

Abstract
Acute respiratory distress syndrome (ARDS) is a clinical syndrome characterized by stereotypic host inflammatory and repair cellular responses; however, mechanisms regulating the resolution of ARDS are poorly understood. Here, we report the isolation and characterization of a novel population of mesenchymal cells from the alveolar space of ARDS patients via fiber-optic bronchoscopy with bronchoalveolar lavage (BAL). BAL was performed on 17 patients during the course of ARDS. Immunofluorescence staining and multiparameter flow cytometric analysis defined a population of alveolar mesenchymal cells (AMCs) that are CD45−/prolyl-4-hydroxylase+/α-smooth muscle actin+/−. AMCs proliferated in ex vivo cell culture for multiple passages; early passage (3–5) cells were subsequently analyzed in 13 patients. AMCs isolated from patients with persistent or nonresolving ARDS (ARDS-NR, n = 4) demonstrate enhanced constitutive activation of prosurvival signaling pathways involving PKB/Akt, FKHR, and BCL-2 family proteins compared with AMCs from patients with resolving ARDS (ARDS-R, n = 9). Exogenous transforming growth factor-β1 markedly induces PKB/Akt activation in AMCs from ARDS-R. ARDS-NR cells are more resistant to serum deprivation-induced apoptosis compared with ARDS-R. This study identifies a novel population of mesenchymal cells that can be isolated from the alveolar spaces of ARDS patients. AMCs in patients with ARDS-NR acquire an activational profile characterized by enhanced prosurvival signaling and an antiapoptotic phenotype. These findings support the concept that apoptosis of mesenchymal cells may be an essential component of normal repair and resolution of ARDS and suggest that dysregulation of this process may contribute to persistent ARDS.
Keywords: acute lung injury, fibrosis, BCL-2 genes, fibroblast, protein kinase B, apoptosis, tissue repair
the pathophysiology of acute respiratory distress syndrome (ARDS) consists of overlapping acute “inflammatory” and delayed “repair/fibrotic” phases (50, 51, 55). Alveolar epithelial and endothelial cell injury leads to loss of alveolar-capillary barrier function and allows influx of proteinaceous fluid, blood, and inflammatory cells into the alveolar compartment. Activation of the coagulation system, complement, and the release of cytokines amplify the inflammatory response and may contribute to further injury. Overlapping with the acute/exudative phase is the process of repair marked by the presence of intra-alveolar mesenchymal cells/fibroblasts. In some cases, repair proceeds normally with complete resolution and reestablishment of the normal alveolar architecture. In other cases, however, mesenchymal cells persist in the interstitial/alveolar spaces and continue to secrete extracellular matrix with resultant fibrosing alveolitis (47, 55). Although clinical resolution following lung injury is typically appreciated several days after the onset of ARDS, biochemical measurements suggest that the repair process is initiated within hours of injury and that the fibroproliferative response increases with the duration of ARDS (14, 17, 21, 39, 57). Moreover, several studies have shown that the fibroproliferative response to ARDS correlates with poor outcomes (14, 15, 30, 39, 40). The mechanisms underlying dysfunctional repair/fibrosis in ARDS remain unclear; although mesenchymal cells are responsible for collagen synthesis in wound repair and fibrosis, little is known about the role of these cells in the pathobiology of persistent, nonresolving ARDS.
Mesenchymal cell apoptosis is necessary for the normal resolution of the repair process following tissue injury in other organ systems; for example, failure of apoptosis results in pathological scar formation after cutaneous wounds (19). Transforming growth factor (TGF)-β1 is a multifunctional cytokine that is critical for wound repair responses and has been implicated in the pathophysiology of tissue fibrosis (7), as well as in the pathobiology of both early and late phases of ARDS (20, 22, 48). Studies have shown increased expression of TGF-β1 mRNA in lungs of patients who died of fibroproliferative ARDS (22) and the presence of activated fibroblasts/myofibroblasts in diffuse alveolar damage of ARDS (37, 45). A recent study demonstrated that bronchoalveolar lavage (BAL) fluid obtained within 48 h of the onset of ARDS contained increased levels of active TGF-β1, which accounts, in part, for the collagen-inducing activity identified in BAL fluid of ARDS patients (10). Mesenchymal cells isolated postmortem from patients with persistent ARDS demonstrate increased proliferative capacity (13, 49). We have previously shown that TGF-β1 stimulates activation of the prosurvival serine-threonine kinase, protein kinase B (PKB)/Akt, in both normal human lung fibroblasts and in lung mesenchymal cells isolated from a patient with ARDS (32). We have also shown that TGF-β1 activation of PKB/Akt protects normal human lung fibroblasts from serum deprivation-induced apoptosis (32).
A major limiting factor in our understanding of the mechanisms underlying normal repair and persistent/nonresolving ARDS is the difficulty in obtaining mesenchymal cells for ex vivo studies. Previous investigations of mesenchymal cells in ARDS have utilized cells isolated from lung tissue obtained postmortem (13, 22). BAL is a diagnostic procedure performed by fiber-optic bronchoscopy as a means of recovering cells and soluble substances from the distal airway and alveolar spaces of the lung (51). Studies in other lung disorders have shown that mesenchymal cells from the alveolar compartment can be isolated from BAL fluid (23, 24, 36, 38). BAL can be performed safely in mechanically ventilated patients with ARDS (29, 53); however, previous BAL studies in humans have focused primarily on assessments of inflammatory cell number/function, measurements of acellular components (proteins and lipids) or the effects of BAL fluid on other (reporter) cells (10, 51).
We hypothesized that resistance to apoptosis of mesenchymal cells within alveolar spaces contributes to the pathobiology of persistent/nonresolving ARDS. This study was undertaken to characterize alveolar mesenchymal cells (AMCs) isolated by BAL of patients with ARDS and to investigate whether differences in prosurvival signaling and apoptotic phenotype exist between AMCs derived from patients with resolving (ARDS-R) vs. nonresolving ARDS (ARDS-NR).
MATERIALS AND METHODS
Isolation and culture of BAL cells from ARDS patients
Patients with ARDS, as defined by the American-European Consensus Conference (AECC) (5), were identified in the critical care medicine unit at the University of Michigan Medical Center. Bronchoscopy with BAL in ARDS patients were performed as previously described (53) within the first week of onset of ARDS or as soon as it was considered safe by the attending physician. Research protocols were approved by the Institutional Review Board at the University of Michigan. Recovered BAL fluid was initially filtered through sterile gauze to remove noncellular particulate material and mucous. Cells were pelleted by centrifugation at 1,000 g for 5 min. The cell pellet was then resuspended in 2 ml of Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Grand Island, NY). Total cell counts were estimated by hemocytometer counting of an aliquot of this cell suspension, and differential counts were obtained by counting 300 cells per high-power field of a fresh cytospin and Wright-Giemsa staining. BAL cells were then plated at a density of 5 × 106 cells per 100-mm cell culture dish. The cells were maintained in medium consisting of DMEM supplemented with 10% fetal calf serum (Sigma, St. Louis, MO), 100 U/ml penicillin-streptomycin (Sigma), and fungizone (GIBCO); medium was changed every 3 days. Cells were incubated at 37°C in 5% CO2-95% air. Foci of proliferating fibroblasts were observed within 7 days, and the number of days to reach a confluent monolayer after the first trypsinization/passage varied between 7 and 25 days. Cells were passaged twice to achieve a homogeneous population of mesenchymal cells (as shown in results) and cryo-preserved in liquid nitrogen before use. All cells were subsequently analyzed at passages 3–5.
