

Abstract
β-Lactamase confers resistance to penicillin-like antibiotics by hydrolyzing their β-lactam bond. To combat these enzymes, inhibitors covalently cross-linking the hydrolytic Ser70 to Ser130 were introduced. In turn, mutant β-lactamases have emerged with decreased susceptibility to these mechanism-based inhibitors. Substituting Ser130 with glycine in the inhibitor-resistant TEM (IRT) mutant TEM-76 (S130G) prevents the irreversible cross-linking step. Since the completely conserved Ser130 is thought to transfer a proton important for catalysis, its substitution might be hypothesized to result in a nonfunctional enzyme; this is clearly not the case. To investigate how TEM-76 remains active, its structure was determined by X-ray crystallography to 1.40 Å resolution. A new water molecule (Wat1023) is observed in the active site, with two configurations located 1.1 and 1.3 Å from the missing Ser130 Oγ; this water molecule likely replaces the Ser130 side-chain hydroxyl in substrate hydrolysis. Intriguingly, this same water molecule is seen in the IRT TEM-32 (M69I/M182T), where Ser130 has moved significantly. TEM-76 shares other structural similarities with various IRTs; like TEM-30 (R244S) and TEM-84 (N276D), the water molecule activating clavulanate for cross-linking (Wat1614) is disordered (in TEM-30 it is actually absent). As expected, TEM-76 has decreased kinetic activity, likely due to the replacement of the Ser130 side-chain hydroxyl with a water molecule. In contrast to the recently determined structure of the S130G mutant in the related SHV-1 β-lactamase, in TEM-76 the key hydrolytic water (Wat1561) is still present. The conservation of similar accommodations among IRT mutants suggests that resistance arises from common mechanisms, despite the disparate locations of the various substitutions.
The catalytic activity of the TEM family of class A β-lactamases is a major resistance mechanism against β-lactam antibiotics, such as penicillins (Figure 1A); hydrolysis of the β-lactam ring of these antibiotics renders them ineffective against bacteria. To combat these enzymes, inhibitors against β-lactamase, such as clavulanate, tazobactam, and sulbactam (Figure 1B–D), were developed. In turn, inhibitor-resistant TEM β-lactamases (IRTs)1 have emerged in the clinic with decreased susceptibility to these mechanism-based inhibitors. These inhibitors function by acylating the enzyme’s active site. After reacting with the catalytic Ser70, these β-lactam compounds can partition into either the hydrolytic pathway common to β-lactam substrates or an inactivation pathway culminating in irreversible modification of Ser130, a completely conserved active site residue (1–5) (Figure 2). Though the cross-linking reaction may occur several hundreds of times less frequently than the competitive hydrolytic pathways, eventually the enzyme is completely and irreversibly inhibited (6–8).
Figure 1.
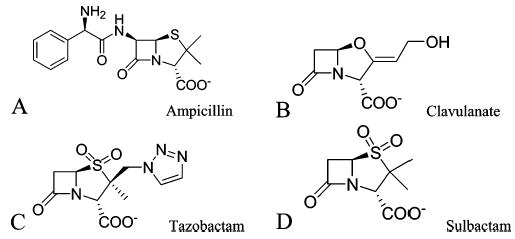
Chemical structures of the TEM β-lactamase substrate ampicillin (A) and mechanism-based inhibitors (B–D).
Figure 2.
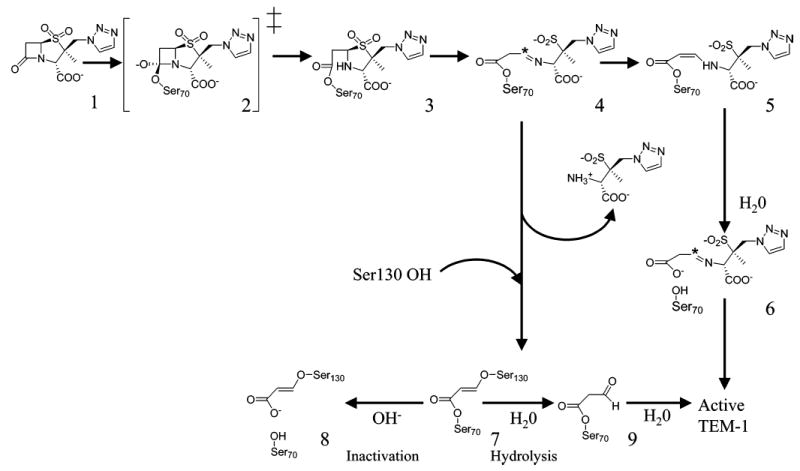
Simplified hydrolytic and inhibition pathways of TEM β-lactamase for tazobactam (1, 11).
The IRT mutant enzymes that have emerged in the clinic typically involve substitutions at several positions, including Met69, Arg275, Asn276, and Arg244 (www.lahey.org/Studies/temtable). To date, atomic resolution structures of five IRT mutants have been determined (9–11). The Arg244Ser substitution found in the TEM-30 mutant results in displacement of a water molecule involved in the inactivation chemistry of clavulanate (3, 10). This same water molecule was only transiently present in the TEM-84 (N276D) IRT mutant as well (9). In both TEM-32 (M69I/M182T) and TEM-34 (M69V), movement of Ser130, a site of covalent attachment, explains the inhibitor-resistant phenotype of these two mutants (10). The structures of the wild-type and the IRT TEM-33 (M69L) were observed to be virtually identical (with the exception of the site of mutation). Differences in the structures of the two enzymes became apparent only in the dynamic motions of the mutant and wild-type enzymes during computational simulations (11). The primary contributing factor to the IRT phenotype of the TEM-33 enzyme was an increase in the apparent dissociation constants of the preacylation complexes with the inhibitors. Though replacing the Ser130 side chain would seem to be the easiest way to prevent mechanism-based inhibition, its substitution is somewhat rare among clinical isolates. Most of the IRT substitutions are relatively distant from this position and, in this sense, seem relatively subtle. For instance, the Cα atoms of Arg244 and Asn276 are located 14 and 17 Å from the Ser130 Cα, respectively. In contrast, the IRT mutant TEM-76 has simply replaced the cross-linking Ser130 with glycine, thus disrupting the inhibition reaction. At first glance, the mechanism of this mutant thus seems fairly obvious. The question here is not why this mutant is resistant to inhibition (though we will see that even here there are some subtleties) but rather how it remains active in its turnover of substrates, since Ser130 is thought to be a key catalytic residue.
