

Abstract
The clinical utility of cisplatin to treat human malignancies is often limited by the development of drug resistance. We have previously shown that cisplatin-resistant human KB adenocarcinoma cells that are cross-resistant to methotrexate and heavy metals have altered endocytic recycling. In this work, we tracked lipids in the endocytic recycling compartment (ERC) and found that the distribution of the ERC is altered in KB-CP.5 cells compared to parental KB-3-1 cells. A tightly clustered ERC is located near the nucleus in parental KB-3-1 cells, but it appears loosely arranged and widely dispersed throughout the cytoplasm in KB-CP.5 cells. The altered distribution of the ERC in KB-CP.5 cells is related to the amount and distribution of stable detyrosinated microtubules (Glu-α-tubulin), as previously shown in CHO B104-5 cells that carry a temperature-sensitive Glu-α-tubulin allele. In addition, B104-5 cells with a dispersed ERC under non-permissive conditions were more resistant to cisplatin compared to B104-5 cells with a clustered ERC under permissive conditions. We conclude that resistance to cisplatin might be due, in part, to reduced uptake of cisplatin resulting from an endocytic defect reflecting defective formation of the ERC, possibly related to a shift in the relative amounts and distributions of stable microtubules.
Keywords: endocytic recycling compartment, cisplatin resistance, detyrosinated microtubules, transferrin
INTRODUCTION
Cisplatin (CP) is widely used in the treatment of solid tumors (1). The mechanism of CP entry into the cell and its intracellular transport from the cytosol to the nucleus are complex. Recent studies suggest that entry requires interaction with membrane proteins (2) and that endocytic recycling is defective in cisplatin-resistant (CP-r) cells (3). After entry, CP becomes aquated, and approximately 1% of the total cellular cisplatin can then interact with macromolecules like DNA to form intrastrand or interstrand adducts (4). The aquated platinum species binds preferentially to the highly nucleophilic N-7 positions of guanine and adenine (5). Apart from DNA and RNA, CP also interacts with intracellular proteins and polypeptides (6) and with negatively charged phospholipids in intact human erythrocytes and tumor cells (7-9). Despite the obvious interactions of CP with membrane components, few studies have been done on entry of CP into target cells and its facilitated transport (10, 11). Since many CP-r cells show decreased uptake of CP, the identification of specific transport pathways responsible for drug resistance should provide targets for therapy aimed at circumventing or decreasing CP resistance.
We have previously shown that CP-r human KB adenocarcinoma cells have altered endocytic recycling, demonstrated by the mislocalization of membrane proteins (12). To study the recycling pathway, the transferrin receptor and its ligand, transferrin, have been used extensively (13). Within early endosomes, Fe3+ dissociates from transferrin and the transferrin receptor-transferrin complexes either return directly to the plasma membrane or reach a network of tubular membranes, called the endocytic recycling compartment (ERC), before returning to the plasma membrane. Transferrin is a bona fide marker of the ERC, and at steady-state, a majority of the internalized transferrin localizes to this compartment (14). The ERC is concentrated in the perinuclear region of many cell types, and most membrane components pass through it along their endocytic recycling itineraries. Some evidence indicates that the ERC has sorting abilities of its own, and is involved in the delivery of membrane proteins to the trans-Golgi network (13). The ERC has been confirmed to be involved in receptor and lipid recycling (13, 15) and is characterized by its tubulovesicular morphology and dependence on intact microtubules for localization (16).
In this study, we utilize fluorescently labeled transferrin, as well as fluorescent lipid analogs, to demonstrate an abnormal morphology of the ERC in CP-r cells. Instead of being concentrated in the perinuclear area, as in the parental cells, the ERC tubules in CP-r cells are distributed throughout the cytoplasm. Our results further show that this altered distribution of the ERC is similar to that seen with increased stable, detyrosinated microtubules (Glu-α-tubulin). Interestingly, mutant Chinese hamster ovary (CHO) B104-5 cells with this microtubule defect are CP-r under non-permissive conditions, compared to mutant cells under permissive conditions or to parental CHO TRVb-1 cells under conditions non-permissive for the mutant. These ERC tubules with abnormal distribution in CP-r cells also show an altered intraorganellar pH, as well as a change in the rate of metabolism of a lipid species, namely sphingomyelin. Furthermore, fluorescently-labeled methotrexate and cisplatin colocalize with the ERC structure labeled by transferrin. We conclude that cisplatin resistance and cross-resistance to methotrexate in our CP-r cells may be due to a defect in the formation and distribution of the ERC.
MATERIALS AND METHODS
Cell lines and culture conditions. KB-3-1 was originally derived from human KB epidermoid carcinoma cells (a variant of HeLa) after two subclonings from the parental cells (17). The CP-r KB-CP.5 cells were selected for resistance to 0.5 μg/ml cisplatin in a single step, and they were maintained in medium containing 0.5 μg/ml cisplatin for these experiments (12). B104-5 cells, a temperature-sensitive mutant CHO cell line, were isolated from parent TRVb-1 cells that lack the endogenous transferrin receptor and stably express the human transferrin receptor. B104-5 cells, unlike the parental TRVb-1 cells, do not grow well at the non-permissive temperature of 39°C. They are normally grown in Ham’s F12 medium containing bicarbonate, 5% Fetal Bovine Serum, 2% penicillin-streptomycin and 200 μgm/ml G418 (Geneticin) at the permissive temperature of 32°C. The mutant TRVb-1 cell line (B104-5) is frequently used to study the function of Glu-α-tubulin (18).
Endocytic compartments labeled with various markers. The KB-3-1 and KB-CP.5 cells were labeled for 1 min at 37°C with NBD-sphingomyelin, DiIC16, DiIC12 or Alexa488-transferrin, rinsed and chased for 30 min at 37°C. All lipid analogs (NBD-SM, DiIC16 and DiIC12) were added from stocks previously loaded on fatty acid free BSA, to ensure efficient transfer of monomers to the plasma membrane (19). Cells labeled with DiIC16, DiIC12 or Alexa488-transferrin were imaged live using wide field microscopy. Cells labeled with NBD-SM were chilled by washing with ice-cold Medium (∼0°C), and held on ice for 10 min for equilibration. Then, the NBD-SM on the plasma membrane was extracted by incubating the cells on ice with medium containing 5% w/v fatty acid-free BSA, a method known as “back exchange”. This was performed four times, for 10 min each (total 40 min) (20, 21). NBD-SM labeled cells were then fixed with 3% paraformaldehyde for 15 min at room temperature, and imaged using wide field microscopy.