The definition of ARDS-NR (≥10 days of ARDS) used in the current study was determined retrospectively. All cellular and biochemical analyses of AMC signaling/phenotype were performed in a blinded manner (i.e., without knowledge of the patient’s clinical status or course of ARDS).
Reagents
Rabbit polyclonal antibodies to phospho-Akt (Ser473), total Akt, phospho-forkhead transcription factor (FKHR, S256), and phospho-ERK1/2 were purchased from Cell Signaling Technology (Beverly MA). Mouse monoclonal anti-prolyl-4-hydroxylase (PH, clone #5B5) and mouse monoclonal anti-α-smooth muscle actin (α-SMA, clone #1A4) were from DAKO Cytomation (Carpinteria, CA). Anti-CD45 antibodies were obtained from PharMingen (San Diego, CA). Monoclonal mouse anti-single stranded (ss) DNA was obtained from Chemicon International (Temecula, CA). Mouse monoclonal antibodies to BCL-2, BAX, α-tubulin, cellular fibronectin (clone IST-4), and vimentin were from Sigma. Mouse monoclonal antibody to cyclin D1 was from Santa Cruz Biotechnology (Santa Cruz, CA). Human platelet-derived TGF-β1 was purchased from R&D Systems (Minneapolis, MN).
Immunofluorescence labeling
Cells grown on tissue culture dishes were initially rinsed with DMEM (GIBCO) for 30 s at ambient temperature, and then fixed in 4% formaldehyde for 10 min. Cells were then washed three times in phosphate-buffered saline (PBS) before permeabilization and after each subsequent step. Permeabilization was performed in buffer consisting of 0.1% Triton in 50 mM PIPES (pH 7.0), 90 mM HEPES (pH 7.0), 0.5 mM MgCl2, 0.5 mM EGTA, and 75 mM KCl for 30 s at room temperature. Cells were sequentially incubated with 1:100 dilutions of mouse monoclonal anti-PH antibody or anti-α-SMA antibody and FITC-labeled anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA), each for 60 min at room temperature. Cells were then visualized and photographed under a Zeiss fluorescence microscope.
Multiparameter flow cytometric analyses
BAL cells were pelleted by centrifugation, and red blood cells were lysed by hypotonic shock. Cells were then fixed in 4% formalin solution. For staining of intracellular proteins, cells were first permeabilized with a commercially available kit (PharMingen). Cells were then incubated with 1.55 μg/ml (1:100 dilutions) of anti-PH antibody or 0.9 μg/ml (1:100 dilution) of anti-α-SMA antibody followed by FITC-labeled detection antibody. For cell surface staining of CD45, cells were incubated with 1:200 dilution of anti-CD45 antibody and analyzed on a FACSCalibur flow cytometer with the CellQuest software program (Becton Dickinson, Franklin Lakes, NJ).
Western immunoblotting
Cells grown on tissue culture plates were gently washed with 5 ml of PBS and lysed in 0.5 ml of cold radioimmunoprecipitation assay lysis buffer (1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.01 M NaH2PO4, 2 mM EDTA, 0.5 mM NaF) containing 2 mM sodium orthovanadate and 1:100 dilution of protease inhibitor mixture III (Calbiochem). Protein concentrations were determined by the bicinchoninic acid protein assay (Pierce). Whole cell lysates were mixed with a 1:5 vol/vol ratio of 6× electrophoresis sample buffer (0.2 M EDTA, 40 mM dithiothreitol, 6% SDS, 0.06 mg/ml pyronin, pH 6.8) and boiled at 95°C for 5 min to denature protein. Sample mixtures were then loaded and subjected to electrophoresis in a 4–20% polyacrylamide gradient gel. Proteins were electrophoretically transferred to polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford, MA) and incubated in blocking buffer containing 75 mM sodium phosphate, 70 mM sodium chloride, and 0.1% Tween 20 (pH 7.4) with 5% bovine serum albumin for 1 h at room temperature. The blot was treated with a 1:1,000 dilution of primary antibody in blocking buffer overnight at 4°C. Three washes with a buffer containing 10 mM Tris, 100 mM NaCl, and 0.1% Tween 20 were then performed before incubation with a secondary antibody conjugated to horseradish peroxidase. The washes were repeated, and membrane was incubated with SuperSignal Substrate Western blotting reagent (Pierce) for 10 min. The blot was then exposed to chemiluminescence-sensitive Kodak XOmat AR film (Eastman Kodak). Image analysis was performed with the public domain NIH Image program available on the internet at http://rsb.info.nih.gov/nih-image/.
Assessment of apoptosis
Apoptosis was assessed by an ELISA-based assay to detect ssDNA, Apoptosis ELISA Kit (Chemicon International) as previously described (32). In brief, AMCs were seeded into a 96-well plate at a density of 5,000 cells per well and cultured for 4 days in the presence of 10% FBS to 60% confluence. The medium was changed to DMEM with 0.1% FBS, and this serum deprivation was maintained for 6 days. The plate was centrifuged to bring down floating cells, the medium was removed, and the cells were fixed in 80% methanol. After fixation, cells were treated with formamide according to the manufacturer’s protocol. After being blocked and washed, cells were exposed to monoclonal mouse anti-ssDNA antibodies (1:100 dilutions), washed, and then exposed to a horseradish peroxidase-conjugated secondary antibody. After washing, 2,2-azino-bis-(3-benzthiazoline-6-sulfonic) was added for 45 min followed by a “stop solution.” Absorbance was measured by an ELISA plate reader at 405 nm. We determined the “apoptosis index” by subtracting the absorbance of negative controls for each cell line and dividing the corrected absorbance by cell counts that were obtained by Coulter counter before fixing the cells.
Assessment of proliferative capacity
AMCs were plated at equal density in 96-well ELISA culture plates, grown to 80% confluence, and growth-arrested for 24 h in DMEM containing 0.01% FBS. BrdU labeling was then performed over the subsequent 24-h period in the presence or absence of 10% FBS, according to manufacturer’s instructions (EMD Biosciences, San Diego, CA).
Statistical analysis
Statistical analysis was performed with Graph-Pad Prism version 3.0 for Windows (GraphPad Prism Software, San Diego, CA, www.graphpad.com). Densitometric analysis of Western blots was performed with the public domain NIH Image program. The density of each band was measured, and the background was subtracted; in most cases, the relative changes in densitometric values were compared with that of controls (unstimulated) normal human lung fibroblasts on the same Western blot. The two-tailed Mann-Whitney test was used for comparisons between two groups. Correlations were determined by linear regression analysis. Analysis of variance (ANOVA) with Bonferroni posttest analysis was used for comparisons between multiple groups.