Ser130 has been implicated as the catalytic acid necessary for protonating the lactam nitrogen leaving group, promoting the opening of the β-lactam ring during substrate hydrolysis (12, 13). Thus substitution of this serine with glycine in TEM-76 might be expected to render the enzyme nonfunctional. As S130G is a naturally evolved mutant enzyme, it is apparent that TEM-76 has somehow compensated for the loss of this active site serine. To understand how this occurs, we have measured the kinetic parameters for activity and inhibition of TEM-76 and have determined its X-ray crystal structure to 1.40 Å resolution. This structure reveals how S130G compensates for the loss of its catalytic acid and reveals some unexpected similarities with the more subtle mutant enzymes TEM-30, TEM-32, and TEM-84.
MATERIALS AND METHODS
Construction and Purification of TEM-1 β-Lactamases
The cloning of the wild-type TEM-1 β-lactamase has been reported. To produce the Ser130Gly mutant of TEM-1 β-lactamase, a previously reported construct was used (14). In this construct the gene for the wild-type (TEM-1) enzyme was fused to the leader sequence of the outer membrane protein A (OmpA) and cloned into the expression vector pET24a(+) to allow secretion of large amounts of β-lactamase into the growth medium. The mutant enzyme was prepared with the QuikChange site-directed mutagenesis kit (Stratagene) using two oligonucleotide primers, TEM130Gly-D, gccataaccatgggtgataacactg (the codon for Gly is in italics; the point mutation is in bold), and TEM130Gly-R, cagtgttatcacccatggttatggc, that introduced a single mutation into the gene for the TEM-1 β-lactamase, resulting in the desired substitution. The mutation was confirmed by sequencing of both strands of the entire gene. The TEM-1 and S130G mutant enzymes were isolated and purified as described previously (11).
Kinetic Measurements
The steady-state kinetic parameters for penicillins and cephalosporins were determined from nonlinear regression plots. The experimental data were fitted using Grafit software. The assays were carried out in 100 mM sodium phosphate buffer, pH 7.0, at room temperature, monitoring hydrolysis of penicillin G at 240 nm (Δε240 = 570 M−1 cm−1), ampicillin at 235 nm (Δε235 = 820 M−1 cm−1), cephaloridine at 267 nm (Δε267 = 1000 M−1 cm−1), and nitrocefin at 482 nm (Δε267 = 17 400 M−1 cm−1). In all cases the substrate concentrations were such that they flanked the Km values.
Inactivation constants for clavulanate, sulbactam, and tazobactam were calculated for the S130G mutant enzyme as per the method described by Imtiaz et al. (3). Inactivation experiments with the mutant enzyme were initiated by the addition of a portion of a stock solution of clavulanate or sulbactam (10–160 μM final concentrations) to the enzyme (1 μM) solutions. For tazobactam the final concentration in the inactivation experiment ranged from 9 to 53 μM. Incubation was stopped by dilution of 10 μL aliquots of the incubation mixture into a 1 mL assay containing 0.8 mM penicillin G, at various time intervals. The residual enzyme activity in the aliquot was monitored until complete depletion of the substrate resulted. A progressive increase of the activity of the enzyme was observed initially, attributed to its recovery from the transiently inhibited species. The highest steady-state rates, in the course of substrate hydrolysis, were used in calculation of the remaining enzyme activity.
The apparent KI values for all three compounds were calculated for the S130G mutant enzyme by the method of Dixon (15). Two concentrations of the substrate penicillin G, typically 400 and 700 or 800 μM, were used. A series of assay mixtures containing both substrate and various concentrations of the inactivators (0.2–2 mM for clavulanate, 52–570 μM for sulbactam, and 11–500 μM for tazobactam) were prepared in 100 mM sodium phosphate buffer, pH 7.0. An aliquot of the stock solution of the enzyme was added to afford a final concentration of 60 nM S130G mutant, in a total volume of 1.0 mL, followed by the immediate measurement of enzyme activity. The rates were measured for the first 5% of substrate turnover.
The partition ratios (kcat/kinact) for clavulanate, sulbactam, and tazobactam were determined by the titration method (16). Several buffered mixtures containing various molar ratios of [I]/[E] of each of the inhibitors with the S130G mutant enzyme were incubated at 4 °C overnight (ca. 20 h). The molar ratio [I]/[E] for the experiment with clavulanate was from 4 to 1300, for sulbactam from 4 to 800, and for tazobactam from 4 to 400. The remaining activity of the enzyme was assayed under conditions of the excess substrate, with penicillin G.
The rate constants for recovery of the enzyme activity (krec) from the transiently inhibited species for the S130G mutant enzyme incubated with each of the compounds were measured at the molar ratio ([I]/[E]) that displayed a long delay prior to the linear steady-state hydrolysis of penicillin G. These experiments were performed under conditions of excess substrate, as described by Koerber and Fink (17). The calculations of the constants were performed according to the method of Glick et al. (18).
Crystallization and Structure Determination
TEM S130G crystals were grown in hanging drops over a well solution of 1.4 M NaH2PO4/K2HPO4, pH 8.3. A volume of 2 μL of 20 mg/mL protein in 50 mM KPi buffer was mixed with 2 μL of well solution, and the resulting 4 μL drop was seeded with microcrystals of TEM M182T (19). Crystals grew within 5 weeks. Crystals were soaked in 25% sucrose in 1.6 M KNaPi as cryoprotectant for 10–30 s and then frozen inliquid nitrogen. Diffraction data were collected from a single crystal on the 8.3.1 beamline at the Advanced Light Source (Berkeley, CA) at a wavelength of 1.0332 Å. A total of 169 frames were integrated, and 149639 reflections were scaled and merged using the DENZO/SCALEPACK package (20). The data are 98.9% complete to 1.40 Å resolution (43334 unique reflections, Table 1). Crystals belong to the space group P212121, with unit cell dimensions of 41.60, 59.71, and 88.36 Å, which are similar to the WT crystals (21). An initial structure was obtained by the molecular replacement program EPMR using the M182T TEM mutant (Protein Data Bank accession code 1JWP) apo structure as an initial search model. Five percent of the data (2126 reflections) was set aside randomly as the test set. Following rigid body refinement in CNS 1.1 (22), the S130G structure was refined by cycles of Cartesian and B-factor refinement. Active site features, such as water molecules and alternate configurations (see Results for further discussion), were modeled in the structure on the basis of strong positive Fo – Fc electron density features at a minimum contour level of 3.0σ. Other water molecules were recognized and placed in the structure by CNS 1.1 on the basis of positive Fo – Fc density at a minimum contour level of 2σ, confirmed by visual inspection, and only those with B-factors <60 Å2 were retained. Multiple configurations of protein side chains and water molecules were added to interpret high significance features (typically at 3.0σ) of Fo – Fc electron density maps.
