

Abstract
Fibroblasts at the tumor-host interface can differentiate into myofibroblasts and pericytes, where they contribute to guidance and stabilization of endothelial sprouts. After intravenous administration of biotin-BSA-GdDTPA-(FAM) to mice with subcutaneous MLS human ovarian carcinoma tumors, distribution of the macromolecular MRI/optical contrast material was confined to blood vessels in normal tissues, while it co-registered with αSMA positive stroma tracks within the tumor. These αSMA positive tumor associated myofibroblasts and pericytes showed uptake of the contrast material into intracellular granules. Thus, we evaluated the use of this contrast material for in vitro labeling of tumor fibroblasts as an approach for tracking their involvement in angiogenesis. Fluorescence microscopy demonstrated internalization of the contrast material and MRI revealed significant increase in R1 relaxation rate of labeled fibroblasts. R1 not only remained elevated for two weeks in culture, but also increased with cell proliferation, indicating prolonged retention of the contrast material and subsequent intracellular processing and redistribution of the material, thereby enhancing MR contrast. Uptake of the contrast material could be suppressed by nystatin, suggesting internalization by caveolae mediated endocytosis. This study shows that labeling of fibroblasts is feasible and may allow non-invasive in-vivo tracking of fibroblasts during tumor angiogenesis and vessel maturation.
Introduction
The integrity of epithelial tissues is highly dependent on the cross talk between the epithelium and its surrounding stroma. The stroma is comprised of fibroblasts, leucocytes and endothelial cells, which reside in an extracellular matrix (ECM) and hence, spatially separated from the epithelium by the basement membrane. When the epithelium transforms into epithelial carcinoma, the stroma inevitably changes as well (1). All solid tumors, regardless of their origin, require stroma for their progression beyond a minimal size of 1–2 mm. Tumor-associated fibroblastic stroma is often referred to as “stromal response” or desmoplasia, which is responsible for the “scirrhous” character of some carcinoma types (2–4). Indeed, the extent of fibroblastic stromal reaction in epithelial solid tumors can account for over 90% of the total tumor mass (5). This desmoplastic reaction is characterized by pronounced alterations in the phenotype and expression profile of the associated fibroblastic cells (3). Myofibroblasts are a dominant cell population in tumor desmoplasia and critically contribute to the abnormal distribution and composition of the extracellular matrix in carcinomas (2,3,6,7). They have been observed in the stroma of various epithelial tumor types such as carcinomas of the colon, breast, lung, liver, prostate and pancreas (4,8–12) and their appearance may precede the invasive tumor stage (1). Myofibroblasts are a unique fibroblastic cell population, which possess contractile qualities through the expression of smooth muscle filaments such as alpha smooth muscle actin. Although their origin remains controversial, fibroblasts are considered to be the main progenitor cells (1,2).
Angiogenesis is a basic requirement for solid tumor growth and progression. Recent studies suggested that fibroblastic tumor stroma, and in particular myofibroblasts, essentially contribute to both the angiogenic process and cancer progression (13,14). It was suggested that fibroblasts at the tumor-host interface differentiate into myofibroblasts and then into pericyte-like cells, which are proposed to guide endothelial sprouts (15). In order to verify this hypothesis and to better understand the impact of fibroblasts and myofibroblasts on the development of the abnormal tumor vasculature, interactions between endothelial cells and fibroblast cell populations in a tumor environment are to be studied. Indeed, it has been shown that matrix-bound fibroblasts enhance angiogenesis as well as endothelial cell motility (16), and co-cultures of ovarian carcinoma SKOV-3 cells with myofibroblasts facilitated invasion of endothelial cells in three-dimensional cultures (13). However, in order to gain deeper insight into the process of tumor angiogenesis, in-vivo monitoring of fibroblast/myofibroblast-endothelial cell interactions is required.
Recent studies suggested the use of MRI for in-vivo cell tracking via paramagnetic and superparamagnetic cell labeling (17,18). One of the most promising approaches is the application of membrane impermeable paramagnetic metal cation compounds. However, the application of these compounds has been limited and so far they were mostly utilized to monitor cell movement in embryogenesis and embryonic cell lineage analysis (19). Covalent attachment of Gd-DOTA to the HIV-Tat peptide was used for lymphocyte labeling by receptor mediated endocytosis (20) and the attachment of Gd-DTPA to poly-L-lysine-transferrin-DNA, enabled labeling of erythroleukemia cell line via co-transport of the contrast material with DNA during transfection (21). At present, most work in the field has been focused on detection of cells belonging to the hematopoietic system. The motivation of the present study was to establish and verify a technique to label fibroblasts in vitro, and to characterize the uptake mechanism, retention and MR visibility of the contrast material. This work provides the basis for future in-vivo MR monitoring, with the intention to detect their interaction with the vessel wall during tumor angiogenesis and vascular remodeling.
We show here that fibroblasts can be efficiently labeled ex-vivo with biotin-BSA-GdDTPA without loss of cell viability thus providing MR detectable contrast. Additionally, in a xenograft model of human fibroblasts and human ovarian cancer cells, after intravenous administration of biotin-BSA-GdDTPA, immunohistochemical stainings detected the contrast material intracellularly in myofibroblasts. This model is of particular interest since in a previous study we reported that the exit of implanted human MLS ovarian carcinoma spheroids from dormancy correlated with infiltration of alpha SMA positive (myo)fibroblasts into the tumor mass, suggesting a critical role for these cells in initiation of tumor progression (14).