RESULTS
Patient characteristics
BAL samples were studied from 17 consecutive patients with ARDS who consented to this research study at the Specialized Center of Clinically Oriented Research (SCCOR) in Acute Lung Injury at the University of Michigan. All patients met AECC criteria for ARDS (6). There were no complications encountered with bronchoscopy or BAL in these patients. The freshly isolated BAL from two patients was analyzed at sequential time points (days 1 and 5 of ARDS) by flow cytometry, and the BAL from another two patients was used to culture AMCs for further characterization by immunofluorescence staining. BAL from the remaining 13 patients was used to isolate mesenchymal cells for protein analysis and assessment of the susceptibility to apoptosis. The clinical characteristics of these 13 patients are summarized in Table 1. The mean age and SD was 43.3 ± 12.7 yr for this cohort. This group included seven men and six women. Nine of the thirteen episodes of ARDS in this patient population were classified as ARDS-R defined as duration of ARDS <10 days. Four of these episodes resulted in duration of ARDS of ≥10 days and were classified as ARDS-NR. All patients in the ARDS-R group (9/9) were alive at 30 days post-ARDS, whereas the 30-day mortality rate in the ARDS-NR group was 50% (2/4). There was no statistically significant difference in the age of patients with ARDS-R compared with ARDS-NR (40.1 ± 9.1 vs. 50.5 ± 17.8 yr, P = 0.18). The majority of ARDS cases (9/13) in this medical intensive care unit patient population were secondary to pneumonia; four patients had hospital-acquired pneumonia, three patients had community-acquired pneumonia, and two patients had aspiration pneumonia. There was one case each of ARDS due to severe sepsis, hemorrhagic shock with multiple transfusions, polysubstance overdose, and necrotizing pancreatitis (Table 1).
Table 1.
Clinical characteristics of ARDS patients
| Patient | Age, yr | Sex | Diagnosis | ARDS Days | MV Days | BAL Day | Outcome |
|---|---|---|---|---|---|---|---|
| 1 | 36 | M | CAP | 9 | 10 | 3 | Survival |
| 2 | 55 | F | Pneumonia (HAP) | 5 | 5 | 2 | Survival |
| 3 | 44 | F | Severe sepsis | 10 | 10 | 2 | Death |
| 4 | 75 | F | Pneumonia (HAP) | 14 | 20 | 7 | Death |
| 5 | 32 | M | Aspiration | 5 | 15 | 5 | Survival |
| 6 | 49 | M | Pneumonia (HAP) | 8 | 8 | 2 | Survival |
| 7 | 33 | M | Pneumonia (CAP) | 11 | 12 | 10 | Survival |
| 8 | 33 | F | Pneumonia (HAP) | 4 | 4 | 2 | Survival |
| 9 | 50 | F | Aspiration | 21 | 24 | 8 | Survival |
| 10 | 48 | M | Hemorrhagic shock | 2 | 6 | 1 | Survival |
| 11 | 29 | F | Pneumonia (CAP) | 7 | 14 | 6 | Survival |
| 12 | 39 | M | Polysubstance overdose | 2 | 6 | 1 | Survival |
| 13 | 43 | M | Necrotizing pancreatitis | 4 | 4 | 3 | Survival |
ARDS days, number of days that patient met American-European Consensus Conference criteria for acute respiratory distress syndronic (ARDS) (PaO2/FiO2 < 200; bilateral pulmonary infiltrates on chest radiograph; presence of risk factor(s) for ARDS; no evidence of left atrial hypertension) starting at day 0 as first day that diagnostic criteria were met. MV days, total number of days that the patient required mechanical ventilation (MV). BAL day, day of ARDS on which bronchoalveolar lavage (BAL) was performed. Survival implies that the patient was alive at 30 days post-ARDS. M, male; F, female; CAP, community-acquired pneumonia; HAP, hospital acquired pneumonia.
Only one of the four patients with ARDS-NR received steroids for ARDS (prednisone 25 mg every 6 h starting on day 7). The BAL for this patient was done before the initiation of steroids. Another patient with ARDS-NR had a remote history of renal transplant and was on 5 mg/day of prednisone. That dose was maintained throughout the hospitalization. In the ARDS-R group, two patients were on chronic steroids and received stress dose steroids during their course of ARDS and were rapidly tapered back to their baseline steroid dosage.
Isolation and in vitro culture of AMCs
AMCs were recovered from all ARDS-BAL samples in sufficiently high numbers that they could be expanded and maintained in cell culture and for multiple passages; all AMC cultures were initially harvested and cells cryogen-preserved at passage 3. Details of the isolation procedure are described in materials and methods. BAL cells plated from a normal volunteer show a collection of primarily “round” cells (Fig. 1A, left) that express the common leukocyte marker, CD45 (Fig. 1B, left), consistent with the known predominance of alveolar macrophages and leukocytes in normal BAL. In contrast, a significant number of “spindle-shaped” cells were observed within 24 h after initial plating of BAL cells from patients with ARDS (Fig. 1A, middle, represents a patient on day 7 postdiagnosis). Over a period of 1–2 wk of initial plating, the spindle-shaped cells in the ARDS-BAL proliferate in cell culture, while most round cells, likely representing inflammatory cells (such as macrophages, neutrophils and lymphocytes), do not appear to proliferate. Subsequent trypsinization and passaging in cell culture eliminates the morphologically round cells and results in a more homogenous population of spindle-shaped cells that are uniformly negative for CD45 by passage 3 (Fig. 1, A and B, right).
Fig. 1.