Table 1.
Data and Refinement Statistics for the S130G Crystal Structure
| unit cell parameters (Å) | 41.60, 59.71, 88.36 |
| resolution (Å) | 20.0–1.40 (1.45–1.40)a |
| unique reflections | 43334 (4225) |
| total observations | 149639 |
| working set/test set | 40386/2126 reflections |
| Rmerge (%) | 4.2 (38.4) |
| completeness (%) | 98.9 (97.8) |
| 〈I〉/〈σ(I)〉 | 15.42 (2.37) |
| no. of residues | 263 |
| no. of water molecules | 327 |
| space group | P212121 |
| RMSD bond lengths (Å) | 0.010 |
| RMSD bond angles (deg) | 1.90 |
| alternate conformations | 14 |
| R-factor (%) | 18.30 |
| Rfree (%) | 22.40 |
Values in parentheses are for the highest resolution shell.
Thermal Denaturation
The S130G enzyme was denatured by raising the temperature in 0.1 °C increments at a ramp rate of 2 °C/min in 200 mM potassium phosphate buffer, pH 7.0, using a Jasco 715 spectropolarimeter with a Peltier effect temperature controller and an in-cell temperature monitor (23). Denaturation was marked by an obvious transition in the far-UV CD (223 nm) signal. Consistent with previous stability work with TEM enzymes, all melts were reversible and apparently two-state, as judged by the sharp transition in the denaturation curve. Temperature of melting (Tm) and van’t Hoff enthalpy of unfolding (ΔHvh) values were calculated using EXAM (24). The free energy of unfolding relative to WT was calculated using the method of Becktel and Schellman: ΔΔGu = ΔTmΔSuWT (25). A positive value of ΔΔGu indicates a stability gain. The ΔSuWT was 0.43 (±0.02) kcal mol−1 K−1 (19).
RESULTS
Kinetic Parameters for Turnover of Substrates and for Inhibition of TEM-1 and S130G β-Lactamases
We investigated the turnover of two penicillins and two cephalosporins with both the TEM-1 and its S130G mutant variant (Table 2). Whereas Km values increased for both ampicillin (6-fold) and benzylpenicillin (14-fold) for the mutant protein, they decreased slightly for nitrocefin (2.5-fold) and cephaloridine (2.3-fold). However, the catalytic rate constants (kcat) of S130G for these β-lactam antibiotics all decreased significantly; the effect ranges from a 9-fold decrease in kcat for ampicillin to a 2300-fold decrease for cephaloridine. This decrease in hydrolytic ability of the mutant enzyme is responsible for the decrease in the pseudo-second-order rate constant kcat/Km for all substrates measured; for instance, the kcat/Km for S130G is only 0.05% that for TEM-1 against benzylpenicillin. Considering that kcat/Km values for the wild-type enzyme were at the high levels of 106 to nearly 108 M−1 s−1, it would appear that some sacrifice in catalysis does take place on mutation of Ser130 to glycine, but the levels of activity are still sufficient for the manifestation of resistance.
Table 2.
Kinetic Parameters for TEM-1 Wild Type and the S130G Mutant
|
Km (mM)
|
kcat (s−1)
|
kcat/Km(M−1 s−1)
|
||||
|---|---|---|---|---|---|---|
| TEM-1 | S130G | TEM-1 | S130G | TEM-1 | S130G | |
| ampicillin | 0.02 | 0.13 ± 0.01 | 1530 | 170 ± 4 | 7.7 × 107 | (1.3 ± 0.1) × 106 |
| benzylpenicillin | 0.02 | 0.28 ± 0.02 | 2000 | 15 ± 1 | 1.0 × 108 | (5 ± 0.4) × 104 |
| nitrocefin | 0.22 | 0.09 ± 0.01 | 920 | 10 ± 1 | 4.2 × 106 | (1.0 ± 0.1) × 105 |
| cephaloridine | 0.93 | 0.39 ± 0.05 | 1500 | 0.7 ± 0.1 | 1.6 × 106 | (1.7 ± 0.2) × 103 |
We also investigated the parameters for interaction with the three clinically used mechanism-based inhibitors clavulanate, tazobactam, and sulbactam (Table 3). These analyses follow the procedures described earlier (3, 9, 11). Somewhat unexpectedly, the kcat values for the turnover of these inhibitors (the compounds serve both as inhibitors and substrates) by the mutant enzyme were actually improved, but the values for the S130G mutant enzyme approach those for typical substrates (Table 2). The first-order rate constants for enzyme inactivation (kinact) were also somewhat improved in each case for the mutant enzyme. The increases in the values for kcat were relatively larger than those for kinact (sulbactam being the exception). As a consequence, the partition ratios (kcat/kinact) for clavulanate and tazobactam were elevated somewhat for the mutant variant, but the parameter for sulbactam actually decreased by 5.3-fold. The partition ratio is an indication of the efficiency of the enzyme inactivation chemistry; the lower the number, the better the inactivation chemistry. Therefore, the mutant enzyme is less prone to inactivation by clavulanate and tazobactam.
Table 3.
