ± 1 mV (n = 6, p > .05).
Similarly, ouabain had no significant effect on steady-state resting arteriolar diameter: prior to ouabain exposure resting diameter was 60 ± 3 μm, during superfusion with 100 μM ouabain, diameter was 62 ± 3 μm (n = 19, p > .5, max diameter = 109 ± 5 μm). However, in the mean, this cardiac glycoside essentially eliminated reactivity to elevated [K+]o (Figure 8): both the hyperpolarization and the dilation were severely attenuated and not significantly different from zero.
Despite this overall average abolition, there were instances where ouabain only blunted the K+-induced dilation at both concentrations. In 11 of 18 experiments, 1 mM ouabain inhibited dilation by greater than 90% (4 experiments) or reversed the K+-induced dilation to constriction (7 experiments) (mean %inhibition = 106 ± 3%). In the remaining 7 vessels, this high concentration of ouabain only attenuated the response by 43 ± 14%. Similar results were observed when a lower concentration of ouabain was used. In 14 of 19 experiments, 100 μM ouabain severely inhibited dilation when [K+]o was elevated from 5 to 20 mM (>88% inhibition in 3 cases, and reversed the responses to constriction in the remaining 11 arterioles − mean %inhibition = 134 ± 12%). However, in the remaining 5 experiments, K+-induced dilation was inhibited by only 26 ± 10%. In one of these latter experiments, Ba2+ (100 μM) was combined with ouabain and this combination reversed both the ouabain-insensitive component of the hyperpolarization and dilation induced by elevated [K+]o to frank depolarization and vasoconstriction. Of the 12 arterioles with ouabain-insensitive responses, 3 showed sustained K+-induced dilation (in the absence or presence of ouabain). In the remaining 9 vessels, ouabain converted transient responses into more sustained dilations. In 7 additional experiments, the effects of combination of 1 mM ouabain and 100 μM BaCl2 were assessed. Control arteriolar diameters were 53 ± 6 μm (maximum = 100 ± 6 μm) and the vessels dilated to 85 ± 5 μm when [K+]o was elevated to 20 mM. Twenty minutes of exposure to the combination of ouabain and BaCl2 resulted in arteriolar constriction to 36 ± 6 μm (p < .05) and near abolition of K+-induced dilation (1.6 ± 0.5 μm) in all experiments. The inhibitory effects of 100 μM ouabain appeared to be selective, because dilation induced by acetylcholine was unaffected by this inhibitor: prior to ouabain exposure, arterioles dilated 35 ± 3 μm in response to 5 μM acetylcholine, whereas during superfusion with 100 μM ouabain, arterioles dilated 32 ± 5 μm (n = 4, p > .05).
Substitution of Na+ with Li+ Inhibits K+-Induced Dilation
The Na+/K+-ATPase can also be inhibited by removal of extracellular Na+ (2). Therefore, experiments were performed in LiCl-PSS in which all of the NaCl was replaced by LiCl, and pH was adjusted with LiOH. Superfusion of arterioles with this solution caused transient hyperpolarization and dilation that faded within 10–20 min of exposure, culminating in depolarization of the resting potential and a trend toward vasoconstriction. Prior to exposure to LiCl-PSS, resting membrane potential was −25 ± 2 mV. After waning of the initial hyperpolarization, membrane potential depolarized to −17 ± 1 mV (n = 5, p < .05, compared to control). In the steady state, arteriolar diameters were not significantly different from control during LiCl superfusion, although there was a tendency toward vasoconstriction: diameters were 51 ± 5 μm during control conditions, and stabilized at 45 ± 3 μm during LiCl-PSS superfusion (n = 5, p > .05). Exposure to LiCl also tended to make membrane potential oscillatory even in 5 mM K+ as membrane potential oscillations appeared during LiCl superfusion in all 5 experiments. Replacement of Na+ with Li+ converted 20 mM K+-induced hyperpolarization and dilation into depolarization and constriction, respectively (Figure 9). This inhibition appeared to be selective because dilation induced by acetylcholine (50 nmol applied as a bolus) was not significantly affected: the control dilation was 23 ± 4 μm, while during LiCl-PSS the dilation was 19 ± 4 μm (n = 5, p > .05).