Material and Methods
Contrast material
Biotin-BSA-GdDTPA-FAM was synthesized as previously described (22). Protein concentration was determined by ultraviolet absorption at 280 nm against standard concentration of BSA. The degree of fluorescence of 5(6)-carboxyfluorescein, succinimidyl ester (FAM, Molecular Probes Inc., Eugene, Oregon) labeling was determined according to the manufacturer protocol (2–4 FAM molecules per BSA). The number of biotin groups bound to BSA was measured using HABA/avidin reagent (Sigma Chemical Co., St. Louis, MO; 1–2 biotin moieties per BSA molecule). Gadolinium (Gd) was measured by inductively coupled plasma – atomic emission spectrometer (ICP-AES, Optima 3300, Perkin Elmer, Norwalk, CT; 27 GdDTPA per BSA).
Longitudinal proton relaxation rate (R1) was determined at room temperature on a 4.7 T Bruker Biospec spectrometer (Karlsruhe, Germany) for a range of concentrations of the contrast material. Longitudinal relaxation rates were measured using spin echo images (flip angle 90°) with 10 repetition times TR 100–2000 ms, TE 10 ms for each concentration. The relaxivity of the contrast material was calculated as previously reported (22) and was found to be 186 mM−1s−1 (6.8 s−1 per mM GdDTPA).
Cell culture
Primary fibroblasts were obtained from normal skin (N1), normal breast (PFN2) and ductal breast carcinoma biopsies (PF40T – from a highly differentiated G1 breast tumor, PF42T – from a poorly differentiated invasive G3 breast tumor) and were characterized and processed as previously described (23). For the present experiments, fibroblasts were cultured from frozen stocks in DMEM (Dulbecco’s Modified Eagle’s Medium) with a CPD (cumulative population doublings) of <40. Assays were performed using primary fibroblasts of passages 3 to 7. Cos-7 cell line was cultured in DMEM medium. MLS, human ovarian epithelial carcinoma cell line, was cultured in alpha MEM (Minimal Essential Medium). All media were supplemented with 10% FCS, 100 u/ml penicillin, 0.1 mg/ml streptomycin, 0.06 mg/ml amphotericin B and 2mM L-glutamine. Media and serum were purchased from Biological Industries, Beit Haemek, Israel.
Ex-vivo cell labeling
Labeling of fibroblasts for MRI measurements was performed on cells grown to confluence in 6 cm culture plates. Fibroblasts were incubated on a rotating board in fresh culture medium supplemented with 10 mg/ml biotin-BSA-GdDTPA. After 48 hours of incubation, labeling was terminated by removal of the medium and four washes with serum free medium.
Retention of the contrast material was assayed using labeled cells (passages 6–7), which were subsequently washed and cultured in fresh medium. In some cases, cells were harvested with trypsin-EDTA following labeling, washed, and re-plated in fresh medium. After an additional period of culture of up to two weeks, cells were analyzed by MRI. The role of caveolae in biotin-BSA-GdDTPA uptake into fibroblasts was assessed by addition of nystatin (50 μM; Sigma) to the labeling media. Correlation between proliferation and relaxivity of the contrast material was determined using cos-7 cell line. Cos-7 cells, grown to sub-confluence in a 10 cm dish, were labeled with 10 mg/ml biotin-BSA-GdDTPA for 24 hours. At each subsequent time point 1*106 cells in log phase were taken for MRI analysis and 2*106 were seeded for further culturing in 10 cm plates.
Fluorescence imaging of labeled cells was performed with cells grown to sub-confluence on 12-mm coverslips. Cells were labeled with the fluorescent (FAM) conjugated contrast material for 48 hours as described above. Cells washed with serum free medium, were fixed (20 minutes in 3% paraformaldehyde), and nuclei were stained (5 μg/ml propidium iodide; PI, Sigma). The contrast material and PI were detected by fluorescence confocal microscopy (Axiovert 100; Zeiss, Goettingen, Germany).
Cell survival was determined for cells cultured in 96 well plates by neutral red accumulation as previously described (24). Mean net optical density (570 nm; after subtraction of assay blanks) was computed (mean ± SEM).
MRI measurements of ex-vivo labeled cells
NMR microscopy measurements were performed on a 400 MHz (9.4 Tesla) vertical wide-bore DMX spectrometer, equipped with a microimaging attachment with a 5 mm Helmholtz radio frequency coil (Bruker). Labeled and control cells (primary human fibroblasts, 3×105 cells; and cos-7 cell line, 1*106 cells) were washed four times in serum free medium, harvested with trypsin-EDTA and suspended in 1% low gelling temperature agarose (Sigma; 200 μl/500 μl for primary human fibroblasts and cos-7 cells respectively), in 5 mm (od) NMR tubes (Wilmad Glass Company, Buena, NJ).
R1 measurements: Spin echo images were acquired with different repetition times (TR values 100–2000 ms); TE 8.8 ms; 128 ×128 pixels; 2 averages; slice thickness of 1mm; field of view (FOV) of 1.5 cm × 0.6 cm; in-plane resolution 117 × 47 μm; spectral width (SW) 50,000 Hz.
MRI data were analyzed using Matlab computing software (The Matlab Work Inc, Natick, MA, USA). Images acquired with 10 different TR values were used for generation of R1 maps as well as for calculation of average R1 values in selected regions of interest (ROI), by non-linear least square pixel-by-pixel fitting to a single exponent. Mean ΔR1 was calculated from the differences in R1 between labeled and unlabeled cells (mean values ± SEM are reported). The student’s t-test (paired, one tail) was used for statistical analysis to compare control and labeled groups.