NBD-sphingomyelin (NBD-SM) to NBD-ceramide (NBD-cer) conversion assay. Parental KB-3-1 and the cisplatin resistant KB-CP.5 cells were grown in 6-well tissue culture plates. Three wells were pooled for each sample. Cells were labeled with NBD-SM, chased and excess plasma membrane NBD-SM was back-exchanged as described in the previous section. After back exchange, cells were washed again with chilled medium, and the total cell lipids were extracted with 2 changes of hexane: isopropanol (3:2 v/v) for 30 min each.
Lipids were dried under Argon and further dried by overnight storage under vacuum. The NBD-SM and any converted NBD-Ceramide were separated by thin layer chromatography using a previously reported solvent system (Chloroform: Methanol: Water: Ammonium hydroxide at 72:48:9:2 v/v) (22). Since the NBD-Ceramide ran very close to the solvent front, the plates were first pre-run in pure chloroform to remove hydrophobic impurities. After the pre-run, the TLC plates were air-dried, and the samples spotted in 20 μl chloroform each. NBD-SM and NBD-cer standards were run on each plate to ascertain the position of the spots in the cell extracts.
After the TLC run, the plates were dried and the spots quantified using a Molecular Dynamics (Sunnyvale, CA) densitometer. The intensities of the spots were quantified using MetaMorph (Molecular Devices, Downingtown, PA) image analysis software. The images from the densitometric scans were first background corrected, and then integrated intensity was measured in a defined area covering the whole spot or a larger area (the same size area was chosen to measure all spots, and its size was chosen so as to completely include the largest spot). All further calculations were performed using the Microsoft Excel program.
Immunoblotting detection of Glu-α-tubulin and Tyr-α-tubulin. Cells were grown in 75 cm2 cell culture flasks until 80% confluence, and washed three times with ice-cold PBS buffer. Using a rubber policeman, cells were scraped into 1 ml of RIPA lysis buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.004% sodium azide in TBS) and passed 30 times through a ball-bearing homogenizer. The cellular fractions were separated as described previously (12). The samples were separated on a 4%-20% Tris-Gly gradient gel and transferred onto nitrocellulose membranes. Subsequently, membranes were subjected to immunostaining with antibodies against Glu-α-tubulin, diluted 1:1,000, for 1 h at RT (Chemicon Inc, Temecula, CA) and against Tyr-α-tubulin under the same conditions (Novus Biologicals, Littleton, CO). Enhanced chemiluminescence (ECL) reagents were used for developing signals as described by the manufacturer (Pierce Biotechnology, Rockford, IL).
Distribution of Glu-α-tubulin and Tyr-α-tubulin detected by immunohistochemistry. B104-5 cells were incubated at 32°C or 39°C individually for 3 days before fixation. The cells (KB-3-1, KB-CP.5, and B104-5 at 32°C or 39°C) were cultured on 18-mm glass coverslips, fixed in 3.3% paraformaldehyde freshly diluted in PBS for 15 min, and then permeabilized with 70% ethanol in PBS at -20°C for 10 min. Cells were subsequently washed with PBS and preblocked in 3% bovine serum albumin in PBS for 30 min, then incubated for 1 h with a primary antibody against Glu-α-tubulin (Chemicon Inc, Temecula, CA), and Tyr-α-tubulin (rat monoclonal YL1/2, Novus Biologicals, Littleton, CO), which was followed by 1 h of incubation with a rhodamine-conjugated secondary antibody (1:50) (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) before being mounted on slides with fluorescence mounting medium. Controls with nonimmune IgG were negative. Cells were processed at room temperature and photographed by immunofluorescence under a laser scanning confocal microscope (MRC-1024 confocal scan head; Bio-Rad, Hercules, CA) at 600x magnification. Background fluorescence was determined by applying only the second antibody, a rhodamine-conjugated AffiniPure goat anti-rat IgG.
In vitro colony formation assay. Briefly, an in vitro colony-forming assay was used to obtain IC50 values for B104-5 and TRVb-1 cells to cisplatin. B104-5 and TRVb-1 cells were seeded at 400 cells per 60-mm dish at 32°C or 39°C. At the time of seeding, different concentrations of individual drugs (0.05-5 μg/ml) were added to the dishes. The medium with different concentrations of drugs was changed every week. After incubation for about 4 weeks, the colonies formed at each concentration of drug were stained with 0.5% methylene blue in 50% methanol and counted. The IC50 for each cell line was calculated based on the drug concentration that reduced the number of colonies to 50% of those in the control, drug-free medium. The values are means of triplicate determinations.
RESULTS
Trafficking of lipid analogs to late endosomes/lysosomes is normal in cisplatin-resistant KB-CP.5 cells. It has been previously reported that CP-r cells exhibit an alteration in endocytosis (23), and more specifically, that their lysosomes exhibit an elevated pH. We thus wanted to determine whether this alteration affects lipid trafficking into late endosomes/lysosomes (LE/LY). Toward this purpose, we labeled both parental KB-3-1 cells and the CP-r KB-CP.5 cells with DiIC16, which is a lipid analog containing long, saturated alkyl chains. We have previously shown that this lipid analog is efficiently trafficked to LE/LY in CHO cells (19). As shown in Fig. 1A, after a half hour of chase, cell-associated DiIC16 is observed on the plasma membranes and in dispersed punctate structures throughout the cells. These punctate structures are LE/LY, since they also contain internalized high molecular weight dextrans (unpublished data). Furthermore, there is no significant difference in the distribution patterns between the parental KB-3-1 and the CP-r KB-CP.5 cells, indicating that at least bulk membrane trafficking from the plasma membranes to LE/LY is not severely affected in CP-r cells.
Figure 1.