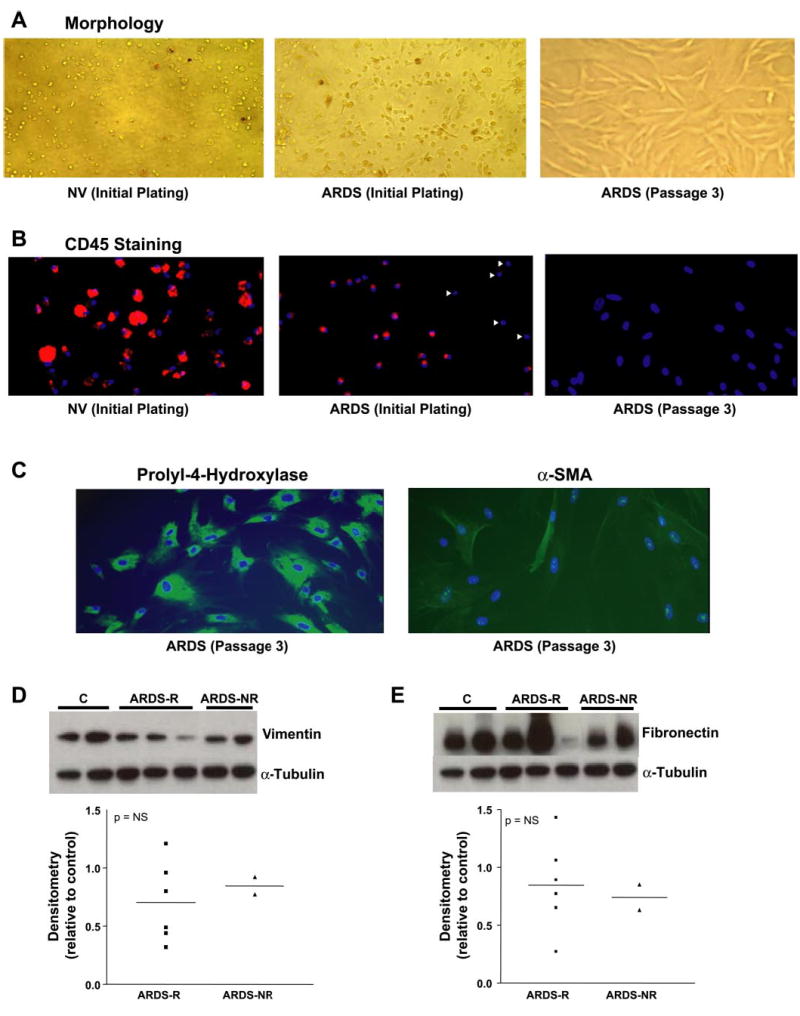
Morphological and phenotypic characterization of cultured alveolar mesenchymal cells (AMCs) from patients with acute respiratory distress syndrome (ARDS). A: morphological appearance (×20 magnification) of adherent cells recovered by bronchoalveolar lavage (BAL) and plated on tissue culture plastic within 24 h from a normal volunteer (NV, left) and from a representative patient with ARDS on day 7 postdiagnosis (middle). Subsequent trypsinization and passage in culture of the cells from ARDS patients produced homogeneous cultures of spindle-shaped cells (right). B: CD45 staining of BAL cells that were processed as described in A. C: AMC cultures at passage 3 were stained for the collagen cross-linking enzyme, prolyl-4-hydroxylase (PH, left) and for α-smooth muscle actin (α-SMA, right). D: AMC cultures at passage 3 were growth-arrested for 24 h. Whole cell lysates were obtained and assessed by Western immunoblotting for vimentin and α-tubulin. E: the Western blot in D was stripped and probed sequentially for cellular fibronectin and α-tubulin. The bottom panels demonstrate the band densitometry, relative to controls, for all AMC cell lines analyzed. ARDS-R, resolving ARDS; ARDS-NR, nonresolving ARDS; C, control; NS, not significant.
Total and cell differential counts in BAL from ARDS-R and ARDS-NR were not significantly different between the groups. Average differential cell counts were as follows: neutrophils 66.1 ±12.6% vs. 72.2 ±19.8%, macrophages 32.8 ±12.4% vs. 26.7 ± 19.4%, lymphocytes 1.2 ± 0.6% vs. 1.0 ± 0.4%, eosinophils 0.1 ± 0.1% vs. 0.25 ± 0.1% for ARDS-R vs. ARDS-NR, respectively.
AMC phenotypic profile
Differentiated cells of the hematopoietic lineage that are normally resident in or recruited into alveolar spaces of the lung are generally positive for the common leukocyte antigen, CD45 (27). Consistent with this, when BAL cells from a normal volunteer were plated on tissue culture plastic, adherent cells stained positively for CD45 by immunofluorescence labeling; significant heterogeneity in cell size and levels of expression of CD45 in these cells were also noted (Fig. 1B, left). Similar processing of cells from a patient on day 7 of ARDS, however, demonstrated a significant number of cells that are negative for CD45 staining (Fig. 1B, middle). CD45− cells appear more spindle shaped than CD45+ cells when correlated with cell morphology (not shown). Subsequent trypsinization and culture in vitro produced a more homogeneous collection of spindle-shaped cells at passage 3 (as shown in Fig. 1A, right) that were uniformly CD45− (Fig. 1B, right). To further characterize the phenotype of these fibroblast-like, CD45− cells, we stained for PH, an enzyme that cross-links collagen fibrils by hydroxylating proline residues on collagen and is expressed specifically in mesenchymal cells (11, 31). The monoclonal antibody used to detect this enzyme in AMCs has been reported not to stain lymphocytes, monocytes, dendritic cells, and granulocytes (35). All CD45− cells, at passage 3, stained positively for PH with varying degrees of intensity, whereas expression of α-SMA, a marker of myofibroblast differentiation, was expressed only in a minority of cells (Fig. 1C, right). This is consistent with the concept that mesenchymal cells/fibroblasts, particularly in injury states, may be phenotypically heterogeneous (52). Further characterization of AMCs demonstrated that these cells express vimentin and cellular fibronectin, typically associated with mesenchymal cells. There were no significant differences between ARDS-R and ARDS-NR in the levels of expression of these cellular proteins by Western blot analyses (Fig. 1, D and E). Collectively, these results confirm the mesenchymal cell phenotype of AMCs and the expected heterogeneity among these cells isolated from injured tissues, particularly with regard to α-SMA expression.
Flow cytometric analysis of AMCs in BAL during the evolution of ARDS
Cellular repair responses following lung injury are likely to be regulated and dynamic during the evolution of ARDS. To determine whether there were differences in the numbers of AMCs during the clinical course of ARDS, BAL was performed on day 1 and day 5 following the initial diagnosis of ARDS in two patients. The number of adherent fibroblast-like cells visualized within 24 h after initial plating was markedly increased on the second BAL (data not shown). To eliminate potential confounding variables/influences of in vitro cell culture/adhesion signaling, freshly isolated BAL cells were analyzed sequentially at the same time points (days 1 and 5 postdiagnosis) in another two ARDS patients by multiparameter flow cytometry. Figure 2 shows a representative analysis from one of these patients. Less than 1% (~0.2%) of all BAL cells isolated on day 1 of ARDS stained negatively (or weakly) for CD45, whereas 99.8% were CD45+ (Fig. 2, left middle). Similar results were obtained from flow cytometric analysis of BAL from two normal volunteers in which <1% of cells were CD45− (data not shown). Analysis of the BAL from the same ARDS patients 4 days later demonstrated a marked increase in the number of CD45− cells (13.7%, Fig. 2, right middle). Further analysis of these CD45− cells revealed a distinct subpopulation that expressed PH (12%; Fig. 2, right bottom). A subgroup of these CD45−/PH+ cells also expressed α-SMA (data not shown). Interestingly, a significant proportion (88%) of the CD45− population was found to be PH− by flow cytometric analysis (Fig. 2, right bottom), suggesting that these CD45−/PH− cells may represent a more “immature” mesenchymal progenitor population. Collectively, these data demonstrate dynamic changes in the number/phenotype of mesenchymal cells isolated from the alveolar space of ARDS patients during the evolution of acute lung injury.
Fig. 2.