Animal experiments
Animal experiments were approved by the Weizmann Institutional Animal Care and Use Committee. CD1-nude mice (female; n=4; 6–10 weeks-old; body weight, 28–30 g) were studied 14–20 days after subcutaneous co-inoculation in the hind limb of MLS ovarian carcinoma cells and PF42T fibroblasts derived from a desmoplastic, poorly differentiated breast tumor (1*106 and 2.5*105 cells/mouse, respectively). Mice were anesthetized by intra-peritoneal injection of 75 mg/kg ketamine (Fort Dodge Animal Health, Fort Dodge, Iowa) and 3 mg/kg xylazin (XYL-M2, V.M.D, Arendox, Belgium), followed by additional subcutaneous injection of half a dose. The tail vein was catheterized and biotin-BSA-GdDTPA was injected intravenously (12 mg/mouse in 0.2 ml PBS). Mice were sacrificed approximately 30 minutes after administration of the contrast material. Tumor samples were removed and fixed in Carnoy’s solution (6:3:1 ethanol/chloroform/acetic acid).
Immunohistochemistry
Fixed tumor samples were embedded in paraffin blocks and serially sectioned (4-μm). Following deparaffinization with xylene and rehydration in ethanol (100%, 95%, 70% and DDW), sections were blocked and incubated with monoclonal anti-alpha smooth muscle actin (αSMA) antibody (alkaline phosphatase (AP) conjugated; 1:30, Sigma). Biotinylated contrast material was detected by incubation with avidin-FITC (1:40, Sigma). Nuclei were counter-stained with Hoechst reagent (1:1000 in PBS, Molecular Probes). Sections were sealed with antifade reagent (Molecular Probes).
RESULTS
In-vivo labeling of tumor stroma
Tumor xenografts were initiated from a human ovarian carcinoma cell line (MLS) co-inoculated with human tumor-derived fibroblasts (PF42T). Mice were sacrificed approximately 30 minutes after intravenous administration of biotin-BSA-GdDTPA. The distribution of the biotinylated contrast material in the tumor mass was detected by avidin-FITC staining of thin histological sections. During the time interval between intravenous administration of the contrast material and sacrifice of the mice, contrast material leaked from blood vessels into the tissue at sites of hyper-permeability (figure 1). Low magnification views (e.g. figure 1a) revealed that the contrast material was localized at the tumor rim delineating the borders of the tumor (figure 1; arrowheads) with channels extending into the tumor inner mass. Images with a higher magnification (figure 1b) showed that the contrast material was not chaotically scattered but rather reflected a pattern associated with specific types of stromal cell tracks.
Figure 1. Distribution of the contrast material in MLS tumors.
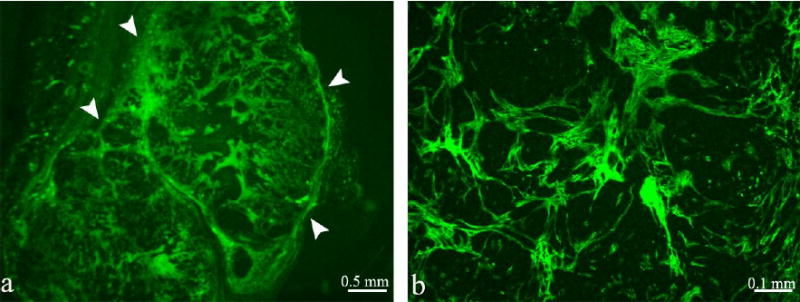
Fluorescence microscopy of representative tumors initiated by co-inoculation of MLS cells and PF42T fibroblasts. Biotin-BSA-GdDTPA was administered intravenously into the tail vein of a tumor bearing mice (16 days after implantation; 30 minutes prior to tumor retrieval). The distribution of biotin-BSA-GdDTPA was detected by staining with Avidin-FITC. Tumor borders are illustrated by arrowheads (a).
Since myofibroblasts are the most abundant cell type in the stroma of epithelial carcinomas, we double stained the tumor specimens with avidin-FITC and anti-alpha smooth muscle actin (αSMA). The results not only verified the association between contrast material and myofibroblasts, but also confirmed that contrast material remained confined to areas of αSMA positive myofibroblasts, without penetrating the ‘nodules’ of tumor cells (figure 2). Some contrast material was detected within intracellular granules in these myofibroblasts (arrow heads). Contrast material could be found localized in alpha SMA positive cells which surround a large blood vessel (Figure 2, Tumor #1; yellow), as well as in alpha SMA negative tracks which were surrounded by alpha SMA positive cells. Thus, we evaluated the potential of exploiting this uptake for development of a new approach for ex vivo labeling of tumor associated fibroblasts and myofibroblasts.
Figure 2. Internalization of contrast material into myofibroblasts in vivo.
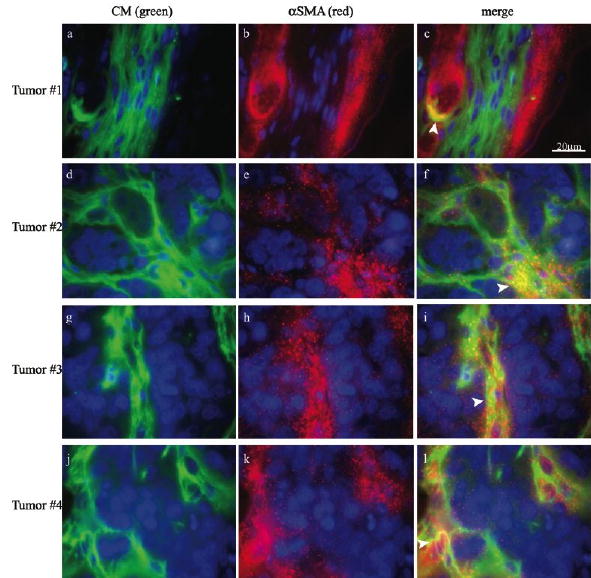
Immunohistochemical fluorescence microscopy of representative images acquired from four individual tumors initiated by co-inoculation of MLS cells and PF42T fibroblasts. Biotinylated contrast material (green; avidin-FITC; a, d, g, j) was intravenously administrated into the tail vein of tumor bearing mice (16 days after implantation; 30 minutes prior to tumor retrieval). αSMA staining of pericytes, vascular smooth muscle cells and myofibroblasts (red; b, e, h, k), merged images (c, f, i, l), and nuclear staining (blue; a-f). Arrowheads indicate intracellular uptake.