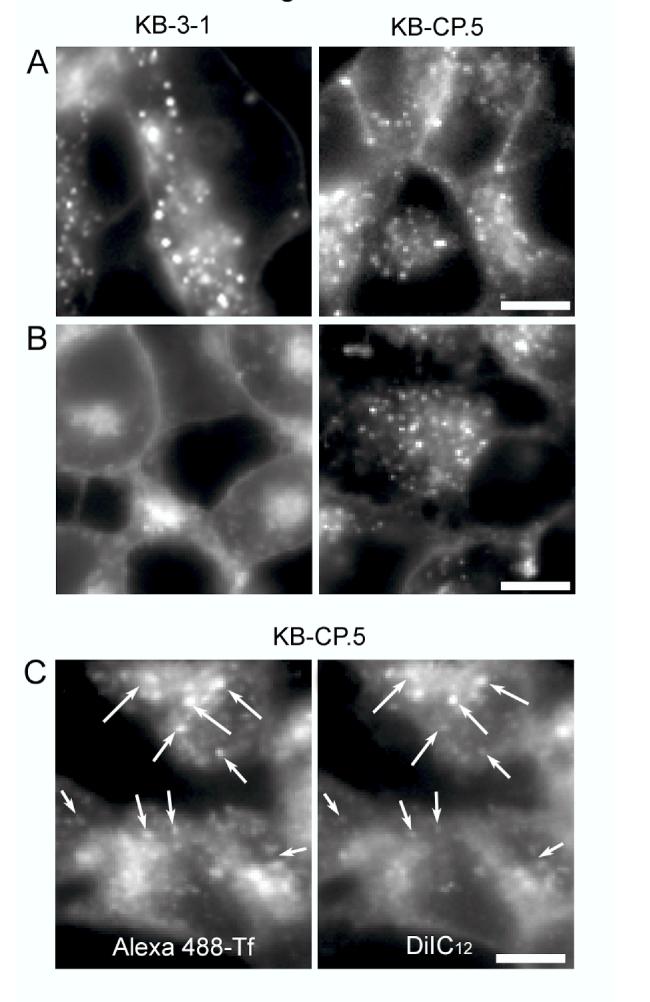
Altered ERC structures in CP-r cells as shown by labeling with lipid probes. A, DiIC16 labeling of KB-3-1 cells and KB-CP.5 cells. The cells were labeled for 1 min at 37°C with DiIC16 loaded on fatty acid free BSA, rinsed and chased for 30 min at 37°C. Cells were imaged live using wide field microscopy. Bar, 10 μm. B DiIC12 labeling of KB-3-1 cells and KB-CP.5 cells. DiIC12 labeling is similar to that of DiIC16 mentioned above. Bar, 10 μm. C, Colocalization of Alexa488-Tf and DiIC12 in KB-CP.5 cells. The cells were labeled for 5 min at 37°C with 10 μg/ml Alexa488-Tf, rinsed, labeled for 1 min with DiIC12 loaded on fatty acid free BSA, rinsed and chased for 30 min at 37°C. Cells were imaged live using wide field microscopy. Arrows point to individual endosomes or clusters of endosomes, which are colocalized. Bar, 10 μm.
Lipid analogs that are normally trafficked to the ERC show altered distribution in cisplatin-resistant KB-CP.5 cells. Since our results, presented in Fig. 1A, suggested that trafficking from the plasma membrane to the LE/LY was relatively unaffected in CP-r cells, we then decided to investigate the fate of lipid analogs that normally traffic via the other arm of endocytosis—namely, lipid analogs that are recycled to the cell surface via the ERC. For this, we carried out experiments identical to those presented in Fig. 1A, except that the lipid analog used was DiIC12, an analog containing a short alkyl chain that has been previously shown to recycle efficiently via the ERC in CHO cells (19). The results presented in Fig. 1B show that unlike DiIC16, DiIC12 exhibits a very different distribution in the cisplatin sensitive vs. resistant KB cells. In the normal parental (cisplatin-sensitive) KB-3-1 cells, the ERC distribution is similar to that of CHO cells. The ERC tubules are collected at the center of the cells near the microtubule organizing center, and appear as a bright patch of fluorescence at the cell center (Fig. 1B). In contrast, in KB-CP.5 cells, the intracellular DiIC12 appears as discrete punctate structures that are more peripherally distributed through the cells (Fig. 1B).
To further explain our observations, we double-labeled KB-CP.5 cells with DiIC12 and the green fluorescent Alexa488-transferrin. Transferrin (Tf) is a bona fide marker of the endocytic recycling pathway, and its primary intracellular localization at steady state is in the ERC. Fig. 1C shows that in the KB-CP.5 cells the Tf distribution is punctate as well, and the spots containing DiIC12 and Tf colocalize rather well in these cells (see Fig. 1C). This suggests that in KB-CP.5 cells, DiIC12 is still trafficked to the ERC, just like the parental cells, but the morphology and distribution of this organelle is altered in these cells.
The structurally altered ERC in KB-CP.5 cells sequesters fluorescently-labeled cisplatin. As shown in Fig. 1, B and C, the ERC tubules in the KB-CP cells no longer accumulate at the cell center, but instead, are now dispersed throughout the cells. A similar distribution was previously observed for fluorescently-labeled MRP1 and cisplatin (12, 24). We next asked whether such a change in ERC distribution had a functional consequence for cisplatin resistance. Toward this end, we double-labeled KB-CP cells with fluorescently labeled Tf and cisplatin, to test where they would localize inside the cells. Interestingly, we found that the intracellular CP accumulated inside these dispersed ERC tubules, which also contained Tf (see Fig. 2). This observation suggests that there is something about the properties of these altered ERC tubules that sequester cisplatin, possibly restricting its delivery to other intracellular and nuclear targets, thereby reducing cell killing, and making the cells resistant to cisplatin. We have since made the observation that these Tf-containing ERC tubules also sequester fluorescently labeled methotrexate, another chemotherapeutic agent to which these cells are cross-resistant.
Figure 2.
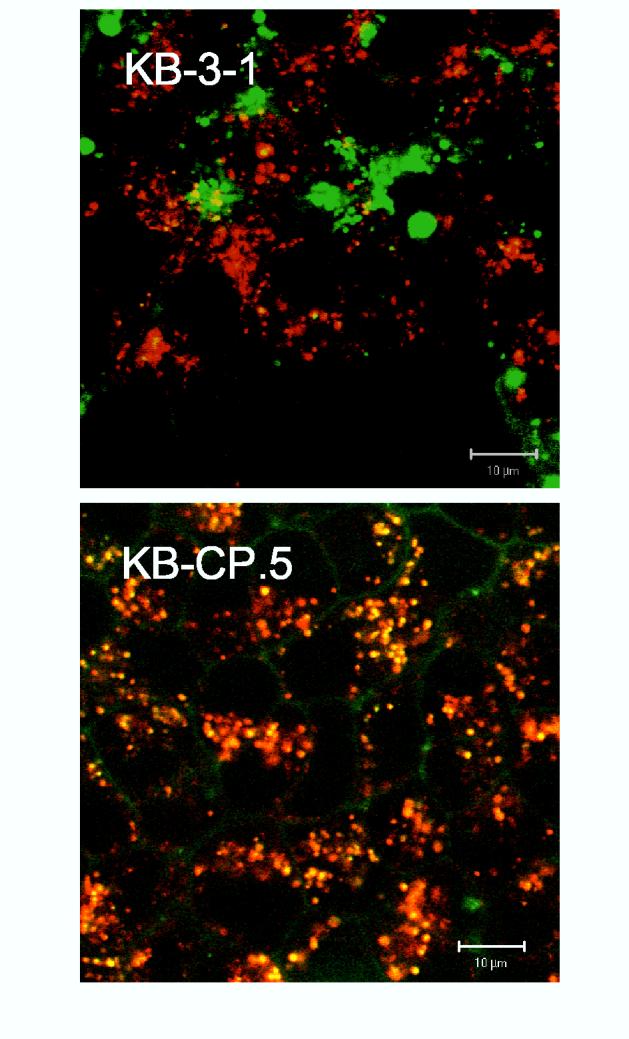
Indirect fluorescence to localize transferrin with fluorescently-labeled cisplatin. Cells were labeled with Texas Red-Transferrin (red color) and Alexa 488-cisplatin (green color) as described in “Materials and Methods”, Labeled cells were excited at 488 nm and 568 nm provided by a krypton-laser as indicated, and fluorescence emissions at 520 nm and 598 nm were used for collecting green and red fluorescence, respectively, while DIC images of the same cells were collected in the third channel using a transmitted light detector. After sequential excitation, red and green fluorescent images of the same cells were merged for co-localization analysis. Colocalization of Texas Red-Transferrin (Red) and Alexa 488-cisplatin (Green) is shown in yellow.