Figure 9.
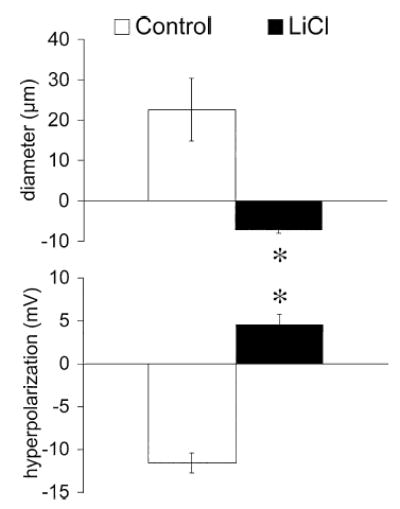
LiCl inhibits K+-induced dilation and hyperpolarization. Data are means ± SE (n = 5), for the peak diameter and membrane potential responses when [K+]o was elevated from 5 to 20 mM, in the absence (control) or presence of PSS in which all NaCl was replaced by LiCl. *Significantly different from control and significantly greater than zero (p < .05).
DISCUSSION
The purpose of the present investigation was to evaluate the mechanism by which elevated [K+]o causes dilation of skeletal muscle arterioles. This was accomplished by studying the responses of isolated, cannulated, pressurized segments of hamster cremasteric arterioles. As studies in the literature suggested that KIR channels (17,19,27,28,31,39) or the Na+/K+ ATPase (10,16,20,35) were likely potential mechanisms, we focused on these 2 pathways. Our results suggest that both mechanisms may contribute to the means by which elevated [K+]o causes arteriolar dilation in skeletal muscle.
We observed that elevated [K+]o caused concentration-dependent dilation, with maximal effects observed at a concentration of 20 mM, and that the dilation to 20 mM K+ was preceded by smooth muscle cell hyperpolarization and a fall in intracellular Ca2+. These findings support and extend results reported in other systems (10,16,17,19,20,27,28,31,35,39). However, in contrast to findings in small cerebral and coronary arteries (17), K+-induced dilation of cremasteric arterioles was often transient. These data are consistent with in vivo, microcirculatory studies of hamster (4) and rat (20) cremasteric arterioles and earlier whole-muscle studies (10,16,35). Our data indicate that the transient nature of the response to elevated [K+]o is an intrinsic property of skeletal muscle arterioles. Most of our data support the hypothesis that elevated [K+]o can contribute to the initiation of functional hyperemia in skeletal muscle, but that it may not be responsible for the maintained phase of vasodilation that is associated with prolonged exercise (10,16,35). However, in 17% of our experiments, exposure to elevated [K+]o caused relatively sustained dilation. These data suggest that the mechanisms responsible for K+-induced dilation may be modulated to produce transient or sustained responses. Thus, there may be instances where elevated [K+]o can contribute to the sustained phase of functional dilation. Further experiments will be required to identify the factors responsible for modulation of the duration and kinetics of the response to elevated [K+]o.
Edwards et al. (5) first proposed that KIR channels were involved in K+-induced dilation in cerebral arterioles. This idea was later championed by Nelson and colleagues, who have provided compelling evidence that Kir channels mediate K+-induced dilation of small arteries (17,27,39). They showed that low micromolar concentrations of Ba2+ could abolish the hyperpolarization and vasodilation induced by elevated [K+]o. In contrast, we found that Ba2+ concentrations up to 100 μM only attenuated K+-induced hyperpolarization and dilation. We do not think that the low of efficacy Ba2+ in the present study resulted from the use of submaximal concentrations of this KiR channel blocker, because the degree of inhibition observed was similar with both 50 and 100 μM concentrations. Our findings clearly indicate that KIR channels are not the sole pathway mediating K+-induced dilation of hamster cremasteric arterioles.