Ex vivo labeling of fibroblasts with biotin-BSA-GdDTPA-FAM
In view of the importance of fibroblasts and myofibroblasts in regulating tumor perfusion, and the intracellular uptake of contrast material into these cells, we assessed the use of biotin-BSA-GdDTPA-FAM for ex-vivo pre-labeling of fibroblasts. Tumor-derived and normal human breast fibroblasts were incubated with biotin-BSA-GdDTPA-FAM (48 hours; 10 mg/ml), followed by extensive washes for removal of excess contrast material. Cells labeled with the fluorescent tagged contrast material, were fixed and counter stained with propidium iodide (5μg/ml) to identify all cell nuclei. In contrast to unlabeled fibroblasts, the cytoplasm of labeled PFN2 normal breast fibroblasts showed high fluorescence (green), demonstrating internalization of the fluorescein labeled contrast material (figure 3a). After 48 hours of incubation, 100% of the cells were fluorescently labeled. Thus, the contrast material was internalized into the cytoplasm of all cells revealing the high efficiency of this labeling procedure.
Figure 3. Viability of fibroblasts after ex-vivo labeling.
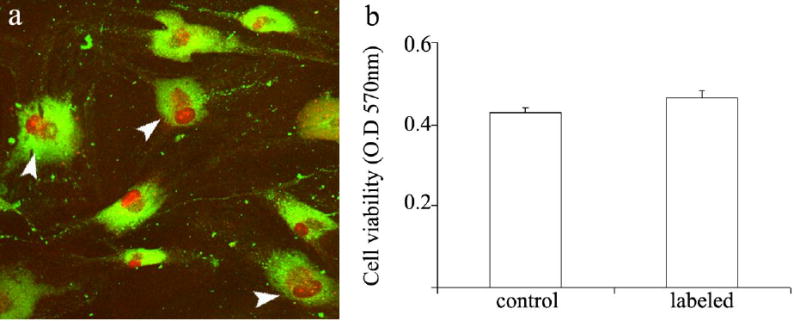
a) Confocal microscopy of PFN2 fibroblasts double labeled with biotin-BSA-GdDTPA-FAM (48 hours; 10 mg/ml) and propidium iodide (nuclear staining). Arrowheads indicate proliferating cells. b) Neutral red incorporation was used to measure cell viability in control unlabeled cells and in labeled cells. Optical density was measured at 570 nm (mean ± SEM; n=9; p=0.12, t-test un-paired, two tail).
Cells remained viable following labeling
The viability of cells treated with biotin-BSA-GdDTPA was evaluated after 48 hours of labeling (10 mg/ml) using the neutral red viability assay (figure 3b). The number of viable cells in labeled fibroblasts did not differ from an unlabeled control. Hence, the cell viability was not affected by the labeling procedure. In addition, confocal microscopy revealed cells which were in the process of cell division (figure 3a, arrowheads).
Labeled cells are MR visible
In order to determine whether the intracellular contrast material is detectable by MRI, we measured R1 relaxation rates of control and labeled cells (48 hours; biotin-BSA-GdDTPA 10 mg/ml). MR images of fibroblasts suspended in agarose (3×105 cells/0.2 ml) were obtained for derivation of R1 relaxation maps (Figure 4 a,b) and calculation of average R1 at selected ROIs (Figure 4 c,d). Data analysis revealed a significant increase in R1 relaxation rates of labeled cells. In detail, R1 values of labeled fibroblasts derived from normal skin, normal breast or breast tumor (N1, PFN2 and PF40T/PF42T, respectively) were compared to R1 values of corresponding unlabeled control fibroblasts. In all cases, incubation with biotin-BSA-GdDTPA significantly elevated R1 value, resulting in positive ΔR1 (figure 4d; ΔR1=R1(labeled)−R1(unlabeled) >0 for all fibroblast types; p<0.03). The increase in R1 relaxation rate is in accord with both intracellular uptake and MR visibility of the intracellular contrast material.
Figure 4. Detectable increase in R1 relaxation rate of fibroblasts labeled with biotin-BSA-GdDTPA.
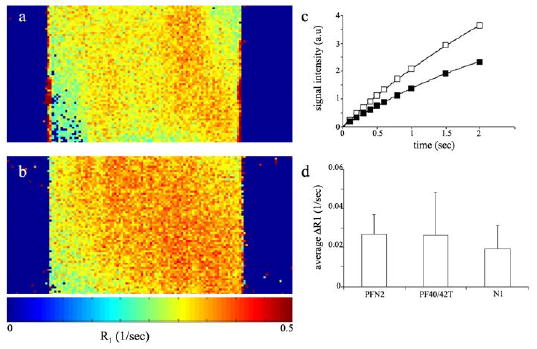
T1 weighted images of control and labeled cells were acquired on a 400 MHz spectrometer. (a) Representative R1 maps of unlabeled cells and (b) cells labeled with biotin-BSA-GdDTPA contrast material (48 hours; 10 mg/ml). (c) Non linear single exponential fittings of signal intensity as a function of TR, used for derivation of R1 for unlabeled cells (close squares) and for labeled cells (open squares). (d) MRI detection of the change in R1 (ΔR1=R1(labeled)−R1(unlabeled), obtained from ROI analysis) of fibroblasts labeled with biotin-BSA-GdDTPA compared with that of unlabeled cells (PFN2 n=4; PF40/42T n=3; N1 n=2; * significant change in R1 of labeled versus unlabeled cells p<0.03, t-test paired, one tail).