The structurally altered ERC in KB-CP.5 cells exhibits rapid sphingomyelin metabolism. To test whether the altered distribution observed with the normal recycling endocytic markers, namely DiIC12 and Tf, extended to other lipid analogs that normally recycle as well, we chose C6-NBD-Sphingomyelin (NBD-SM) as another model lipid. NBD-SM has one short (6-carbon) acyl chain, which is conjugated at the terminal position with the fluorescent label, NBD. We and others have previously shown that this lipid analog recycles efficiently in CHO cells (20, 21). Indeed, this is what was observed when the parental KB cells were labeled with NBD-SM and chased for a half hour to reach steady state (Fig. 3A). These cells show only the intracellular NBD-SM, since the NBD-SM on the cell surface, which normally interferes with the observation of intracellular distribution, has been back exchanged using excess fatty acid free BSA, using established procedures (20, 21).
Figure 3.
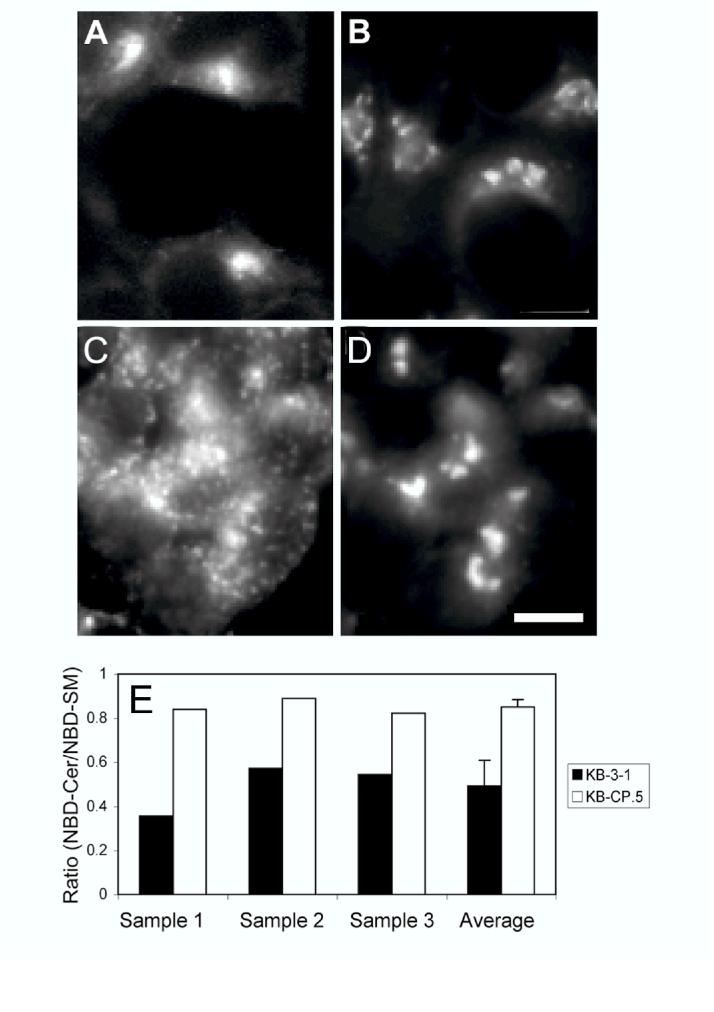
Sphingomyelin trafficking and metabolism measured in KB-3-1 and KB-CP.5 cells. A and B: NBD-SM labeling of KB-3-1 (panel A) and KB-CP.5 (panel B) cells. The cells were labeled with NBD-SM at 37° for 1 minute, chased for 30 min to achieve steady state distribution, back exchanged on ice to remove excess cell surface label, and then imaged using wide field microscopy. Bar, 10 μm. C and D: KB-CP.5 cells double-labeled with Alexa546-Tf (panel C) and NBD-SM (panel D). Negligible colocalization is observed. Bar, 10 μm. E: Ratio of NBD-Ceramide to NBD-SM in KB-3-1 cells (black bars) and KB-CP.5 cells (white bars), after excess NBD-SM on the plasma membrane has been removed by back exchange. The NBD lipid ratio thus represents primarily the NBD-lipid proportions in the ERC (since most of the NBD-SM from the plasma membranes is removed by the back exchange procedure). The protocol used for the NBD lipid ratio assay is explained in Materials and Methods. The first three sets of bars in the figure show three independent measurements, while the fourth set of bars show the average of these measurements, along with the standard deviation.
However, when the same experiment was carried out in the KB-CP.5 cells, we found that the distribution of the NBD fluorescence in these cells was rather unusual, and although located at the cell center, this distribution did not resemble that of the ERC. As shown in KB-CP.5 cells, the distribution of Alexa 546-Tf shows no overlap with that of NBD-SM (compare Fig. 3, C and D). In fact, the distribution of NBD-SM is very similar to that obtained when these cells were directly labeled with NBD-Cer (data not shown).
Lipsky and Pagano (25, 26) have shown that NBD-SM can be metabolized to NBD-Cer, which then localizes in the trans-Golgi network (TGN). We therefore postulated that this change of distribution of cells labeled with NBD-SM in the KB-CP.5 cells was due to a very fast metabolism of NBD-SM, such that NBD-SM in the ERC was rapidly being converted to NBD-Cer, which was what we observed to accumulate at the cell center, inside the TGN. To test this hypothesis, we labeled cells with NBD-SM, chased for 30 min, and back exchanged excess plasma membrane NBD-SM in a manner identical to experiments shown in Fig. 3. We then extracted the whole cell lipids using organic solvents and ran them out on TLC plates. These plates also contained NBD-SM and NBD-Cer standards for comparison. We quantified the fluorescent bands from the parental KB-3-1 and KB-CP.5 cells (see Fig. 3G), and found that the KB-CP.5 cells were indeed metabolizing NBD-SM to NBD-Cer more rapidly (∼1.7-fold faster).
Thus, the altered distribution of the ERC also appears to accompany a change in its properties, which leads to more rapid sphingomyelin metabolism, as compared to the parental cells.