Inhibition Parameters for TEM-1 Wild Type and the S130G Mutant
| enzyme | inhibitor | kcat (s−1) | kinact (s−1) | KI (μM) | kcat/kinact | krec (s−1) |
|---|---|---|---|---|---|---|
| wild typea | clavulanate | 0.21 ± 0.03 | (1.7 ± 0.03) × 10−3 | 0.4 | 125 ± 36 | (8.4 ± 0.1) × 10−3 |
| sulbactam | 1.95 ± 0.15 | 2.0 × 10−4 | 1.6 | 1 × 104 | (1.12 ± 0.02) × 10−2 | |
| tazobactam | 1.4 ± 0.9 | (3 ± 2) × 10−3 | (2 ± 1) × 10−2 | 475 ± 42 | (7.23 ± 0.05) × 10−3 | |
| S130G mutant | clavulanate | 4.6 ± 1.0 | (2.9 ± 0.6) × 10−3 | 105 ± 20 | 1600 ± 100 | (1.2 ± 01) × 10−2 |
| sulbactam | 49 ± 22 | (2.6 ± 1.0) × 10−2 | 220 ± 15 | 1900 ± 450 | (9.5 ± 0.1) × 10−4 | |
| tazobactam | 22 ± 13 | (3.5 ± 1.3) × 10−2 | 95 ± 15 | 630 ± 90 | (5.2 ± 0.1) × 10−3 |
See ref 4.
Furthermore, the apparent KI values for the noncovalent preacylation complexes measured for the S130G mutant enzyme were consistently and significantly elevated over those measured for the wild-type enzyme (Table 3). This observation indicated that saturation of the active site of the mutant enzyme by these agents is substantially less likely to take place for the S130G enzyme. We acknowledge that the mathematical expressions for kcat and kinact for the cases of mechanism-based inhibitors are complicated, made up of multiple microscopic rate constants that define the various steps. Similarly, Km is a complex factor. For examples of these mathematical expressions, please consult Mobashery et al. (26). It is conceivable that within the short period of measurements for the competition experiments, a significant catalytic commitment to turnover would take place, and indeed what is measured as KI may actually approximate the Km for the given inhibitor. An increased apparent KI value might thus result from an increased deacylation vs acylation rate constants, a true increase in the dissociation constant, or both. Regardless of whether it is the KI or the Km that is being evaluated, the consequence is the same: the IRT β-lactamase does not experience saturation with the given inhibitor until a higher concentration of the inhibitor is achieved, with the attendant manifestations of poorer inhibition of the enzyme. Therefore, it is plausible that elevated apparent KI values play an important role in manifestation of the IRT phenotype in organisms that harbor the S130G substitution.
We parenthetically comment here that the substitution at position 130 is not influencing the recovery from the portion of the enzyme that has been inhibited by the transiently inhibited species (5, Figure 2). The recovery from inhibition (krec) by this species in each case (Table 3) is insignificantly influenced by the mutation.
X-ray Crystallography
The crystallographic structure of TEM-76 was determined to 1.40 Å resolution. The quality of the data and refinement are described in Table 1. The structure closely resembles that of the wild-type and pseudo-wild-type proteins (the isofunctional stabilized M182T variant, WT*, used here for structural comparisons) (19); the root mean squared deviation of all Cα atoms was 0.26 Å to the latter. Ramachandran analysis of the structure showed that 93% of the residues were in most favored regions, with 6.6% in additionally allowed regions and 0.4% in generously allowed regions. The final R-factors for the working set (Rcryst) and the test set (Rfree) are 18.3% and 22.4%, respectively. At this resolution, interesting features in the density can be seen. Among these are holes in the density of aromatic side chains (Figure 3A) and the appearance of alternate configurations in the electron density maps; 14 residues were accordingly modeled in two configurations (see Materials and Methods) based on positive Fo – Fc density (Figure 3B).
Figure 3.
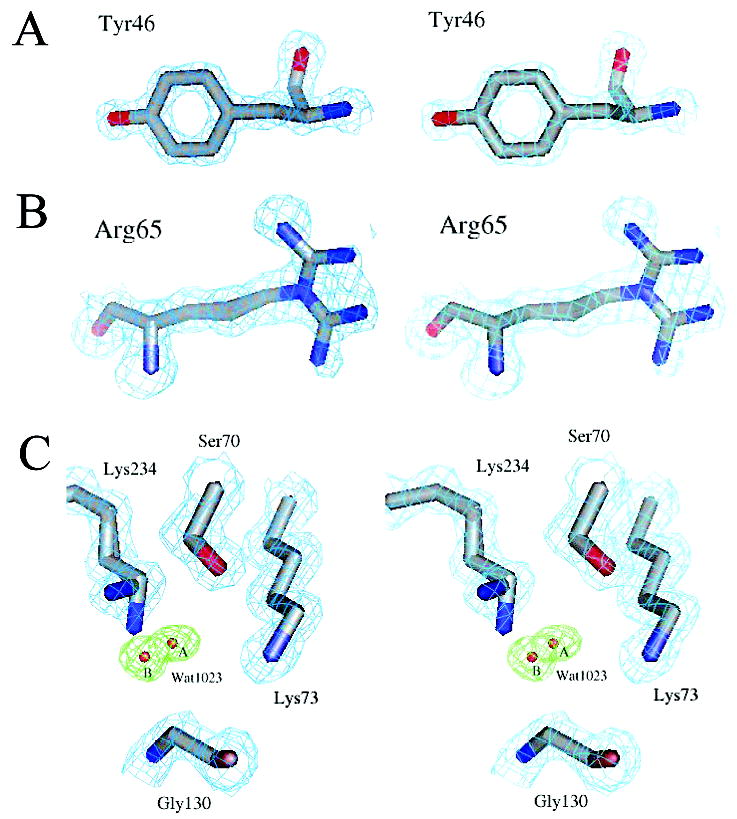
Electron density of the characteristic regions of the S130G structure. Carbon, nitrogen, and oxygen atoms are colored gray, blue, and red, respectively. (A–C) Quality of the S130G β-lactamase mutant crystal structure at 1.40 Å resolution. (A) Stereoview of the Tyr46 structure and 2Fo – Fc electron density contoured at 2σ (cyan). (B) Stereoview of the alternative conformations modeled for Arg65 superimposed on the 2Fo – Fc electron density map (cyan) at 1σ. (C) Electron density for the S130G active site. The final 2Fo – Fc map (cyan) contoured at 1.5σ is overlaid with simulated annealing omit electron density at 4.0σ (green) for the new active site water (Wat1023A/Wat1023B).