Barium ions have been shown to block KATP channels as well as KIR channels (28). To eliminate the possibility that Ba2+ was affecting responses to elevated [K+]o by blocking KATP channels, the effects of glibenclamide, a KATP channel blocker (28), on K+-induced dilation were assessed. Glibenclamide (1 μM) eliminated vasodilation to the KATP channel agonist cromakalim (10 μM) (28). However, glibenclamide did not inhibit K+-induced dilation of cremasteric arterioles. Thus, KATP channels are not involved in the arteriolar response to elevated [K+]o in this tissue.
We observed K+-activated inward currents inhibited by Ba2+ in arteriolar muscle cells supporting the presence of KIR channels in these cells. However, under the conditions of these patch clamp experiments (room temperature, no pressure-induced tone), K+-induced activation of these channels did not produce outward, hyperpolarizing currents in voltage clamp experiments as would be necessary to cause dilation. Membrane hyperpolarization also was not seen in current clamp experiments. To see if the lack of pressure-induced tone might account for these results, we assessed the effects of elevated [K+]o on membrane potential of smooth muscle cells in unpressurized arterioles at 34°C. While a small, transient-2 mV hyperpolarization was observed, this was significantly smaller than the consistent −10 mV response seen in pressurized vessels (p < .05). Thus, substantial K+-induced hyperpolarization appears to require significant smooth muscle tone. We did not examine whether this was specific to pressure-induced tone, as that was beyond the scope of the present study. Nonetheless, our data suggest that caution must be observed when extrapolating between experiments in which conditions are not identical. These data also support our hypothesis that the mechanisms responsible for K+-induced hyperpolarization and dilation may be modulated.
Exposure of cremasteric arterioles to Ba2+ caused a sustained depolarization and a small increase in tone. We cannot completely rule out nonspecific effects due to this depolarization and constriction. However, dilation induced by acetylcholine was not inhibited by Ba2+, suggesting that the effects of the blocker were selective. Therefore, we conclude that KIR channels may contribute to a portion of the response to elevated [K+]o. We did find that Ba2+ reduced the duration of the K+-induced dilation. Thus, KIR channels may play a role in modulating the duration and kinetics of responses to elevated [K+]o.
Early studies of the mechanism by which elevated [K+]o causes vasodilation in skeletal muscle proposed that the Na+/K+ ATPase may be involved (10,16,35). More recently, Lombard and Stekiel (20) supported this hypothesis by showing that ouabain (1 mM) inhibits K+-induced dilation of rat cremasteric arterioles in vivo. Our findings buttress their conclusions. We found that both ouabain (100 μM or 1 mM) and replacement of Na+ with Li+, both known inhibitors of the Na+/K+ ATPase (2,10,11), substantially inhibited K+-induced hyperpolarization and dilation of cremasteric arterioles in vitro. Furthermore, this effect appeared to be specific because acetylcholine-induced dilation was not altered. Thus, our data support a major role for the Na+/K+ ATPase in K+-induced dilation of skeletal muscle arterioles.
Activation of the Na+/K+ ATPase by elevated [K+]o should be transient and also should cause a transitory hyperpolarization as Na+ is pumped out of the cells and intracellular [Na+] decreases (10,11). This is consistent with the transient dilation and hyperpolarization that we observed and inhibition of the electrophysiological and mechanical effects of elevated [K+]o by both ouabain and replacement of Na+ by Li+. We also observed in 3 of the 12 experiments where ouabain had little effect that the K+-induced dilation was sustained. In the remaining 9 experiments, the transient responses to K+ were converted into more sustained responses in the presence of ouabain. These data support the hypothesis that K+-induced activation of the Na+/K+ ATPase contributes to the transient nature of hyperpolarization and vasodilation that we observed. However, additional mechanisms also may be involved. Potassium-induced hyperpolarization and vasodilation were sustained in all 5 experiments in which LiCl abolished these responses. These data suggest that there may be other factors that contribute to the transient nature of the K+-induced responses in skeletal muscle arterioles. Alternatively, Li+ may have other effects in addition to inhibiting the Na+/K+ ATPase.