The abnormally distributed ERC tubules in KB-CP.5 cells have lower internal pH compared to parental KB-3-1 cells. As shown above, our results indicate that the abnormal distribution of the ERC tubules in the KB-CP.5 cells has functional consequences—both in terms of abnormal metabolism of lipids such as sphingomyelin, as well as in terms of the enhanced sequestration of labeled cisplatin. We next attempted to address the mechanism behind these observations. We reasoned that a change in pH inside the ERC tubules could account for both observations. A lowered intra-ERC pH could change the protonation status of cisplatin, thereby making it more hydrophobic. Similarly, a lowered pH would make acid sphingomyelinases more active, thereby increasing sphingomyelin metabolism, and its conversion to ceramide.
In order to test this possibility, we labeled human Tf simultaneously with both Alexa546 and fluorescein. The idea was to have both red (Alexa 546) and green (fluorescein) labels on the same Tf molecule. Fluorescein fluorescence intensity is pH sensitive, with the intensity decreasing with the lowering of pH. On the contrary, Alexa546 fluorescence is pH-insensitive. Thus, when cells are labeled with the double-labeled Tf, and the Tf is allowed to chase into the ERC, it’s green fluorescence intensity will be indicative of the pH within the compartment, whereas the red fluorescence will provide an internal control of the degree of loading of the particular tubule. We labeled both KB-CP.5 cells and the parental KB-3-1 controls with the double-labeled Tf, and let them reach steady-state. We then imaged these cells in both the red and the green channels, and determined the red/green fluorescence intensity ratio (Fig. 4). We found that the ERCs of cisplatin resistant KB-CP cells exhibited an increased R/F ratio as compared to parental KB cells, indicating thereby that KB-CP cells, on average, have lower pH in the ERC tubules, relative to parental controls. This observation points to a possible mechanism by which the KB-CP ERCs are able to sequester CP, as well as have increased sphingomyelin metabolism.
Figure 4.

Frequency distribution of the ratios of Alexa546 to Fluorescein (R/F ratio) in Tf-labeled KB-3-1 cells (black bars) and KB-CP.5 cells (grey bars). The cells were labeled for 30 min at 37°C with Tf double labeled with Alexa546 (red fluorescence; pH insensitive) and Fluorescein (green fluorescence; pH sensitive). They were then rinsed and chased for an additional 10 min at 37°C in the presence of deferroxamine (an iron chelator) and 10-fold excess unlabeled Tf, to prevent rebinding of labeled Tf. The cells were then fixed with 3% paraformaldehyde for 2 min at room temperature, and imaged using wide field microscopy. The Alexa 546/Fluorescein (R/F) ratio was measured after background correction. Increasing R/F ratio indicates increasing pH values.
The altered distribution of the ERC tubules in CP-r cells is dependent on the expression of Glu-α-tubulin. We next addressed the molecular mechanism that might cause the abnormal distribution of the ERC tubules in KB-CP.5 cells. Another cell line that has been extensively studied in our laboratory, and has dispersed the ERC under specialized conditions, is B104-5. Under normal permissive conditions, this cell line behaves identically to its parental cell line, TRVb-1, a CHO cell line that lacks endogenous Tf receptors, and instead, expresses transfected human Tf receptors. The B104-5 cells have a temperature-dependent increase in the level of Glu-α-tubulin (27). The cells were incubated at two different temperatures, 32°C (permissive) and 39°C (non-permissive) for 3 days before fixation, then permeabilized and immunostained for Glu-α-tubulin. B104-5 cells incubated at 32°C have relatively low levels of stable Glu-α-tubulin, which is found as a network aggregated in linear, polygonal or irregular-shaped plaques of high fluorescence density near the nuclei of the cells (Fig. 5A). An elevated level of Glu-α-tubulin, which appeared loosely arranged and spread throughout the cytoplasm, is detected in the B104-5 cells after 3 days at 39°C, the non-permissive temperature (Fig. 5A). However, the expression level of Tyr-α-tubulin in B104-5 cells grown at 39°C was not significantly different from cells incubated at the permissive 32°C (Fig. 5A). No alterations in the structural appearance of the Tyr-α-tubulin could be detected and the overall appearance was fairly similar in B104-5 cells cultured at both temperatures. This is consistent with previous results indicating that the elevated expression of Glu-α-tubulin at the restrictive temperature could return to the lower level of Glu-α-tubulin by lowering the temperature of B104-5 cells (18, 27).
Figure 5.

Expression of Glu-α-tubulin and Tyr-α-tubulin by Western blotting and confocal microscopy. A, Expression of Glu-α-tubulin and Tyr-α-tubulin in B104-5 cells under permissive and non-permissive condition. B104-5 cells were incubated under permissive 32°C and non-permissive 39°C conditions for 3 days in 5% CO2, then cells were fixed and permeabilized as described in “Materials and Methods”. Several fields of cells were captured and one representative field of each is shown. Scale bar, 20 μm. B, Detection of Glu-α-tubulin and Tyr-α-tubulin in isolated fractions of KB-3-1 and KB-CP.5 cells. Fractions (W, whole cell lysis; M, plasma membrane enriched fraction; C, cytosol fraction) were isolated by different types of ultracentrifugation as described in “Materials and Methods.” C, Immunofluorescence pictures of KB-3-1 and KB-CP.5 cells stained with antibodies against Glu-α-tubulin and Tyr-α-tubulin. A pronounced aggregation of Glu-α-tubulin near the nuclei in KB-3-1 cells is observed, compared with KB-CP.5 cells. No changes in the cytoplasmic distribution pattern of the Tyr-α-tubulin are detectable. A representative image from at least 6 is shown.
The distribution of the ERC and Glu-α-tubulin was then examined in KB-3-1 and KBCP.5 cells. The expression of Glu-α-tubulin was found to be increased in KB-CP.5 cells compared with parental KB-3-1 cells, as detected by Western blotting. As shown in Fig. 5B, there is a higher level of Glu-α-tubulin expression in KB-CP.5 cells compared to parental KB-3-1 cells. We did not detect any major difference in the expression of Tyr-α-tubulin between the CP-r KB-CP.5 and sensitive KB-3-1 cells. Based on immunohistochemistry, parental KB-3-1 cells have a relatively clustered distribution of down-regulated, stable Glu-α-tubulin near the nuclei of the cells. An elevated level of Glu-α-tubulin was also detected in the CP-r KB-CP.5 cells, but with a dispersed distribution of Glu-α-tubulin throughout the cytoplasm (Fig. 5C). However, there was no significant alteration of the appearance or in the expression level of Tyr-α-tubulin in the resistant KB-CP.5 cells (Fig. 5C), which was similar to results with B104-5 cells, mentioned above. The results indicate that there is a correlation between the levels of stable Glu-α-tubulin and the appearance of the ERC and suggest that stable microtubules containing elevated levels of Glu-α-tubulin might play a role in vesicle recycling or transportation.