Whereas the S130G structure resembles that of the WT* overall, in the active site several changes at key positions were apparent in the difference electron density maps. A new water molecule (Wat1023) was modeled into the active site (Figure 3C) based on strong positive Fo – Fc electron density features at a contour level of 3σ. Wat1023 was modeled in two configurations (Wat1023A and Wat1023B) due to residual positive Fo – Fc electron density at a contour level of 3σ after refinement. The two configurations of this water molecule are 1.5 Å apart. Using this refined model, simulated annealing omit Fo – Fc electron density maps were then calculated for the alternate configurations of Wat1023. These showed strong electron density for this water molecule at a contour level of 4.0σ (Figure 3C), with distinct density still present at a contour level of 5.0σ. The B-factors of these waters are as follows: Wat1023A = 9.9 Å2 and Wat1023B = 12.4 Å2. The two conformations of the new active site water molecule, Wat1023A and Wat1023B, are located 1.1 and 1.3 Å, respectively, from the position that the missing Oγ of Ser130 occupies in the wild-type structure. A second conformation of the active site residue Lys234 was modeled on the basis of unambiguous features in Fo – Fc electron density maps at a contour level of 3.0σ. This second conformation is rotated relative to the canonical wild-type conformation; this moves the Nζ atom of this new conformation 1.5 Å away from the position observed in the WT* structure. Correspondingly, the Nζ atom of Lys73 has moved 0.5 Å relative to the WT* TEM structure; this increases the Lys73 Nζ–Ser70 Oγ distance from 2.9 Å in WT* to 3.4 Å in TEM-76, breaking this key hydrogen bond (Table 4). A similarity with the WT* structure was the presence of the hydrolytic water (Wat1561), which was modeled into Fo – Fc positive electron density visible at a 3σ contour level (Figure 4); in the refined structure, the B-factor of this water was 29.2 Å2. On the basis of results with the analogous S130G mutant in SHV, the presence of this water was surprising (see Discussion). In addition, the Ser130Gly substitution results in a local expansion of the binding site; the Ser70–Ser130 Cα–Cα distance has increased from 6.7 Å in WT* to 7.3 Å in S130G, a difference of 0.6 Å.
Table 4.
Selected Distances in TEM-76 (S130G), TEM-32 (M69I/M182T), and WT*
| distance (Å)
|
|||
|---|---|---|---|
| TEM-76 | TEM-32 | WT* a | |
| catalytic waterb–Ser70 Oγ | 2.6 | 2.8 | 2.8 |
| catalytic water–Glu166 Oε2 | 2.3 | 2.5 | 2.7 |
| catalytic water–Asn170 Nδ | 3.2 | 2.7 | 2.6 |
| oxyanion hole waterc–Ser70 Oγ | 2.8 | 3.0 | 2.7 |
| oxyanion hole water–Ser70 N | 3.0 | 2.9 | 2.8 |
| oxyanion hole water–Ala237 N | 2.9 | 3.2 | 3.1 |
| oxyanion hole water–Ala237 O | 2.7 | 2.6 | 2.8 |
| C3 waterd–Arg244 NH2 | 2.7 | 2.9 | 2.8 |
| C3 water–Ser235 Oγ | 3.7 | 3.1 | 2.8 |
| Ser70 Oγ–Lys73 Nζ | 3.4 | 3.1 | 2.8 |
| Ser70 Oγ–Ser130 Oγ | NPe | 5.5 | 3.2 |
| Ser130 Oγ–Lys73 Nζ | NP | 5.4 | 3.8 |
| Ser130 Oγ–Lys234 Nζ | NP | 3.3 | 2.8 |
| active site waterf–Ser70 Oγ | 2.9 | 2.7 (first)g
3.2 (second) |
NP |
| active site water–Lys73 Nζ | 2.8 | 2.9 | NP |
| active site water–Lys234 Nζ | 3.0 | 2.9 | NP |
| active site water–Lys234 Nζ(B) | 2.9 | NP | NP |
| active site water(B)–Ser70 Oγ | 3.1 | NP | NP |
| active site water(B)–Lys73 Nζ | 3.8 | NP | NP |
| active site water(B)–Lys234 Nζ | 3.7 | NP | NP |
| active site water(B)–Lys234 Nζ(B) | 4.0 | NP | NP |
| Lys73 Nζ–Asn132 Oδ1 | 3.0 | 3.0 | 3.0 |
PDB entry 1JWP.
The catalytic water is numbered Wat1561 in TEM-76, Wat6 in TEM-32, and Wat57 in WT*.
The oxyanion hole water is numbered Wat1012 in TEM-76, Wat5 in TEM-32, and Wat196 in WT*.
This water, expected to interact with the C3 carboxylate of β-lactams, is numbered Wat1614 in TEM-76, Wat7 in TEM-32, and Wat99A in WT*.
Not present.
The active site water is numbered Wat1023A/Wat1023B in TEM-76 and Wat85 in TEM-32.
Measured from the alternate conformations of Ser70 seen in TEM-32.
Figure 4.
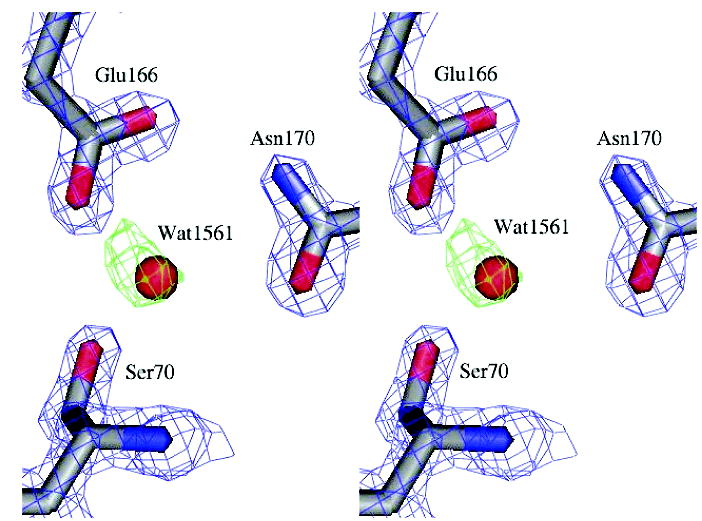
Electron density for the S130G catalytic water. The final 2Fo – Fc map (blue) at 2σ for the enzyme side chains is overlaid with the Fo – Fc electron density map (green, 3σ) prior to modeling the catalytic water.