Caveolae mediated internalization of biotin-BSA-GdDTPA
Most cells internalize albumin via endo- or pinocytosis in a process that could be mediated through caveolae (25). The mechanism of contrast material internalization into fibroblasts was evaluated by the addition of nystatin to the labeling media. Nystatin, a general inhibitor of caveolae mediated endocytosis, is known to precipitate cholesterol in the plasma membrane of the cell, thereby disrupting caveolae function (26). Fibroblasts were labeled with biotin-BSA-GdDTPA (48 hours; 10mg/ml) with or without the addition of nystatin (50 μM). MRI measurements of R1 showed that uptake of biotin-BSA-GdDTPA was significantly suppressed by the presence of nystatin in the labeling media (figure 5a, p=0.006). In all fibroblast types, the elevation in R1 was considerably restrained by nystatin, and in PFN2 fibroblasts ΔR1 was almost abolished. Cells that were grown in the presence of nystatin remained intact as was visualized by microscopy prior to MRI assays, and viable, as demonstrated by neutral red viability assay, performed with nystatin in the labeling medium (figure 5b). The inhibition of labeling of cells by nystatin, emphasizes the role of specific endocytosis and caveolae in the internalization of biotin-BSA-GdDTPA into fibroblasts ex-vivo.
Figure 5. Nystatin inhibits internalization of biotin-BSA-GdDTPA into fibroblasts.
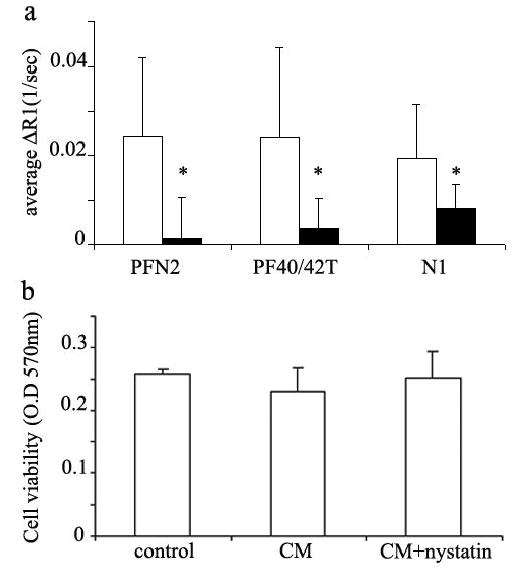
a) Various primary fibroblasts were labeled with biotin-BSA-GdDTPA (48 hours; 10 mg/ml) in the presence (close bars) or absence (open bars) of the caveolae inhibitor nystatin (50 μM). (PFN2 n=3; PF40/42T n=2; N1 n=2; * p=0.006 for nystatin effect t-test paired, one tail). b) Neutral red incorporation was used to measure cell viability in cos-7 cells after incubation with CM and nystatin. Optical density was measured at 570 nm (mean ± SEM; n=10 p>0.5, t-test un-paired, two tail).
MR signal was retained in labeled cells for at least two weeks in culture
For in-vivo follow-up of pre-labeled cells, the contrast material should remain detectable in the cells or their progeny over a sufficient period. Thus, retention of biotin-BSA-GdDTPA was determined in both normal PFN2 and tumor-derived PF42T fibroblasts. Also, the ability of labeled cells to re-adhere to the surface of the culture plate was assessed. After incubation with biotin-BSA-GdDTPA (48 hours; 10 mg/ml) and extensive washes, cells were either incubated in fresh medium for an additional culturing period (up to two weeks), or harvested with trypsin-EDTA before re-plating them in fresh medium for further culturing. After two weeks in culture, cells were harvested, counted, suspended in agarose (3 × 105 cells/0.2 ml), and R1 was assessed (figure 6a). MRI measurements showed that even after two weeks of subsequent culturing, biotin-BSA-GdDTPA labeled cells maintained significantly higher R1 values than unlabeled control cells (p<0.01). In addition, the ability of the cells to re-adhere to culture plates was not affected by labeling treatment, and vise versa, harvesting and re-plating did not affect R1 values of labeled cells. Thus, MR detectable contrast material was retained in the cells for at least two weeks.
Figure 6. Prolonged intracellular retention and enhanced visibility of the contrast material during cell proliferation.