B104-5 cells are relatively resistant to cisplatin under non-permissive conditions. To determine whether the formation of an irregular ERC might lead to CP resistance, we compared the CP toxicity of B104-5 cells at permissive and non-permissive temperatures. B104-5 cells were incubated with various CP concentrations at 32°C (permissive) or 39°C (non-permissive) for 4 weeks. Based on a colony-forming assay, there was a 2.0-fold difference in CP resistance between the B104-5 cells at the non-permissive temperature (0.31μg/ml) and B104-5 cells incubated at the permissive temperature (0.15μg/ml) (Fig. 6A). Similar results were found using a 72-hour cell proliferation assay, with about 4-fold resistance to CP at the non-permissive temperature. However, there was no significant difference in CP resistance between the TRVb-1 CHO parental cells at the permissive temperature (32°C) and non-permissive temperature (39°C) (Fig. 6B). Therefore, temperature-sensitive B104-5 cells with overexpressed Glu-α-tubulin were more resistant to CP at the non-permissive temperature compared to parental TRVb-1 CHO cells.
Figure 6.

Cytotoxicity of cisplatin measured in cells with different ERC structures. A, The cytotoxicity of cisplatin in B104-5 cells incubated at permissive or non-permissive temperature was determined by colony forming assays, 4 weeks after continuous exposure to CP at different concentrations. The results are expressed as the percent of viable CP-treated cells compared with untreated cells. Each value represents the mean ± SEM of triplicate measurements. B, Colony forming assay to measure the cisplatin resistance of parental TRVb-1 CHO cells. After the cells were incubated in medium with various concentrations of CP, cells were treated and counted as described in Materials and Methods.
DISCUSSION
Cellular resistance to CP is multifactorial and may consist of mechanisms limiting the formation of DNA adducts, repair mechanisms and alterations that promote cell survival (28, 29). The formation of DNA adducts by CP can be limited by reduced drug accumulation, enhanced drug efflux, sequestration of drugs within the cytoplasm, and inactivation of platinum drugs by coordination of sulfur-containing compounds (30). Details of the mechanisms underlying the antitumor activity of CP or resistance to this drug are not entirely understood despite intensive research in this area (31). However, reduced accumulation of CP appears to be a common mechanism of resistance (32). In this work, we monitored the trafficking of the bona fide ERC marker, transferrin, bound to its receptor, as well as several fluorescent membrane lipid analogs, through the ERC. Our previous study and the work of others have shown that altered uptake of CP occurs because of reduced membrane-binding/transport proteins and reduced endocytosis (2, 33). Here, we show that there is a defect in the localization and probably the function of the ERC consistent with a demonstrated alteration in the level of Glu-α-tubulin (in stable, detyrosinated microtubules).
In most cell types, about 95% of endocytosed membrane is recycled to the plasma membrane. This recycling is important for the maintenance of membrane composition and for cell surface expression of receptors involved in nutrient uptake as well as for several other cellular processes (34, 35). We used fluorescently-labeled Tf to study the lipid recycling pathway, and found that the ERC is altered in KB-CP.5 cells compared to KB-3-1 cells. This was confirmed by other known ERC markers. A tightly-clustered ERC is located near the nucleus in parental KB-3-1 cells, but it appears loosely arranged and widely dispersed throughout the cytoplasm in KB-CP.5 cells. The overall distribution of the ERC in these cells is related to the amount and distribution of stable detyrosinated microtubules. The structural and functional integrity of the ERC in part depends on the cytoskeleton, which appears to be involved in regulating molecular sorting and vesicular transport of the ERC with microtubules (36). In this work, we found that B104-5 cells sensitive to temperature are more resistant to cisplatin, with an altered ERC due to stable Glu-α-tubulin-containing microtubules at non-permissive temperature compared to permissive temperature.
There are only a few hints to a possible influence of the cytoskeleton on CP resistance. CP can affect cytoplasmic actin filaments, as seen in qualitative histological and ultrastructural findings (37). Biochemical studies performed with isolated tubulin molecules showed that CP was able to inhibit irreversibly the polymerization of microtubules by covalent binding to sulfhydryl groups in tubulin dimers (38). Most mammalian cells possess two subsets of microtubules: dynamic microtubules with a half-life of 5-10 min, and stable microtubules that have a half-life of hours (39). Dynamic microtubules contain mainly tyrosinated tubulin (Tyr-α-tubulin). Stable microtubules contain various modified tubulins including detyrosinated tubulin (Glu-α-tubulin), which accumulate in stable microtubules but do not cause microtubule stabilization (40). The best-characterized modification of tubulin is detyrosination, which involves the reversible removal of the carboxy-terminal Tyr residue from α-tubulin, exposing a Glu residue as the new C-terminus (41, 42). Stable microtubules are important in the distribution of the ERC (27).
We chose B104-5 cells to study the effects of the distribution and expression pattern of tubulin on cisplatin resistance. B104-5 has a striking temperature-induced alteration of Glu-α-tubulin associated with the morphology of its endocytic recycling compartment. Under permissive conditions, a tightly-clustered ERC is located near the Golgi complex in B104-5 cells cultured at 32°C. Under non-permissive conditions at 39°C, this compartment appears fragmented and widely dispersed. Surprisingly, this alteration in the morphology of the recycling compartment has no effect on the kinetics of receptor internalization and recycling (18). The wild-type endocytic compartment is closely aligned with the microtubule-organizing center and the Golgi apparatus, and like the Golgi, its clustered appearance is dependent upon intact microtubules. Lin et al. (27) found that treatment with taxol could increase expression of stable Glu-α-tubulin in the B104-5 cells, which resulted in a dispersed ERC structure similar to when cells were incubated at 39°C. In this work, we show that B104-5 cells with a dispersed ERC incubated under non-permissive temperature were more resistant to CP compared to B104-5 cells with a more clustered ERC structure under incubation at permissive temperature. It is very possible that resistance to CP is due to the formation of the ERC, which accumulates active CP inside its vesicles, reduces trafficking of CP from the cell surface into the cell, and limits the possibility of CP forming DNA adducts in the nucleus.
It should, however, be noted that B104-5 cells have been previously shown to overexpress certain heat shock proteins (see discussion in Ref. 18). It is thus possible that increased CP resistance of B104-5 cells under nonpermissive conditions is related more to a general upregulation of heat shock type response, than to a direct effect of the altered ERC. Could the increased CP-r of KB-CP.5 cells also be due to some other phenomenon which may be upstream from the ERC defect? Although this is a possibility, the finding of cisplatin and methotrexate colocalized with the altered ERC structures argues in favor of a direct effect of the altered ERC on cisplatin resistance.