Thermal Stability of the S130G Mutant
Ser130 is hypothesized to have both a kinetic and a structural role in class A β-lactamases (21, 27, 28); the Ser130Gly substitution in Streptomyces albus G β-lactamase resulted in decreased stability, though this was not a thermodynamic measurement (28). TEM-1 has previously been shown to reversibly denature in a two-state fashion (23), and the thermal stability of the TEM S130G mutant was determined in a similar manner. TEM-76 has a melting temperature of 52.3 °C, a stabilization of 0.8 deg relative to wild-type enzyme, and a stability gain of 0.34 kcal/mol, using the method of Schellman (25). The van’t Hoff enthalpy has decreased from 139.5 kcal/mol in wild-type TEM-1 (19) to 115.0 kcal/mol for S130G, which slightly complicates the interpretation of this stability result. Nevertheless, it seems clear that this mutant is more stable in the region of its Tm than the WT enzyme.
DISCUSSION
Over 25 inhibitor-resistant TEM β-lactamase mutants have been clinically isolated to date, many involving substitutions distant from the catalytic center. The Ser130Gly substitution might conceptually be the simplest to understand; eliminating the Ser130 Oγ prevents irreversible covalent attachment of the inhibitor to this residue, thus disrupting the inactivation pathway. In nature, however, substitution of the strictly conserved Ser130 occurs in only 3 out of 25 IRTs, and then only to a glycine residue (www.lahey.org/Studies/temtable), consistent with a mechanistic role for this residue. What we would like to address here is how the S130G mutant remains functional at all and how the structural perturbations caused by truncating the cross-linking residue resemble those seen in more subtle, and arguably more successful, inhibitor-resistant variants.
Like other IRT variants, the inhibitor resistance gained by S130G comes with decreased hydrolytic ability against β-lactam substrates (29–32). If Ser130 acts as the catalytic acid in substrate hydrolysis, how does S130G remain active? The two configurations of the new active site water, Wat1023 (Wat1023A/Wat1023B), are located 1.1 and 1.3 Å from the missing Ser130 Oγ, respectively (Figure 3C), and likely replace Ser130 as the proton source. Replacement of this Ser130 Oγ by a water molecule previously had been predicted on the basis of molecular modeling results (33). Though S130G is less efficient than TEM-1 at hydrolyzing β-lactam substrates (the rate of catalysis has decreased 10-fold for ampicillin and almost 150-fold for benzylpenicillin), what remains of the catalytic competence is sufficient to confer antibiotic resistance on the host bacteria in vivo. This could explain why only Ser130 substitutions to glycine naturally occur, since a substitution to any other residue would be too large to easily accommodate a new water molecule in this position in the active site; in fact, the S130A mutant is much less active than the S130G enzyme, as evidenced by their relative kcat/Km values (28).
One basis for the resistance conferred by Ser130Gly seems obvious: the deletion of the cross-linking residue. Unexpectedly, this “most obvious” of IRT mutants shares mechanistic and structural similarities with several of the more subtle IRTs previously characterized. Like TEM-33 (M69L), the apparent equilibrium KI values have increased dramatically for S130G (Table 3), reflecting diminished active site saturation by the inhibitors. Structurally, the mutant resembles some of the accommodations observed in TEM-32 (Figure 5, Table 4). In TEM-32 (M69I/M182T), the χ1 angle of Ser130 has changed by 64 deg, placing the Oγ of Ser130 2.2 Å away from the canonical Ser130 Oγ conformation. This rotation of Ser130 away from the active site allows incorporation of a new water molecule. Interestingly, this new TEM-32 water molecule is only 0.20 Å from one configuration of the new TEM-76 water molecule (Wat1023A); both of these mutants have acquired the same water molecule to offset displacement, or replacement, of Ser130. In addition, Lys234 has moved relative to the wild-type protein in both TEM-32 and TEM-76. In class A β-lactamases, the Lys234 side chain normally hydrogen bonds to the hydroxyl group of Ser130. The Ser130Gly substitution causes Lys234 to adopt a novel second conformation not present in the pseudo-wild-type structure at 0.85 Å resolution (34). In the primary Lys234 conformation, the side chain hydrogen bonds to the new active site water molecule replacing the Ser130 hydroxyl group. Due to the partial occupancy of this new water molecule, the Lys234 side chain adopts a second conformation where it hydrogen bonds to the backbone carbonyl of Ile127; this Lys234–Ile127 hydrogen bond is present in TEM-32 as well.
Figure 5.
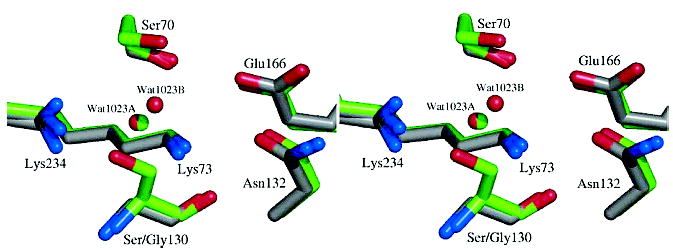
Active site overlay of the S130G mutant (carbons in gray) with TEM-32 (M69I/M182T) (carbons in green). Nitrogen and oxygen atoms are colored blue and red, respectively. Active site waters are shown as spheres in red (S130G) or green (TEM-32).
Another such structural perturbation causing inhibitor resistance is increased disorder of the conserved water molecule (Wat1614) hydrogen-bonded to the guanidinium group of Arg244 and the carbonyl group of Val216; this water molecule is the source of the proton necessary for ring opening in clavulanate inactivation (Figure 2, Table 4) (2, 3). In TEM-76, the B-factor of this water molecule has increased relative to the stabilized wild-type apo structure (33.2 Å2 in S130G, compared to 17.1 Å2 in WT* at 1.75 Å resolution). At least two other IRT mutants appear to have gained inhibitor resistance by similar means; in both the TEM-84 (N276D) and TEM-30 (R244S) X-ray structures, this water molecule could not be crystallographically refined (9, 10). Disrupting the inactivation pathway for mechanism-based inhibitors likely increases partitioning of the inhibitors through the hydrolytic pathway, reducing overall inhibition.