Activation of KIR channels should lead to a sustained hyperpolarization, and hence dilation, as has been reported by others (17,27,31). Consistent with this hypothesis, we found that Ba2+ significantly reduced the duration of responses to elevated K+. We found that Ba2+ decreased the t½ for diameter recovery in 20 experiments by 37%. Furthermore, in 6 vessels that displayed sustained dilation when [K+]o was elevated, Ba2+ converted these to transient responses. In addition, in one experiment in which ouabain-insensitive sustained hyperpolarization and dilation were observed, addition of Ba2+ eliminated these responses. These data suggest that KIR channels, in part, may be responsible for the sustained component of K+-induced hyperpolarization and dilation.
We did not examine, in detail, the ionic basis of the membrane potential spikes that followed the initial hyperpolarization during exposure to elevated [K+]o. However, there appeared to be a strong Ca2+ component to these spikes, as Ca2+ transients often were observed in Fura 2-loaded arterioles. These Ca2+ transients preceded, and were well correlated with, oscillatory decreases in arteriolar diameter (see Results). Most likely the membrane potential spikes and Ca2+ transients involved activation of L-type voltage-gated Ca2+ channels that are the dominant voltage-gated channels conducting inward current in these cells (3), in a fashion similar to that proposed by Bartlett et al. (1) to explain arteriolar vasomotion.
There is cellular communication between vascular smooth muscle and endothelium. Endothelial cells have been shown to respond to different stimuli and to cause relaxation of smooth muscle by releasing vasodilating factors such as prostacyclin and nitric oxide (21,37). In addition, endothelial cells may be electrically coupled to smooth muscle cells by myoendothelial gap junctions (33) and endothelial cells have inward rectifier potassium channels (25). Therefore, we assessed the role played by the endothelium in the dilator response to elevated [K+]o. Removal of the endothelium by air-bubble perfusion, verified by loss of reactivity to acetylcholine, did not eliminate K+-induced dilation in the present study. These data indicate that K+-induced dilation of skeletal muscle arterioles does not require the presence of a functional endothelium and are consistent with observations in other systems (17). We cannot exclude the possibility that a functional endothelium may contribute to, or modulate the arteriolar response to elevated [K+]o. However, our data indicate that arteriolar muscle cells can respond independently to this stimulus.
We found that K+-induced dilation was preceded by membrane hyperpolarization and a fall in [Ca2+]i, respectively, and that voltage-gated Ca2+ channels substantially contribute to resting tone in cremasteric arterioles in vitro. These data strongly support the hypothesis that K+-induced dilation is mediated by membrane hyperpolarization (5,10,11,17), closure of voltage-gated Ca2+ channels (13), and a fall in [Ca2+]i.
We propose that K+-induced dilation of cremasteric arterioles involves activation of both KIR channels and the Na+/K+ ATPase. Supporting this hypothesis, we found that consistent inhibition of vasodilation induced by elevated [K+]o required exposure to both ouabain and Ba2+. Similar conclusions have been drawn in rat cerebral arteries (22) and rat hepatic arteries (6). Our studies may also resolve the differences between the findings of Lombard and Stekiel (20) supporting a role for the Na+/K+ ATPase in rat cremasteric arterioles, and Loeb et al. (19), who suggest that KIR channels are involved in the same tissue. In both studies, inhibition of K+-induced dilation by ouabain (20) or Ba2+ (19) was only partial. Thus, dual mechanisms, as we propose in the present study, may have been involved. We hypothesize that initiation of the hyperpolarization and dilation depends on the Na+/K+ ATPase because both ouabain and LiCl effectively eliminated responses to elevated K+ in the majority (84%) of experiments. Activation of KIR channels may play a more supportive or modulatory role, impacting primarily the duration and kinetics of the response to elevated [K+]o.