It is well established that organelles along both the secretory and endocytic pathways experience a gradient of decreasing pH (13). We found that the pH of the ERC is more acidic in CP-r KB-CP.5 cells compared to parental KB-3-1 cells. This was determined by directly measuring the pH of late endosomes/lysosomes by ratio fluorescence imaging. Acidification of endosomes is necessary for a variety of essential eukaryotic cellular functions (13). However, the mechanisms that regulate pH in these organelles are complex, and not yet completely understood. In general, endosomal acidity is generated by the membrane-bound ATP-dependent proton pump (vacuolar H+-ATPase), and the degree of acidification is modulated by a complex interplay of this pump and several other pumps or transporters, including the Na+-K+-ATPase and chloride channels (13). Further investigation will be required to pinpoint precisely which of these protein(s) is/are affected in the KB-CP-r cells.
pH changes in the ERC in the CP-r cells might cause CP to be “trapped” in the ERC due to the low permeability of the charged CP to vesicular membranes which would diminish the toxicity of CP in KB-CP.5 cells. In addition, the normal distribution of the ERC and intracellular trafficking through this organelle could be disrupted by an acidic alteration. It is therefore possible that CP-r cells could sequester CP in an acidic ERC and expel it from the cells by exocytosis. However, in previous work, we found alkalinization of lysosomes in CP-r cells (23). Why the pH differs in lysosomes and the ERC in CP-r cells is still unclear. These pH changes in the endocytic pathway need further study. Also, we have found in this study that NBD-SM, the bulk of whose intracellular pool localizes to the ERC at steady state, is metabolized significantly faster into NBD-cer in CP-r KB-CP.5 cells. It will be interesting to explore whether this acceleration of sphingomyelin metabolism is correlated with increased activation of acid sphingomyelinases in the ERC of CP-r cells due to a lowering of the pH. Since ceramide is a known second messenger, we speculate that such increased production of ceramide in the ERC might play a role in the carcinogenesis process.
The dispersed ERC localization and the elevated expression of Glu-α-tubulin in KB-CP.5 cells permit a better examination of the association of the ERC with CP resistance in B104-5 cells. Reports of platinum-rich phagocytic granules (platinosomes), which may be related to lysosomes or the ERC, suggested that CP might localize in these intracellular lysosome-like organelles. In addition, relatively high concentrations of CP were found in microsomal fractions of liver and kidney tissues following CP administration (43). Using confocal microscopy, we found nearly all of the internalized Alexa-488 cisplatin in the ERC labeled with Texas-red Tf in KB-CP.5 cells, similar to internalized Alexa 546-labeled methotrexate (Fig. 4). Therefore, it is probable that alterations in the cytoskeleton related to Glu-α-tubulin are sufficient to alter the spatial distribution of intracellular CP. It is worth mentioning that, following treatment with CP, the number of platinum atoms bound covalently to cytoplasmic proteins quantitatively exceeds the number of platination sites on the nuclear DNA (44). These results suggest that nuclear DNA is not the only target of the intracellular action of CP and reveal that additional molecular lesions must occur independently of the molecular attachment to DNA, which could result, for example, in structural alterations to the ERC. As mentioned earlier, we have found that NBD-C6-sphingomyelin was metabolized faster into NBD-C6-ceramide in CP-r KB-CP.5 cells. Therefore, altered phospholipid metabolism or trafficking in tumor cells that are CP-r may represent a novel target for new anticancer agents.
ACKNOWLEDGEMENTS
The authors would like to thank George Leiman for his assistance with editing this manuscript, and Drs. Susan H. Garfield and Stephen M. Wincovitch for their technical assistance with confocal microscopy. This research was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Drs. F. Maxfield and S. Mukherjee were supported by grant DK27083 from the National Institutes of Health.
Footnotes
- CP
- cisplatin
- CP-r
- cisplatin resistant
- ERC
- endocytic recycling compartment
- CHO
- Chinese hamster ovary
- NBD-SM
- NBD-sphingomyelin
- NBD-cer
- NBD-ceramide
- LE/LY
- late endosomes/lysomes
- Tf
- transferrin
References
- Wang G, Reed E, Li QQ. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer. Oncol Rep. 2004;12:955–965. [PubMed] [Google Scholar]
- Safaei R, Howell SB. Copper transporters regulate the cellular pharmacology and sensitivity to Pt drugs. Crit Rev Oncol Hematol. 2005;53:13–23. doi: 10.1016/j.critrevonc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Shen DW, Su A, Liang XJ, Pai-Panandiker A, Gottesman MM. Reduced expression of small GTPases and hypermethylation of the folate binding protein gene in cisplatin-resistant cells. Br J Cancer. 2004;91:270–6. doi: 10.1038/sj.bjc.6601956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AL, Lippard SJ. Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim Biophys Acta. 1985;780:167–180. doi: 10.1016/0304-419x(85)90001-0. [DOI] [PubMed] [Google Scholar]
- Chu G. Cellular response to cisplatin: The roles of DNA binding proteins and DNA repair. J Biol Chem. 1994;269:787–90. [PubMed] [Google Scholar]
- Siddik ZH. Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat Res. 2002;112:263–84. doi: 10.1007/978-1-4615-1173-1_13. [DOI] [PubMed] [Google Scholar]
- Speelmans G, Sips WH, Grisel RJ, et al. The interaction of the anti-cancer drug cisplatin with phospholipids is specific for negatively charged phospholipids and takes place at low chloride ion concentration. Biochim Biophys Acta. 1996;1283:60–6. doi: 10.1016/0005-2736(96)00080-6. [DOI] [PubMed] [Google Scholar]
- Speelmans G, Staffhorst RW, Versluis K, Reedijk J, de Kruijff B. Cisplatin complexes with phosphatidylserine in membranes. Biochemistry. 1997;36:10545–50. doi: 10.1021/bi9703047. [DOI] [PubMed] [Google Scholar]
- Burger KN, Staffhorst RW, De Kruijff B. Interaction of the anti-cancer drug cisplatin with phosphatidylserine in intact and semi-intact cells. Biochim Biophys Acta. 1999;1419:43–54. doi: 10.1016/s0005-2736(99)00052-8. [DOI] [PubMed] [Google Scholar]
- Liang XJ, Yin JJ, Zhou JW, Wang PC, Taylor B, Cardarelli C, Kozar M, Forte R, Aszalos A, Gottesman MM. Changes in biophysical parameters of plasma membranes influence cisplatin resistance of sensitive and resistant epidermal carcinoma cells. Exp Cell Res. 2004;293:283–91. doi: 10.1016/j.yexcr.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Maheswari KU, Ramachandran T, Rajaji D. Interaction of cisplatin with planar model membranes-does dependent change in electrical characteristics. Biochim Biophys Acta. 2000;1463:230–40. doi: 10.1016/s0005-2736(99)00189-3. [DOI] [PubMed] [Google Scholar]