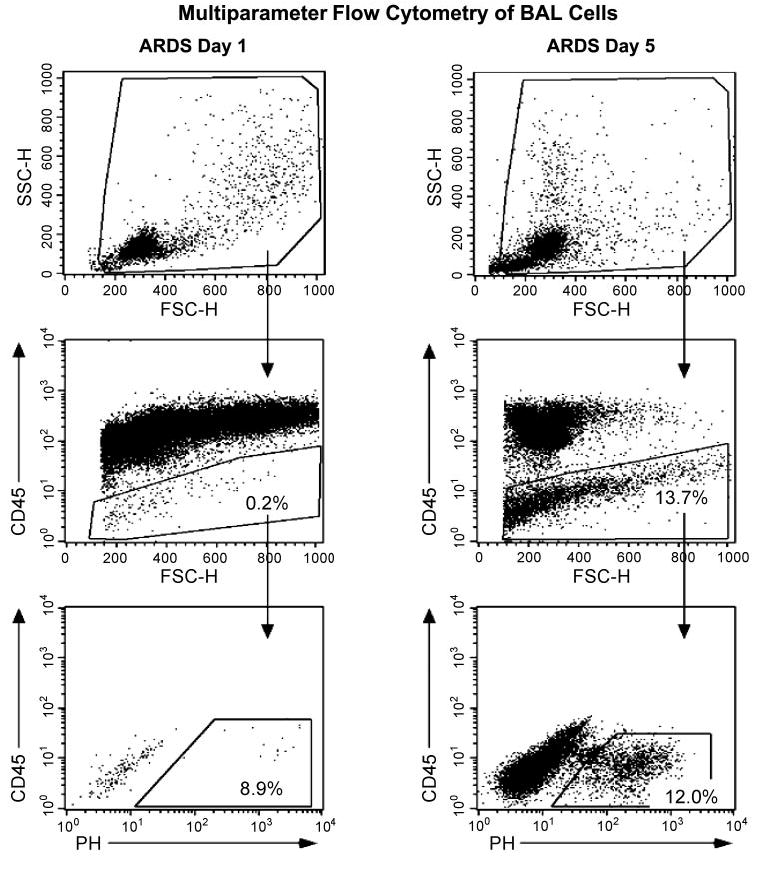
Serial morphological and flow cytometric analyses of freshly isolated BAL cells from patients during early and late ARDS. Freshly isolated BAL cells from 2 representative ARDS patients were analyzed immediately (without plating) by multiparameter flow cytometry with similar results. BAL cells were stained for expression of CD45 and PH on day 1 postdiagnosis (left), repeat bronchoscopy was performed, and BAL cells were reanalyzed on the same patient 4 days later (day 5, right). Top: forward (FSC) vs. side scatter (SSC) gates used to determine expression of CD45 and PH. Middle: CD45 expression vs. FSC. Essentially all BAL cells expressed the common leukocyte antigen, CD45, on day 1 (left). BAL cells from the same patient 4 days later contained a significant number of CD45− cells (right). Bottom: PH expression on CD45− gated BAL cells. Immunophenotyping of BAL cells are described in materials and methods.
Constitutive activation of PKB/Akt in AMCs isolated from patients with ARDS-NR
The phosphatidylinositol 3-kinase (PI3K)/PKB/Akt pathway has been linked with prosurvival/antiapoptotic signaling in a variety of cells (16). To determine whether there were differences in the constitutive activation of PKB/Akt in AMCs from patients with ARDS, we examined the expression of activated (phosphorylated) PKB/Akt by Western immunoblotting on all AMCs cell lines established from patients with both ARDS-R and ARDS-NR. Constitutive PKB/Akt phosphorylation varied among different AMC cell lines, and cells isolated from ARDS-NR appeared to express higher levels of phosphorylated PKB/Akt (Fig. 3A). When the activational state of PKB/Akt was quantitated (by densitometric analysis), constitutive activation of PKB/Akt was found to be significantly increased in ARDS-NR vs. ARDS-R (P = 0.003, Fig. 3B). This suggests that, in patients with persistent/nonresolving ARDS, alveolar microenvironmental factor(s) and/or intrinsic genetic factors may influence the phenotype of AMCs by inducing stable, enhanced activation of PKB/Akt signaling.
Fig. 3.
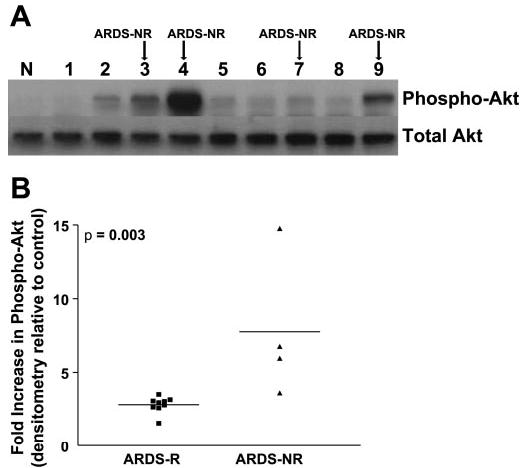
Constitutive activation of the antiapoptotic protein kinase, PKB/Akt, in AMCs from patients with ARDS-NR. A: AMCs isolated from 13 patients with ARDS were grown to 80% confluence in media containing 10% FBS. Whole cell lysates were collected and analyzed with SDS-polyacrylamide gel electrophoresis and immunoblotting with an antibody against phosphorylated (Ser473) PKB/Akt. The blot was then stripped and probed for total Akt. Representative samples from the first 9 patients studied are shown. Lysates from untreated normal human lung fibroblasts that were cultured under identical conditions were used for normalization (N: normal). B: densitometric analysis was done of the Western immunoblots from all 13 ARDS patients. The density of each band was measured and compared with that from untreated normal (control) human lung fibroblasts. Fold increases over control for each patient with ARDS-R and ARDS-NR are shown. P = 0.003 for ARDS-R vs. ARDS-NR.
When patients were classified based on a more relaxed definition of ARDS-NR (≥7 days of ARDS, ARDSNet LaSRS definition), a statistically significant difference in the measured end point of constitutive Akt activation was maintained; however, the level of significance was greater with the ≥10 days vs. ≥7 days definition (P = 0.003 vs. P = 0.05, respectively). Additionally, we found significant correlations between constitutive Akt phosphorylation vs. number of ARDS days (r2 = 0.30, P = 0.05) and between constitutive Akt phosphorylation vs. number of mechanical ventilation (MV) days (r2 = 0.31, P = 0.05). In contrast, the day of ARDS on which BAL was performed did not correlate with levels of constitutive Akt activation (r2 = 0.10, P = 0.29). Positive correlations between constitutive/activated Akt in AMCs (as a biological end point) and clinically relevant end points (number of ARDS days and MV days) suggest that a higher activational state of Akt in AMCs may be associated with nonresolving/persistent ARDS.
Exogenous TGF-β1 induces PKB/Akt activation in AMCs isolated from patients with ARDS-R
We have previously shown that the profibrotic cytokine, TGF-β1, induces the activation of this pathway in human lung fibroblasts/mes-enchymal cells and protects these cells from serum deprivation-induced apoptosis (32). Recent studies have demonstrated higher levels of active TGF-β1 in the BAL fluid of patients with persistent ARDS (10). To determine whether exogenous TGF-β1 is capable of inducing PKB/Akt activation in AMCs, cells from both ARDS-R and ARDS-NR were exposed to TGF-β1 (2 ng/ml) for 16 h before Western immunoblotting for phospho-Akt. TGF-β1 induced PKB/Akt phosphorylation in AMCs from both ARDS-R and ARDS-NR; however, this effect appears to be greater in ARDS-R (Fig. 4, A and B). The magnitude of Akt phosphorylation induced by TGF-β1 in AMCs of ARDS-R approaches the high constitutive (basal) levels seen in ARDS-NR (Fig. 4B). Inducibility of PKB/Akt phosphorylation by TGF-β1 was significantly greater in ARDS-R vs. ARDS-NR (7.17 ± 2.08-fold vs. 1.62 ± 0.23-fold, respectively; P = 0.035) (Fig. 4C). These results suggest that TGF-β1 may represent an important candidate soluble growth factor that influences PKB/Akt activation in mesenchymal cells present within the alveolar milieu of ARDS patients.