The structure of the S130G mutant was recently determined to 1.8 Å in SHV β-lactamase (35), which has 68% sequence identity to TEM-1 and is closely related structurally; their structures have a Cα RMSD of 1.4 Å, and the catalytically important residues have an RMSD of only 0.23 Å (36, 37). Despite these similarities, the Ser130Gly substitution in TEM leads to related, but nevertheless different, conformational changes compared to SHV S130G. Similar to TEM-76, a new water molecule is found less than 1 Å away from the missing Ser130 hydroxyl group in SHV S130G. The Ser130Gly substitution in both TEM and SHV results in a binding site expansion, as evidenced by the 0.6 Å increase in both the Ser70–Ser130 and Lys73–Ser130 Cα–Cα distances. Substitution of Ser130 with glycine in SHV leads to a rotation of the Ser70 hydroxyl group so that it breaks highly conserved hydrogen bonds to Lys73 and the catalytic water molecule and hydrogen bonds to the water molecule within the oxyanion hole instead. No such reorientation of Ser70 is seen in TEM-76; however, rotation of Lys73 achieves some of the same effect, breaking the hydrogen bond with Ser70. Though the TEM-1 and SHV-1 binding sites align closely, Ser70 has a different orientation in each. Interestingly, rotation of Ser70 in SHV S130G causes it to adopt the same conformation seen in wild-type TEM, though different from native SHV-1; perhaps this explains why this catalytic serine does not further reorient in the TEM S130G mutant.
One of the most striking observations in the SHV S130G mutant is the departure of the Glu166-activated water molecule, which is thought to be critical for catalysis. In the TEM S130G structure reported here, this key catalytic water is clearly present (Figure 4), though perhaps less ordered than in the WT* structure. Overall, this key water is still preorganized for hydrolysis and still contacts its primary activating residues, including the deacylation catalytic base Glu166.
In closing, we return to our initial questions: how does S130G tolerate the loss of a key catalytic functionality, and what are the structural bases for loss of activity versus both substrates and inhibitors? The structural replacement of the Oγ of Ser130 by Wat1023 explains how the enzyme remains active at all and offers another example of the extraordinary chemical plasticity of enzymes. The enzyme appears to lose activity based on the relative inefficiency of a bound water versus a serine Oγ and movement of Lys73. The decreased activity of S130G does not appear to result from diminished overall stability of the folded enzyme, which, if anything, is actually somewhat stabilized relative to WT. What is intriguing is that the accommodations observed in S130G are qualitatively similar to those observed in IRT mutants whose substitutions often occur at sites less directly correlated with the sites of cross-linking. Like TEM-33 (M69L), S130G suffers from elevated apparent KI values. TEM-30 (R244S) also results in a displacement of Wat1614, as does TEM-84 (N276D). Similarly, TEM-32 (M69I/M182T) results in a displacement of Ser130, the appearance of a water molecule equivalent to Wat1023A, and a conformational change in Lys234. However, TEM-32 has acquired resistance to inhibitors with only a modest (5-fold) reduction in kcat for substrates compared to S130G. In S130G, nature would appear to have made the most intuitive mutation, but also among the least successful based on catalytic activity and prevalence. The bases of action of the many other action-at-a-distance IRTs, such as TEM-38 (M69V/R275L) and TEM-39 (M69L/W165R/N276D) which, like TEM-30 and TEM-32, result in relatively resistant, relatively active IRTs, appear ever more interesting in light of the conserved accommodations of this simplest of IRT mutants, S130G.
Acknowledgments
We thank Yu Chen, Ruth Brenk, Federica Morandi, and Alan Graves for reading the manuscript and Yu Chen for crystallographic advice and assistance.
Footnotes
This work was supported by NIH Grants GM63815 (to B.K.S.) and AI33170 (to S.M.) and by the Achievement Rewards for College Scientists Foundation and a National Science Foundation predoctoral fellowship (to V.L.T.).
‡ Atomic coordinates and structure factors have been deposited in the Protein Data Bank at the Research Collaboratory for Structural Bioinformatics at Rutgers University (entry 1YT4).
Abbreviations: IRT, inhibitor-resistant TEM; WT, wild type; WT*, isofunctional stabilized wild type; Tm, melting temperature.
References
- 1.Kuzin AP, Nukaga M, Nukaga Y, Hujer A, Bonomo RA, Knox JR. Inhibition of the SHV-1 beta-lactamase by sulfones: crystallographic observation of two reaction intermediates with tazobactam. Biochemistry. 2001;40:1861–1866. doi: 10.1021/bi0022745. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Janota K, Tabei K, Huang N, Siegel MM, Lin YI, Rasmussen BA, Shlaes DM. Mechanism of inhibition of the class A beta-lactamases PC1 and TEM-1 by tazobactam. Observation of reaction products by electrospray ionization mass spectrometry. J Biol Chem. 2000;275:26674–26682. doi: 10.1074/jbc.M002369200. [DOI] [PubMed] [Google Scholar]
- 3.Imtiaz U, Billings EM, Knox JR, Manavathu EK, Lerner SA, Mobashery S. Inactivation of class A beta-lactamases by clavulanic acid: the role of arginine-244 in a proposed nonconcerted sequence of events. J Am Chem Soc. 1993;115:4435–4442. [Google Scholar]
- 4.Brown RP, Aplin RT, Schofield CJ. Inhibition of TEM-2 beta-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry, . Biochemistry. 1996;35:12421–12432. doi: 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- 5.Bush K, Mobashery S. How beta-lactamases have driven pharmaceutical drug discovery. From mechanistic knowledge to clinical circumvention. Adv Exp Med Biol. 1998;456:71–98. [PubMed] [Google Scholar]
- 6.Knowles JR. Penicillin Resistance–The chemistry of beta-lactamase inhibition. Acc Chem Res. 1985;18:97–104. [Google Scholar]
- 7.Fisher J, Charnas RL, Knowles JR. Kinetic studies on the inactivation of Escherichia coli RTEM beta-lactamase by clavulanic acid. Biochemistry. 1978;17:2180–2184. doi: 10.1021/bi00604a024. [DOI] [PubMed] [Google Scholar]
- 8.Charnas RL, Knowles JR. Inactivation of RTEM beta-lactamase from Escherichia coli by clavulanic acid and 9-deoxyclavulanic acid. Biochemistry. 1981;20:3214–3219. doi: 10.1021/bi00514a035. [DOI] [PubMed] [Google Scholar]
- 9.Swaren P, Golemi D, Cabantous S, Bulychev A, Maveyraud L, Mobashery S, Samama JP. X-ray structure of the Asn276Asp variant of the Escherichia coli TEM-1 beta-lactamase: direct observation of electrostatic modulation in resistance to inactivation by clavulanic acid. Biochemistry. 1999;38:9570–9576. doi: 10.1021/bi990758z. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Minasov G, Shoichet BK. The structural bases of antibiotic resistance in the clinically derived mutant beta-lactamases TEM-30, TEM-32, and TEM-34. J Biol Chem. 2002;277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 11.Meroueh SO, Roblin P, Golemi D, Maveyraud L, Vakulenko SB, Zhang Y, Samama JP, Mobashery S. Molecular dynamics at the root of expansion of function in the M69L inhibitor-resistant TEM beta-lactamase from Escherichia coli. J Am Chem Soc. 2002;124:9422–9430. doi: 10.1021/ja026547q. [DOI] [PubMed] [Google Scholar]
- 12.Lamotte-Brasseur J, Dive G, Dideberg O, Charlier P, Frere JM, Ghuysen JM. Mechanism of acyl transfer by the class A serine beta-lactamase of Streptomyces albus G. Biochem J. 1991;279:213–221. doi: 10.1042/bj2790213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atanasov BP, Mustafi D, Makinen MW. Protonation of the beta-lactam nitrogen is the trigger event in the catalytic action of class A beta-lactamases. Proc Natl Acad Sci USA. 2000;97:3160–3165. doi: 10.1073/pnas.060027897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vakulenko S, Golemi D. Mutant TEM beta-lactamase producing resistance to ceftazidime, ampicillins, and beta-lactamase inhibitors. Antimicrob Agents Chemother. 2002;46:646–653. doi: 10.1128/AAC.46.3.646-653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon M. The determination of enzyme inhibitor constants. Biochem J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman, R. (1988) in Mechanism-based Enzyme Inactivation: Chemistry and Enzymology, p 22, CRC Press, Boca Raton, FL.