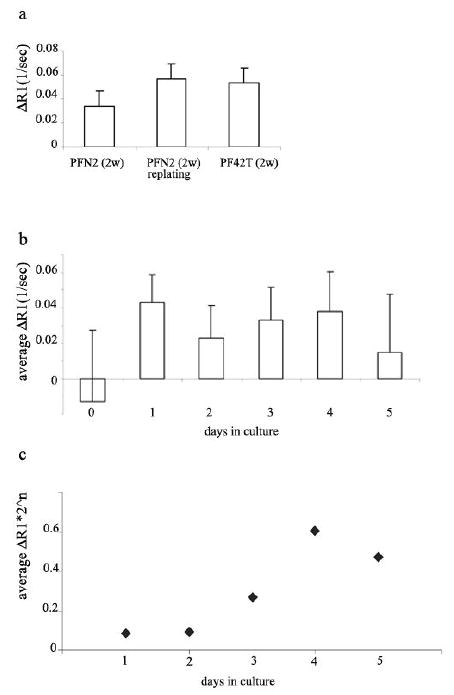
a) PFN2 and PF42T fibroblasts were labeled with biotin-BSA-GdDTPA (48 hours; 10 mg/ml), and incubated in fresh medium for up to two weeks (PFN2 2w; PF42T 2w), or harvested prior to additional incubation (PFN2 2w re-plating). ΔR1 was measured after two weeks of further culturing (* p<0.01 for labeling retention; t-test paired, one tail). b) Cos-7 cells were labeled with the contrast material (24 hours; 10 mg/ml). ΔR1 was measured daily, each time for new 1×106 cells harvested from log phase monolayer culture (* p<0.03 for labeling retention- ΔR1>0 relative to unlabeled cells; t-test paired, one tail). c) Effective relaxivity (Ref) of the contrast material was derived from ΔR1 (b) according to the equation: Ref α ΔR1 * 2n (where n is the number of cell divisions)
Relaxivity of the intracellular contrast material increased with cell proliferation
Unexpected, further enhancement of the labeling contrast was observed with time after labeling, despite the dilution of label by cell proliferation (the same number of cells was taken for each measurement; figures 6a and 4d). Thus, after additional incubation of the cells, the observed ΔR1 was greater than the ΔR1 measured immediately after labeling. In order to evaluate the impact of cell proliferation on the relaxivity of the contrast material, we conducted an additional experiment using cos-7 cell line. As opposed to primary fibroblasts which are characterized by a very moderate proliferation rate and undergo senescence after a few cell divisions, cos-7 cells divide every 24 hours. After incubation with biotin-BSA-GdDTPA (24 hours; 10 mg/ml) excess contrast material was removed by four washes. After harvesting with trypsin, a fraction of cells was immediately analyzed by MRI (1*106 cells) while the remaining cells were re-plated for further divisions (2*106 cells/10 cm plate). R1 was determined on a daily basis for log phase cells, suspended in agarose (each time 1*106 cells/0.5 ml agarose). The results as are shown in figure 6b revealed that ΔR1 persists throughout the four initial cell cycles. Hence, although cells proliferated resulting in 2 fold dilution of the contrast material every 24 h, no corresponding decrease in ΔR1 could be observed. The effective relaxivity of the intracellular contrast material (Ref ), derived by assuming 24 h doubling time (Ref = ΔR1 * 2n), highlights the increased visibility of Gd in the labeled cells after a few cell divisions (Figure 6c).
DISCUSSION
MRI is emerging as a powerful tool for cell tracking via paramagnetic and superparamagnetic cell labeling (27–34). Superparamagnetic iron oxide (SPIO) particles have been used both clinically (27) and in research studies of various biological processes such as detection of stem cells, tumor cells and tumor associated macrophages; migration and homing of stem cells to bone marrow, lymphocytes in the liver, spleen and pancreas, as well as for detection of T-cells and THP-1 phagocytes (28–32). More recently ultra small superparamagnetic iron oxide (USPIO) particles have been used to perform lymphography and detection of T-cells in rat testicles and macrophages within the central nervous system (33,34). However, paramagnetic metal cation compounds which are membrane impermeable were mostly utilized by microinjection, by covalent attachment to specific peptides or in conjugation with transfection agents (19,20).
In the study reported here, we demonstrated that administration of multi-modality triply labeled albumin-based contrast material results in its spontaneous internalization into tumor associated myofibroblasts, where it appears to be localized in intracellular granules. This was established both ex-vivo by incubation of cells with the contrast material, as well as in vivo by intravenous administration of the contrast material to tumor bearing mice. Ex-vivo, incubation of human fibroblasts derived from skin, normal breast and breast tumor tissues with the fluorescent-conjugated contrast material confirmed uptake of biotin-BSA-GdDTPA-FAM into cytoplasmic granules, presumably lysosomes. The physiological effects of labeling were evaluated in order to verify that uptake of the contrast material does not impact cellular function. The parameters tested included the ability of cells to adhere to the surface of culture plate, cell membrane integrity (viability) and cell division. Dividing cells were visualized by confocal microscopy revealing multiple labeled cells in mitosis. Similarly, adhesion of cells after trypsinization and the number of viable cells after continuous culture were unaffected by the labeling procedure and uptake, indicating an unaltered balance in cell death and proliferation characteristics of the fibroblast cultures.
Internalization of the contrast material was detectable not only by fluorescence microscopy but also by MRI. R1 relaxation rates of the three primary human fibroblast types were significantly elevated after incubation with the contrast material. Thus, not only was the contrast material internalized into the cells, but it was MR visible as well. Uptake and internalization of the contrast material, as manifested by elevated R1 relaxation rate, was effectively suppressed by treatment of cells with nystatin, a cholesterol-sequestering agent, suggested as a specific inhibitor of caveolae mediated endocytosis. Thus, these results implicate caveolae in mediating endocytosis and accumulation of the contrast material in fibroblasts. The role of caveolae was further supported by the preliminary finding that all three fibroblast types assessed in this study, express caveolin-1, the most abundant component of caveolae (data not shown).
Retention of labeling was demonstrated in fibroblasts, by culturing cells for up to two weeks following the labeling process. R1 relaxation rates of the labeled cells remained considerably elevated relative to unlabeled control cells. Moreover, relaxation rates increased with time in culture. Following proliferation, despite dilution of the contrast material, the change in R1 was similar throughout the first four cell cycles for the same number of suspended cells. Derivation of the effective relaxivity of the contrast material showed increased visibility of Gd during the first four cell cycles. Thus, we conclude that the effective relaxivity of the contrast material increased with cell proliferation. This finding is consistent with initial internalization of the contrast material into intracellular granules, which was previously demonstrated to limit its detection by MRI (35,36). Cell proliferation along with catabolism of albumin through lysosomal degradation would result in redistribution of the contrast material in intracellular compartments, hence increasing its effect on water relaxation. The early sequestration of the contrast material in granules along with the finding that relaxivity may be enhanced with proliferation, would improve sensitivity for in vivo detection of proliferation of fibroblasts in tumors, and furthermore suggests that it might be possible to develop ways that would allow to determine the number of cell divisions from the relative changes in R1 and R2 relaxation rates.