Fig. 4.
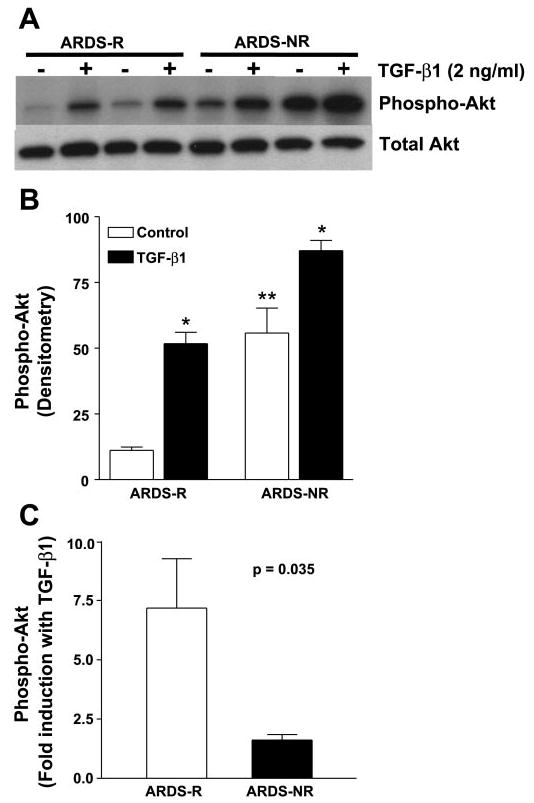
Exogenous transforming growth factor (TGF)-α1 markedly upregulates PKB/Akt activation in AMCs from patients with ARDS-R. A: AMCs isolated from patients with ARDS-R (n = 5) and ARDS-NR (n = 3) were isolated and grown to 80% confluence as described in materials and methods. Cells were then growth-arrested in media containing 0.01% FBS for 24 h and treated with or without TGF-β1 (2 ng/ml) for 16 h. Whole cell lysates were subjected to Western immunoblotting with a phospho-specific antibody to phosphorylated (Ser473) PKB/Akt. The blot was then stripped and probed for total Akt. A representative blot is shown. B: band densities from the Western blots in A were measured using NIH imaging software, and the background density was subtracted. The band densities for the induction of phospho-Akt by TGF-β1 in ARDS-R and ARDS-NR are shown. Statistical analysis was done by a 1-way ANOVA with Bonferroni posttest. *P < 0.001 for TGF-β1 induction compared with the respective baseline/constitutive levels of phosphorylated (active) Akt. **P < 0.001 for baseline/constitutive level of phosphorylated (active) Akt in ARDS-NR vs. ARDS-NR. C: for each set of AMCs from ARDS-R and ARDS-NR, band densities of TGF-β1-induced Akt phos-phorylation were divided by the band densities of the respective untreated/basal values to determine the fold induction with TGF-β1. P = 0.035 for ARDS-R vs. ARDS-NR.
Activation of the prosurvival PI3K/Akt pathway is associated with downstream antiapoptotic signaling and an antiapoptotic phenotype of mesenchymal cells in ARDS-NR
PKB/Akt has been shown to exert its antiapoptotic effects through several mechanisms. Among these downstream actions include the phosphorylation of forkhead family transcription factors leading to their sequestration in the cytoplasm and inhibition of their proapoptotic signaling (18) and the regulation of BCL-2 family proteins (3). We found that phosphorylation of FKHR was significantly increased in ARDS-NR compared with ARDS-R (P = 0.01; Fig. 5, A and B). Moreover, the phosphorylation of PKB/Akt was strongly correlated with FKHR phosphorylation (r2 = 0.88, Fig. 5C). We also examined the relative expression of the antiapoptotic BCL-2 protein and the proapoptotic BAX protein in AMCs from ARDS-R vs. ARDS-NR (Fig. 5D); the BCL-2/BAX ratio was significantly higher in ARDS-NR vs. ARDS-R (Fig. 5E, P = 0.01 by ANOVA with Bonferroni), supporting an antiapoptotic phenotype in mesenchymal cells isolated from ARDS-NR. To determine whether the activation of the PI3K-PKB/Akt and its potential downstream antiapoptotic pathways confer apoptotic resistance to AMCs, we compared the susceptibility to apoptosis under conditions of serum deprivation in all four different AMC lines of ARDS-NR to four AMC lines from ARDS-R. After 6 days of serum deprivation, AMCs from ARDS-R patients demonstrated a more than twofold increase in apoptosis compared with ARDS-NR (apoptosis index of 4.79 ± 0.48 vs. 2.28 ± 0.25; P = 0.028) (Fig. 6).
Fig. 5.
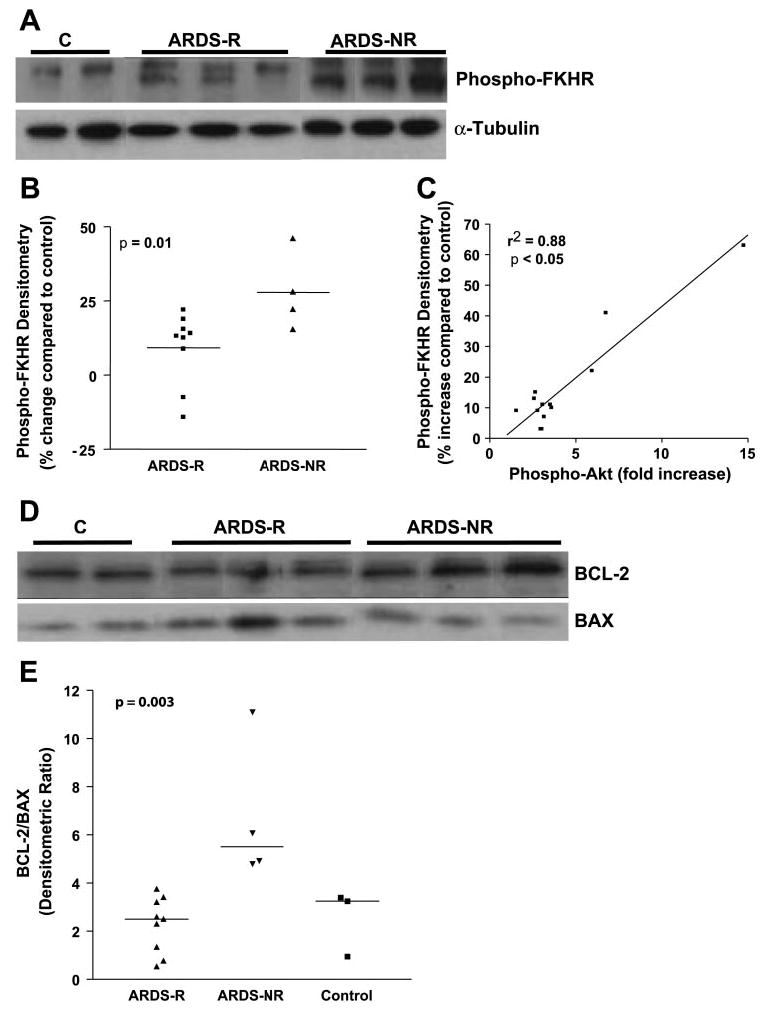
Activation of PKB/Akt-dependent antiapoptotic signaling pathways in AMCs from patients with ARDS-NR. A: mesenchymal cells were isolated from 13 patients with ARDS and cultured as described in materials and methods. Whole cell lysates were obtained and analyzed by Western immunoblotting for phosphorylation (S256) of the forkhead family transcription factor FKHR. The blot was then stripped and probed for α-tubulin. Representative samples from normal, unstimulated fibroblasts and AMCs from patients with ARDS-R and ARDS-NR are shown. B: densitometric analysis of the Western blots described in A on all 13 AMC cell lines were compared with normal controls (on the same blot). The percent change in density over controls was plotted for each of the patients with ARDS-R and ARDS-NR. P = 0.01 for ARDS-R vs. ARDS-NR. C: relative fold-increase in phospho-Akt for each AMC cell line (from Fig. 3B) was plotted against the change in phospho-FKHR (from Fig. 4A) and assessed for correlation by regression analysis (r2 = 0.88; P < 0.05). D: AMCs from ARDS-R and ARDS-NR patients and control fibroblasts were analyzed by Western immunoblotting for BCL-2 and then stripped and probed for BAX. A representative blot is shown. E: densitometric ratios for BCL-2/BAX were determined for ARDS-R fibroblasts (n = 9), for ARDS-NR (n = 4), and for normal control fibroblasts (n = 3). P < 0.01 for ARDS-NR vs. ARDS-R using ANOVA by Bonferroni’s posttest analysis.