Acknowledgments
The authors are grateful for the outstanding technical assistance of Susan K. Kovats. This research was supported by Public Health Service grant HL32469 and Western Michigan University Faculty Research and Creative Activities Support Fund grant 02-016 to Dr. Jackson, and American Heart Association Postdoctoral Fellowship 0120607Z to Dr. Cohen.
References
- 1.Bartlett IS, Crane GJ, Neild TO, Segal SS. Electrophysiological basis of arteriolar vasomotion in vivo. J Vasc Res. 2000;37:568–575. doi: 10.1159/000054090. [DOI] [PubMed] [Google Scholar]
- 2.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275(5 Pt 2):F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 3.Cohen KD, Jackson WF. Hypoxia inhibits contraction but not calcium channel currents or changes in intracellular calcium in arteriolar muscle cells. Microcirculation. 2003;10:133–141. doi: 10.1038/sj/mn.7800178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duling BR. Effects of potassium ion on the microcirculation of the hamster. Circ Res. 1975;37:325–332. doi: 10.1161/01.res.37.3.325. [DOI] [PubMed] [Google Scholar]
- 5.Edwards FR, Hirst GDS, Silverberg GD. Inward rectification in rat cerebral arterioles: involvement of potassium ions in autoregulation. J Physiol. 1988;404:455–466. doi: 10.1113/jphysiol.1988.sp017299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- 7.Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res. 2000;87:474–479. doi: 10.1161/01.res.87.6.474. [DOI] [PubMed] [Google Scholar]
- 8.Fredricks KT, Liu Y, Rusch NJ, Lombard JH. Role of endothelium and arterial K+ channels in mediating hypoxic dilation of middle cerebral arteries. Am J Physiol. 1994;267(2 Pt 2):H580–H586. doi: 10.1152/ajpheart.1994.267.2.H580. [DOI] [PubMed] [Google Scholar]
- 9.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 10.Haddy FJ. Potassium effects on contraction in arterial smooth muscle mediated by Na+, K+-ATPase. Fed Proc. 1983;42:239–245. [PubMed] [Google Scholar]
- 11.Hermsmeyer K. Sodium pump hyperpolari zation–relaxation in rat caudal artery. Fed Proc. 1983;42:246–252. [PubMed] [Google Scholar]
- 12.Jackson WF. Hypoxia does not activate ATP-sensitive K+ channels in arteriolar muscle cells. Microcirculation. 2000;7:137–145. [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson WF. Ion channels and vascular tone. Hypertension. 2000;35(1 Pt 2):173–178. doi: 10.1161/01.hyp.35.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson WF, Duling BR. Toxic effects of silver-silver chloride electrodes on vascular smooth muscle. Circ Res. 1983;53:105–108. doi: 10.1161/01.res.53.1.105. [DOI] [PubMed] [Google Scholar]
- 15.Jackson WF, Huebner JM, Rusch NJ. Enzymatic isolation and characterization of single vascular smooth muscle cells from cremasteric arterioles. Microcirculation. 1997;4:35–50. doi: 10.3109/10739689709148316. [DOI] [PubMed] [Google Scholar]
- 16.Johansson B, Somlyo AP. (1980). Electrophysiology and excitation–contraction coupling. In Handbook of Physiology, Section 2: The Cardiovascular System, Vol. 2: Vascular Smooth Muscle (DF Bohr, AP Somlyo, and HV Sparks, Eds.). Bethesda, MD: American Physiological Society, 301–323.
- 17.Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492:419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laughlin MH, Korthuis RJ, Duncker DJ, Bache RJ. (1996). Control of blood flow to cardiac and skeletal muscle during exercise. In Exercise: Regulation and Integration of Multiple Systems (LB Rowell and JT Shepherd, Eds.). New York: Oxford University Press, 705–769.