In summary, triply labeled albumin was developed here for detection of tumor associated fibroblasts by MRI and optical imaging. Uptake of the contrast material via caveolae mediated endocytosis into intracellular granules resulted in significant enhancement in R1 relaxation rate. These results provide the basis for future use of MRI to analyze the recruitment of fibroblasts, pre-labeled with biotin-BSA-GdDTPA-FAM, during tumor progression. Tracking fibroblasts in tumor models could provide insight into the mechanism of how fibroblasts differentiate in tumors and help support tumor angiogenesis and vessel maturation. Indeed, while in vitro co-culture models of endothelial cells and myofibroblasts suggested their fundamental impact on tumor angiogenesis (1,13,15), the interactions that occur in vivo are still not well understood and the role of fibroblasts in vessel formation and maturation is still ambiguous. It is well known, however, that both endothelial cells and fibroblasts are located in close association in many epithelial tumors, i.e. in those expressing a desmoplastic response. We recently reported the critical role of myofibroblasts in the initiation of growth of dormant MLS ovarian epithelial tumor xenografts (14). These tumor xenografts are not only massively infiltrated by αSMA positive stromal cells (myofibroblasts/pericytes) but they are also characterized by a moderate and non-leaky vasculature. Fibroblasts pre-labeled with biotin-BSA-GdDTPA-FAM in combination with MRI and the xenograft model could provide a fundamental tool that would help in elucidating the role of fibroblasts in tumor angiogenesis.
Acknowledgments
This work was supported by the Mark Family Foundation, by the Israel Science Foundation and by the USA NCI RO1 CA75334.
References
- 1.De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200(4):429–447. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- 2.Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264(1):169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 3.Kunz-Schughart LA, Knuechel R. Tumor-associated fibroblasts (part II): Functional impact on tumor tissue. Histol Histopathol. 2002;17(2):623–637. doi: 10.14670/HH-17.623. [DOI] [PubMed] [Google Scholar]
- 4.Kunz-Schughart LA, Knuechel R. Tumor-associated fibroblasts (part I): Active stromal participants in tumor development and progression? Histol Histopathol. 2002;17(2):599–621. doi: 10.14670/HH-17.599. [DOI] [PubMed] [Google Scholar]
- 5.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 6.Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76(1):69–125. doi: 10.1152/physrev.1996.76.1.69. [DOI] [PubMed] [Google Scholar]
- 7.Sappino AP, Skalli O, Jackson B, Schurch W, Gabbiani G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int J Cancer. 1988;41(5):707–712. doi: 10.1002/ijc.2910410512. [DOI] [PubMed] [Google Scholar]
- 8.Miura S, Kodaira S, Hosoda Y. Immunohistologic analysis of the extracellular matrix components of the fibrous stroma of human colon cancer. J Surg Oncol. 1993;53(1):36–42. doi: 10.1002/jso.2930530111. [DOI] [PubMed] [Google Scholar]
- 9.Neaud V, Faouzi S, Guirouilh J, Le Bail B, Balabaud C, Bioulac-Sage P, Rosenbaum J. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: evidence for a role of hepatocyte growth factor. Hepatology. 1997;26(6):1458–1466. doi: 10.1053/jhep.1997.v26.pm0009397985. [DOI] [PubMed] [Google Scholar]
- 10.Doucet C, Jasmin C, Azzarone B. Unusual interleukin-4 and -13 signaling in human normal and tumor lung fibroblasts. Oncogene. 2000;19(51):5898–5905. doi: 10.1038/sj.onc.1203933. [DOI] [PubMed] [Google Scholar]
- 11.Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, Jesnowski R. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61(2):550–555. [PubMed] [Google Scholar]
- 12.Rowley DR. What might a stromal response mean to prostate cancer progression? Cancer Metastasis Rev. 1998;17(4):411–419. doi: 10.1023/a:1006129420005. [DOI] [PubMed] [Google Scholar]
- 13.Walter-Yohrling J, Pratt BM, Ledbetter S, Teicher BA. Myofibroblasts enable invasion of endothelial cells into three-dimensional tumor cell clusters: a novel in vitro tumor model. Cancer Chemother Pharmacol. 2003;52(4):263–269. doi: 10.1007/s00280-003-0664-2. [DOI] [PubMed] [Google Scholar]
- 14.Gilead A, Meir G, Neeman M. The role of angiogenesis, vascular maturation, regression and stroma infiltration in dormancy and growth of implanted MLS ovarian carcinoma spheroids. Int J Cancer. 2004;108(4):524–531. doi: 10.1002/ijc.11583. [DOI] [PubMed] [Google Scholar]
- 15.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 16.Martin TA, Harding KG, Jiang WG. Regulation of angiogenesis and endothelial cell motility by matrix-bound fibroblasts. Angiogenesis. 1999;3(1):69–76. doi: 10.1023/a:1009004212357. [DOI] [PubMed] [Google Scholar]
- 17.Kircher MF, Allport JR, Graves EE, Love V, Josephson L, Lichtman AH, Weissleder R. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003;63(20):6838–6846. [PubMed] [Google Scholar]