- 17.Koerber SC, Fink AL. The analysis of enzyme progress curves by numerical differentiation, including competitive product inhibition and enzyme reactivation. Anal Biochem. 1987;165:75 –87. doi: 10.1016/0003-2697(87)90203-x. [DOI] [PubMed] [Google Scholar]
- 18.Glick BR, Brubacher LJ, Leggett DJ. A graphical method for extracting rate constants from some enzyme-catalyzed reactions not monitored to completion. Can J Biochem. 1978;56:1055–1057. doi: 10.1139/o78-166. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 Å resolution. Nature. 1992;359:700–705. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 22.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A newsoftware suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Minasov G, Shoichet BK. Noncovalent interaction energies in covalent complexes: TEM-1 beta-lactamase and beta-lactams. Proteins. 2002;47:86–96. [PubMed] [Google Scholar]
- 24.Kirchhoff, W. H. (1993) NIST.
- 25.Becktel WJ, Schellman JA. Protein stability curves. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- 26.Mobashery S, Ghosh SS, Tamura SY, Kaiser ET. Design of an effective mechanism-based inactivator for a zinc protease. Proc Natl Acad Sci USA. 1990;87:578–582. doi: 10.1073/pnas.87.2.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juteau JM, Billings E, Knox JR, Levesque RC. Site-saturation mutagenesis and three-dimensional modelling of ROB-1 define a substrate binding role of Ser130 in class A beta-lactamases. Protein Eng. 1992;5:693–701. doi: 10.1093/protein/5.7.693. [DOI] [PubMed] [Google Scholar]
- 28.Jacob F, Joris B, Lepage S, Dusart J, Frere JM. Role of the conserved amino acids of the “SDN” loop (Ser130, Asp131, and Asn132) in a class A beta-lactamase studied by site-directed mutagenesis. Biochem J. 1990;271:399–406. doi: 10.1042/bj2710399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aumeran C, Chanal C, Labia R, Sirot D, Sirot J, Bonnet R. Effects of Ser130Gly and Asp240Lys substitutions in extended-spectrum beta-lactamase CTX-M-9. Antimicrob Agents Chemother. 2003;47:2958–2961. doi: 10.1128/AAC.47.9.2958-2961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaire M, Labia R, Samama JP, Masson JM. Site-directed mutagenesis at the active site of Escherichia coli TEM-1 beta-lactamase. Suicide inhibitor-resistant mutants reveal the role of arginine 244 and methionine 69 in catalysis. J Biol Chem. 1992;267:20600–20606. [PubMed] [Google Scholar]
- 31.Chaibi EB, Sirot D, Paul G, Labia R. Inhibitor-resistant TEM beta-lactamases: phenotypic, genetic and biochemical characteristics. J Antimicrob Chemother. 1999;43:447–458. doi: 10.1093/jac/43.4.447. [DOI] [PubMed] [Google Scholar]
- 32.Helfand MS, Bethel CR, Hujer AM, Hujer KM, Anderson VE, Bonomo RA. Understanding resistance to beta-lactams and beta-lactamase inhibitors in the SHV beta-lactamase: lessons from the mutagenesis of SER-130. J Biol Chem. 2003;278:52724–52729. doi: 10.1074/jbc.M306059200. [DOI] [PubMed] [Google Scholar]
- 33.Lamotte-Brasseur J, Jacob-Dubuisson F, Dive G, Frere JM, Ghuysen JM. Streptomyces albus G serine beta-lactamase. Probing of the catalytic mechanism via molecular modelling of mutant enzymes. Biochem J. 1992;282:189–195. doi: 10.1042/bj2820189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minasov G, Wang X, Shoichet BK. An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J Am Chem Soc. 2002;124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- 35.Sun T, Bethel CR, Bonomo RA, Knox JR. Inhibitor-resistant class A beta-lactamases: consequences of the Ser130-to-Gly mutation seen in Apo and tazobactam structures of the SHV-1 variant. Biochemistry. 2004;43:14111–14117. doi: 10.1021/bi0487903. [DOI] [PubMed] [Google Scholar]
- 36.Kuzin AP, Nukaga M, Nukaga Y, Hujer AM, Bonomo RA, Knox JR. Structure of the SHV-1 beta-lactamase. Biochemistry. 1999;38:5720–5727. doi: 10.1021/bi990136d. [DOI] [PubMed] [Google Scholar]
- 37.Jelsch C, Mourey L, Masson JM, Samama JP. Crystal structure of Escherichia coli TEM1 beta-lactamase at 1.8 Å resolution. Proteins. 1993;16:364–383. doi: 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]