- Liang XJ, Shen DW, Garfield S, Gottesman MM. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003;63:5909–16. [PubMed] [Google Scholar]
- Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–32. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- Nichols BJ, Kenworthy AK, Polishchuk RS, et al. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–41. doi: 10.1083/jcb.153.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CR, Trowbridge IS. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J Cell Biol. 1983;97:508–21. doi: 10.1083/jcb.97.2.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somatic Cell Mol Genet. 1985;11:117–26. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- McGraw TE, Dunn KW, Maxfield FR. Isolation of a temperature-sensitive variant Chinese hamster ovary cell line with a morphologically altered endocytic recycling compartment. J Cell Physiol. 1993;155:579–94. doi: 10.1002/jcp.1041550316. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Soe TT, Maxfield FR. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J Cell Biol. 1999;144:71–1284. doi: 10.1083/jcb.144.6.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M, Pagano RE. Lipid recycling between the plasma membrane and intracellular compartments: transport and metabolism of fluorescent sphingomyelin analogues in cultured fibroblasts. J Cell Biol. 1989;108:2169–81. doi: 10.1083/jcb.108.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Presley JF, Maxfield FR. Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J Cell Biol. 1993;12:1257–69. doi: 10.1083/jcb.121.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano RE, Watanabe R, Wheatley C, Dominguez M. Applications of BODIPY-sphingolipid analogs to study lipid traffic and metabolism in cells. Methods Enzymol. 2003;88:1327–34. doi: 10.1016/s0076-6879(00)12937-4. [DOI] [PubMed] [Google Scholar]
- Chauhan SS, Liang XJ, Su AW, et al. Reduced endocytosis and altered lysosome function in cisplatin-resistant cell lines. Br J Cancer. 2003;88:1327–34. doi: 10.1038/sj.bjc.6600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XJ, Shen DW, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. Trafficking and localization of platinum complexes in cisplatin-resistant cell lines monitored by fluorescence-labeled platinum. J Cell Physiol. 2005;202:635–41. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- Lipsky NG, Pagano RE. Sphingolipid metabolism in cultured fibroblasts: microscopic and biochemical studies employing a fluorescent ceramide analogue. Proc Natl Acad Sci USA. 1983;80:2608–12. doi: 10.1073/pnas.80.9.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky NG, Pagano RE. Intracellular translocation of fluorescent sphingolipids in cultured fibroblasts: endogenously synthesized sphingomyelin and glucocerebroside analogues pass through the Golgi apparatus en route to the plasma membrane. J Cell Biol. 1985;100:27–34. doi: 10.1083/jcb.100.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SX, Gundersen GG, Maxfield FR. Export from pericentriolar endocytic recycling compartment to cell surface depends on stable, detyrosinated (glu) microtubules and kinesin. Mol Biol Cell. 2002;13:96–109. doi: 10.1091/mbc.01-05-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews PA, Howell SB. Cellular pharmacology of cisplatin: perspectives on mechanisms of acquired resistance. Cancer Cells. 1990;2:35–43. [PubMed] [Google Scholar]
- Stein WD, Litman T, Fojo T, Bates SE. Differential expression of cell adhesion genes: Implications for drug resistance. Int J Cancer. 2005;113:861–5. doi: 10.1002/ijc.20671. [DOI] [PubMed] [Google Scholar]
- Brabec V, Kasparkova J. Molecular aspects of resistance to antitumor platinum drugs. Drug Resist Update. 2002;5:147–61. doi: 10.1016/s1368-7646(02)00047-x. [DOI] [PubMed] [Google Scholar]
- Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–16. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- Shen DW, Goldenberg S, Pastan I, Gottesman MM. Decreased accumulation of [4C]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J Cell Physiol. 2000;183:108–16. doi: 10.1002/(SICI)1097-4652(200004)183:1<108::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Gruenberg J. Lipids in endocytic membrane transport and sorting. Curr Opin Cell Biol. 2003;15:382–8. doi: 10.1016/s0955-0674(03)00078-4. [DOI] [PubMed] [Google Scholar]
- Liang XJ, Shen DW, Gottesman MM. A pleiotropic defect reducing drug accumulation in cisplatin-resistant cells. J Inorg Biochem. 2004;98:1599–606. doi: 10.1016/j.jinorgbio.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–23. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Coumailleau F, Das V, Alcover A, et al. Over-expression of Rififylin, a new RING finger and FYVE-like domain-containing protein, inhibits recycling from the endocytic recycling compartment. Mol Biol Cell. 2004;15:4444–56. doi: 10.1091/mbc.E04-04-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen DW, Liang XJ, Gawinowicz MA, Gottesman MM. Identification of cytoskeletal [14C]carboplatin-binding proteins reveals reduced expression and disorganization of actin and filamin in cisplatin-resistant cell lines. Mol Pharmacol. 2004;66:789–93. doi: 10.1124/mol.66.4.. [DOI] [PubMed] [Google Scholar]
- Tulub AA, Stefanov VE. Cisplatin stops tubulin assembly into microtubules. A new insight into the mechanism of antitumor activity of platinum complexes. Int J Biol Macromol. 2001;28:191–8. doi: 10.1016/s0141-8130(00)00159-8. [DOI] [PubMed] [Google Scholar]
- Palazzo A, Ackerman B, Gundersen GG. Cell biology: Tubulin acetylation and cell motility. Nature. 2003;421:230. doi: 10.1038/421230a. [DOI] [PubMed] [Google Scholar]
- Webster DR, Wehland J, Weber K, Borisy GG. Detyrosination of α-tubulin does not stabilize microtubules in vivo. J Cell Biol. 1990;111:113–22. doi: 10.1083/jcb.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehland J, Weber K. Turnover of the carboxy-terminal tyrosine of alpha-tubulin and means of reaching elevated levels of detyrosination in living cells. J Cell Sci. 1987;88:185–203. doi: 10.1242/jcs.88.2.185. [DOI] [PubMed] [Google Scholar]
- Gundersen GG, Kalnoski MH, Bulinski JC. Distinct populations of microtubules: tyrosinated and nontyrosinated α-tubulin are distributed differently in vivo. Cell. 1984;38:779–89. doi: 10.1016/0092-8674(84)90273-3. [DOI] [PubMed] [Google Scholar]
- Aggarwal SK, Whitehouse MW, Ramachandran C. Ultrastructural effects of cisplatin. In: Prestayko AW, Crooke ST, Carter SK, editors. Cisplatin: Current Status and New Development. Academic Press; New York: 1980. [Google Scholar]
- Kopf-maier P, Huhlhausen SK. Changes in the cytoskeleton pattern of tumor cells by cisplatin in vitro. Chem-Biol Interactions. 1992;82:295–316. doi: 10.1016/0009-2797(92)90002-3. [DOI] [PubMed] [Google Scholar]