- 19.Loeb AL, Godeny I, Longnecker DE. Functional evidence for inward-rectifier potassium channels in rat cremaster muscle arterioles. Microvasc Res. 2000;59:1–6. doi: 10.1006/mvre.1999.2187. [DOI] [PubMed] [Google Scholar]
- 20.Lombard JH, Stekiel WJ. Responses of cremasteric arterioles of spontaneously hypertensive rats to changes in extracellular K+ concentration. Microcirculation. 1995;2:355–362. doi: 10.3109/10739689509148279. [DOI] [PubMed] [Google Scholar]
- 21.Luscher TF, Barton M. (1997). Biology of the endothelium. Clin Cardiol 20(11, Suppl 2): II-3–II-10. [PubMed]
- 22.McCarron JG, Halpern W. Potassium dilates rat cerebral arteries by two independent mechanisms. Am J Physiol. 1990;259(3 Pt 2):H902–H908. doi: 10.1152/ajpheart.1990.259.3.H902. [DOI] [PubMed] [Google Scholar]
- 23.Meininger GA, Zawieja DC, Falcone JC, Hill MA, Davey JP. Calcium measurement in isolated arterioles during myogenic and agonist stimulation. Am J Physiol. 1991;261(3 Pt 2):H950–H959. doi: 10.1152/ajpheart.1991.261.3.H950. [DOI] [PubMed] [Google Scholar]
- 24.Neild TO. Measurement of arteriole diameter changes by analysis of television images. Blood Vessels. 1989;26:48–52. [PubMed] [Google Scholar]
- 25.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 26.Ott L. (1988). An Introduction to Statistical Methods and Data Analysis, 3rd ed. Boston: PWS-Kent, 835.
- 27.Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol. 1993;265(5 Pt 1):C1363–C1370. doi: 10.1152/ajpcell.1993.265.5.C1363. [DOI] [PubMed] [Google Scholar]
- 28.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- 29.Rae JS, Cooper KE, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 30.Renkin EM. (1984). Control of Microcirculation and blood-tissue exchange. In Handbook of Physiology, sec. 2: The Cardiovascular System, Vol. 4: Microcirculation, part 2, (EM Renkin and CC Michel, Eds). American Physiological Society, 627–687.
- 31.Robertson BE, Bonev AD, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat coronary arteries: block by Mg2+, Ca2+, and Ba2+ Am J Physiol. 1996;271(2 Pt 2):H696–H705. doi: 10.1152/ajpheart.1996.271.2.H696. [DOI] [PubMed] [Google Scholar]
- 32.Rowell LB. (1986). Human Circulation New York: Oxford University Press, 96–116.
- 33.Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res. 2000;86:341–346. doi: 10.1161/01.res.86.3.341. [DOI] [PubMed] [Google Scholar]
- 34.Shepherd JT. (1983). Circulation to skeletal muscle. In The Cardiovascular System (JT Shepherd and FM Abboud, Eds.). Bethesda, MD: American Physiological Society; 319–370.
- 35.Sparks HV. (1980). Effect of local metabolic factors on vascular smooth muscle. In Handbook of Physiology; sec. 2: The Cardiovascular System, Vol. 2: Microcirculation, part 2 (DF Bohr, AP Somlyo, and HV Sparks, Eds.). Bethesda, MD: American Physiological Society, 181–309.
- 36.Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79:1089–1125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- 37.Vanhoutte PM. Other endothelium-derived vasoactive factors. Circulation. 1993;87 (Suppl 5):V9–V17. [Google Scholar]
- 38.Welsh DG, Jackson WF, Segal SS. Oxygen induces electromechanical coupling in arteriolar smooth muscle cells: a role for L-type Ca2+ channels. Am J Physiol. 1998;274(6 Pt 2):H2018–H2024. doi: 10.1152/ajpheart.1998.274.6.H2018. [DOI] [PubMed] [Google Scholar]
- 39.Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87:160–166. doi: 10.1161/01.res.87.2.160. [DOI] [PubMed] [Google Scholar]