Fig. 6.
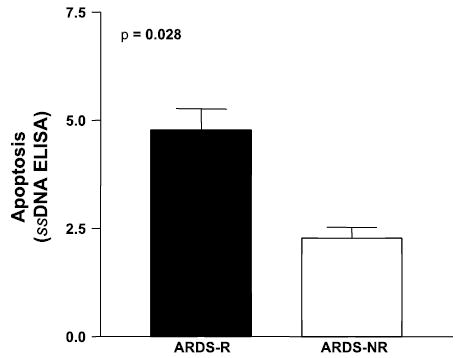
Susceptibility to serum deprivation-induced apoptosis in AMCs from ARDS-R vs. ARDS-NR. AMCs isolated from all 4 patients with ARDS-NR and 4 patients with ARDS-R were plated at an equal density in a 96-well plate. Cells were cultured in DMEM with 10% FBS for 4 days, and the medium was changed to DMEM with 0.1% FBS. Cells were cultured under serum-deprivation conditions for 6 days, and apoptosis was assessed with an ELISA for single-stranded (ss) DNA as described in materials and methods. P = 0.028 by nonparametric Mann-Whitney test; n = 4 for each AMC cell line. The experiment was repeated 3 times with similar results.
To determine whether there were differences in proliferative capacity between AMCs derived from ARDS-R vs. ARDS-NR, we first growth-arrested cells for 24 h and assessed proliferative rates in the presence or absence of 10% FBS by BrdU incorporation assay. Although some trends in higher proliferative capacity were observed in ARDS-NR, they were not statistically significant (data not shown). Furthermore, expression levels of cyclin D1 and phospho-ERK1/2 were also not significantly different between these groups (data not shown). Together, these results demonstrate that the constitutive activation of PKB/Akt in AMCs from patients with ARDS-NR is associated with downstream prosurvival signaling (FKHR and BCL-2 proteins) and resistance to apoptosis; this suggests that apoptotic resistance of mesenchymal cells may contribute to the dysregulated repair/fibrosis of persistent ARDS.
DISCUSSION
ARDS is a clinical syndrome caused by a variety of direct and indirect injurious insults and carries a mortality rate approximating 30% (51, 55). ARDS is associated pathologically with diffuse alveolar damage and manifests physiologically as acute respiratory failure with severe impairments in gas exchange and reduced lung compliance (50, 55). Diffuse alveolar damage leads to an early inflammatory phase and a more subtle, but overlapping, fibroproliferative response. Resolution of ARDS requires the regulated apoptotic clearance from the alveolar space of both inflammatory and mesenchymal cells involved in the reparative response to lung injury. ARDS-NR is marked by persistent abnormalities in gas exchange and lung compliance due to the development of fibrosing alveolitis (2, 4, 55). However, our current understanding of the recruitment, proliferation, differentiation, or apoptosis of mesenchymal cells/fibroblasts in response to lung injury is limited. In this study, we describe the isolation and characterization of mesenchymal cells from the alveolar space of ARDS patients by fiber-optic bronchoscopy with BAL. Phenotypic characterization of these cells, which we term alveolar mesenchymal cells (AMCs), demonstrate stable differences in prosurvival signaling and apoptotic susceptibility in patients with ARDS-R vs. ARDS-NR.
This study demonstrates, for the first time, that mesenchymal cells can be safely and consistently isolated from the BAL of patients with ARDS. Mesenchymal cells isolated in this manner have a unique phenotype, with the absence of the leukocyte marker CD45, the presence of the mesenchymal cell markers PH, vimentin, and fibronectin, and variable expression of the myofibroblast marker α-SMA. As with most mesenchymal cells/fibroblasts isolated from injured tissues, there is significant heterogeneity among these cells, particularly with regard to α-SMA; however, they are uniformly CD45− and PH+. AMCs that are CD45−/PH+/α-SMA± proliferate in cell culture for multiple passages, a property that is not typically associated with differentiated CD45+ leukocyte populations. Although AMCs represent a distinct mesenchymal cell population within the alveolar spaces of ARDS patients, the precise origin(s) of these cells is not addressed by the current study.
Recent studies in murine models of lung injury have demonstrated that lung mesenchymal cells may be recruited from bone marrow-derived/circulating fibrocytes (43, 46) or potentially from another bone marrow-derived mesenchymal cell precursor (28). Alternatively, tissue-specific stem cells or progenitor cells may give rise to mesenchymal cells in the setting of an acute lung injury (34). Recent studies also support the possibility that mesenchymal cells may derive from local alveolar epithelial cells by a transdifferentiation process known as epithelial-mesenchymal transition (33, 56). Finally, AMCs may represent interstitial fibroblasts that have migrated across a damaged alveolar basement membrane (25). Although the precise source(s) of AMCs identified in our studies is unclear, stable changes in cellular signaling/phenotype indicate that these cells may provide valuable information regarding the cellular microenvironment and the regulation of the repair response within the alveolar spaces of the injured lung.
Our study clearly demonstrates stable alterations in prosurvival signaling in AMCs isolated from ARDS patients with different clinical courses. Although the number of patients studied is relatively small, we observed significant increases in constitutive PKB/Akt activation in mesenchymal cells isolated from patients with persistent/nonresolving vs. resolving ARDS. We have observed that AMCs maintain this signaling phenotype even after repeated subculturing ex vivo, suggesting stable alterations in gene regulation that likely relate to their in vivo derivation/microenvironment. This is consistent with previous reports of topographic differentiation and positional memory (even after multiple passages ex vivo) demonstrated by gene expression profiling of human fibroblasts derived from diverse tissue sites in vivo (12).