- 18.Modo M, Mellodew K, Cash D, Fraser SE, Meade TJ, Price J, Williams SC. Mapping transplanted stem cell migration after a stroke: a serial, in vivo magnetic resonance imaging study. Neuroimage. 2004;21(1):311–317. doi: 10.1016/j.neuroimage.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 19.Huber MM, Staubli AB, Kustedjo K, Gray MH, Shih J, Fraser SE, Jacobs RE, Meade TJ. Fluorescently detectable magnetic resonance imaging agents. Bioconjug Chem. 1998;9(2):242–249. doi: 10.1021/bc970153k. [DOI] [PubMed] [Google Scholar]
- 20.Bhorade R, Weissleder R, Nakakoshi T, Moore A, Tung CH. Macrocyclic chelators with paramagnetic cations are internalized into mammalian cells via a HIV-tat derived membrane translocation peptide. Bioconjug Chem. 2000;11(3):301–305. doi: 10.1021/bc990168d. [DOI] [PubMed] [Google Scholar]
- 21.Kayyem JF, Kumar RM, Fraser SE, Meade TJ. Receptor-targeted co-transport of DNA and magnetic resonance contrast agents. Chem Biol. 1995;2(9):615–620. doi: 10.1016/1074-5521(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 22.Dafni H, Israely T, Bhujwalla ZM, Benjamin LE, Neeman M. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer Res. 2002;62(22):6731–6739. [PubMed] [Google Scholar]
- 23.Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, Kunz-Schughart LA. Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. Eur J Immunol. 2003;33(5):1311–1320. doi: 10.1002/eji.200323057. [DOI] [PubMed] [Google Scholar]
- 24.Borenfreund E, Puerner JA. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol Lett. 1985;24(2–3):119–124. doi: 10.1016/0378-4274(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 25.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H, Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276(41):38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 26.Stuart ES, Webley WC, Norkin LC. Lipid rafts, caveolae, caveolin-1, and entry by Chlamydiae into host cells. Exp Cell Res. 2003;287(1):67–78. doi: 10.1016/s0014-4827(03)00059-4. [DOI] [PubMed] [Google Scholar]
- 27.Ferrucci JT, Stark DD. Iron oxide-enhanced MR imaging of the liver and spleen: review of the first 5 years. AJR Am J Roentgenol. 1990;155(5):943–950. doi: 10.2214/ajr.155.5.2120963. [DOI] [PubMed] [Google Scholar]
- 28.Ahrens ET, Feili-Hariri M, Xu H, Genove G, Morel PA. Receptor-mediated endocytosis of iron-oxide particles provides efficient labeling of dendritic cells for in vivo MR imaging. Magn Reson Med. 2003;49(6):1006–1013. doi: 10.1002/mrm.10465. [DOI] [PubMed] [Google Scholar]
- 29.Hinds KA, Hill JM, Shapiro EM, Laukkanen MO, Silva AC, Combs CA, Varney TR, Balaban RS, Koretsky AP, Dunbar CE. Highly efficient endosomal labeling of progenitor and stem cells with large magnetic particles allows magnetic resonance imaging of single cells. Blood. 2003;102(3):867–872. doi: 10.1182/blood-2002-12-3669. [DOI] [PubMed] [Google Scholar]
- 30.Bulte JW, Duncan ID, Frank JA. In vivo magnetic resonance tracking of magnetically labeled cells after transplantation. J Cereb Blood Flow Metab. 2002;22(8):899–907. doi: 10.1097/00004647-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Moore A, Sun PZ, Cory D, Hogemann D, Weissleder R, Lipes MA. MRI of insulitis in autoimmune diabetes. Magn Reson Med. 2002;47(4):751–758. doi: 10.1002/mrm.10110. [DOI] [PubMed] [Google Scholar]
- 32.Foster-Gareau P, Heyn C, Alejski A, Rutt BK. Imaging single mammalian cells with a 1.5 T clinical MRI scanner. Magn Reson Med. 2003;49(5):968–971. doi: 10.1002/mrm.10417. [DOI] [PubMed] [Google Scholar]
- 33.Weissleder R, Elizondo G, Josephson L, Compton CC, Fretz CJ, Stark DD, Ferrucci JT. Experimental lymph node metastases: enhanced detection with MR lymphography. Radiology. 1989;171(3):835–839. doi: 10.1148/radiology.171.3.2717761. [DOI] [PubMed] [Google Scholar]
- 34.Dousset V, Delalande C, Ballarino L, Quesson B, Seilhan D, Coussemacq M, Thiaudiere E, Brochet B, Canioni P, Caille JM. In vivo macrophage activity imaging in the central nervous system detected by magnetic resonance. Magn Reson Med. 1999;41(2):329–333. doi: 10.1002/(sici)1522-2594(199902)41:2<329::aid-mrm17>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi H, Kawamoto S, Saga T, Sato N, Ishimori T, Konishi J, Ono K, Togashi K, Brechbiel MW. Avidin-dendrimer-(1B4M-Gd)(254): a tumor-targeting therapeutic agent for gadolinium neutron capture therapy of intraperitoneal disseminated tumor which can be monitored by MRI. Bioconjug Chem. 2001;12(4):587–593. doi: 10.1021/bc010002o. [DOI] [PubMed] [Google Scholar]
- 36.Geninatti Crich S, Barge A, Battistini E, Cabella C, Coluccia S, Longo D, Mainero V, Tarone G, Aime S. Magnetic resonance imaging visualization of targeted cells by the internalization of supramolecular adducts formed between avidin and biotinylated Gd(3+) chelates. J Biol Inorg Chem. 2005;10(1):78–86. doi: 10.1007/s00775-004-0616-2. [DOI] [PubMed] [Google Scholar]