A limitation of our study is that the day of bronchoscopy/BAL was not uniform in all patients. BAL was performed within 3 days of ARDS onset in 8 out of the 13 patients in whom AMC signaling/phenotypes were studied and at a later time point in 5 patients (3/4 in nonresolving ARDS and 2/9 in resolving ARDS). Thus a disproportionate number of these late BALs were performed in ARDS-NR vs. ARDS-R (for clinical reasons or because patients were transferred from another medical facility). It is possible that the enhanced prosurvival/antiapoptotic phenotype of AMCs in ARDS-NR may reflect this late time point in recovery of AMCs. However, we found no correlation between levels of Akt phosphorylation/activation and the ARDS day on which BAL was performed, although positive correlations were noted with number of ARDS days and MV days. Indeed, alveolar microenvironmental factor(s) at later stages of the disease may induce, promote, or perpetuate a prosurvival/antiapoptotic AMC phenotype. However, the current study does not prove that such a cellular phenotype predicts worse clinical outcomes; additional studies of AMCs derived from both early and late ARDS in larger groups of patients are required to better define cause-effect relationships.
A universally accepted definition for ARDS-NR does not currently exist. In the National Institutes of Health ARDS Network LaSRS (late steroid rescue study), criterion for enrollment was ARDS ≥7 days from onset, although the rationale for selecting this time point is not well defined. In our current study, we found that classification of these groups based on a later time point (ARDS ≥10 days vs. 7 days) was of greater statistical significance in relation to the constitutive levels of phosphorylated/activated Akt in AMCs. Several studies support that a ≥10 days’ definition of ARDS-NR may indeed be more biologically and, perhaps clinically, relevant. In a pathologic study of 17 patients with ARDS, no patient who was mechanically ventilated for <10 days had “fibroproliferative diffuse alveolar damage” (37). Another more recent study defined “prolonged mechanical ventilation” as 14 days of ARDS, which, interestingly, correlated positively with higher levels of active TGF-β1 in BAL fluid of ARDS patients (10). It is likely that fibrogenic responses represent a continuum that begins early during the process of lung injury and repair (14, 15). Regulated early repair responses are likely to be appropriate or “adaptive,” whereas persistent or exaggerated mesenchymal cell activity in later stages of ARDS may be maladaptive. This concept is supported by our finding that higher activational states of AMCs correlate with continuous variables such as the number of ARDS days and MV days that may be more indicative of persistent ARDS in this cohort of patients. A prospective study utilizing the definition of nonresolving ARDS as ≥10 days (as supported by the current study) is required to further evaluate whether constitutive Akt activation in AMCs is predictive of persistent/nonresolving ARDS.
It must be emphasized that the current study does not establish a “causal link” between a prosurvival/antiapoptotic AMC phenotype and ARDS-NR. However, we believe that the emergence of an apoptosis-resistant phenotype of AMCs may represent one plausible mechanism for “nonresolution” of the repair phase of ARDS. Apoptosis is a well-defined physiological process for normal tissue remodeling events in embryonic development and in the decreased cellularity that accompanies resolution of cutaneous wound healing (19, 44). Furthermore, a previous study by our group demonstrated enhanced activation of Akt and focal adhesion kinase in fibrotic regions of the lung in a murine model of bleomycin-induced injury and fibrosis; systemic administration of a protein kinase inhibitor that targets these prosurvival protein kinases diminished the accumulation of myofibroblasts and attenuated fibrotic tissue responses (54). The precise relationships between activation of prosurvival pathways in AMCs, fibroproliferation, and persistent ARDS in humans warrants further investigation.
A number of soluble and insoluble factors are known to regulate the PI3K-PKB/Akt pathway (8, 16). Our previous studies demonstrated that TGF-β1 induces activation of the PI3K-PKB/Akt pathway in human lung fibroblasts by a p38 MAPK-dependent mechanism (32). The current study demonstrates that active TGF-β1, present in increased concentrations in BAL fluid of patients with persistent ARDS (10), is a strong inducer of PKB/Akt phosphorylation in mesenchymal cells that express low levels at baseline. This suggests that TGF-β1, which mediates proapoptotic effects on epithelial cells (26, 42), may be a critical factor that promotes the prosurvival/antiapoptotic phenotype of mesenchymal cells during the fibrotic repair process associated with persistent/nonresolving ARDS.
Activation of PKB/Akt regulates a number of downstream signaling events that promotes cell survival, including phosphorylation of FKHR and BCL-2 family proteins (16). Studies of BCL-2 family proteins have demonstrated that hyperoxia-induced epithelial cell death is associated with increased expression of BAX (9) and that interleukin-6-mediated protection from hypoxia-induced cell death is associated with increased BCL-2 expression (41). However, no studies have investigated the roles of PKB/Akt, FKHR, or the BCL-2 family proteins in mesenchymal cells derived from human ARDS. Our data show that FKHR phosphorylation correlates with PKB/Akt phosphorylation in AMCs from patients with ARDS, and that mesenchymal cells from patients with nonresolving ARDS have increased BCL-2/BAX ratios. Importantly, AMCs from patients with ARDS-NR demonstrate increased resistance to serum deprivation-induced apoptosis compared with AMCs isolated from patients with resolving ARDS. Together, these findings are consistent with downstream effects of enhanced PI3K-PKB/Akt signaling in mesenchymal cells and suggest that AMCs from patients with ARDS-NR may contribute to the dysregulated repair and fibrosis that characterizes persistent/nonresolving ARDS.
This study establishes, for the first time, that mesenchymal cells can be isolated from the alveolar spaces of patients with ARDS and that a prosurvival/antiapoptotic mesenchymal cell phenotype is associated with persistent/nonresolving ARDS. Alveolar microenvironmental factors, such as TGF-β1, other mitogens/growth factors, and hyperoxia, may contribute to PI3K-PKB/Akt activation and acquired apoptosis resistance in mesenchymal cells/fibroblasts of ARDS-NR. Further studies are required to determine the specific role(s) of AMCs and the mechanisms that regulate their cell fates in the evolution of lung injury and repair.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grants K08 HL-81059 (to J. C. Horowitz), SCCOR in Translational Research in Acute Lung Injury Grant P50 HL-74024, and R01 HL-67967 (to V. J. Thannickal).
References
- 1.American Thoracic Society/European Respiratory Society. International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 2.Anderson WR, Thielen K. Correlative study of adult respiratory distress syndrome by light, scanning, and transmission electron microscopy. Ultrastruct Pathol. 1992;16:615–628. doi: 10.3109/01913129209023751. [DOI] [PubMed] [Google Scholar]
- 3.Asnaghi L, Calastretti A, Bevilacqua A, D’Agnano I, Gatti G, Canti G, Delia D, Capaccioli S, Nicolin A. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene. 2004;23:5781–5791. doi: 10.1038/sj.onc.1207698. [DOI] [PubMed] [Google Scholar]
- 4.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3:35–56. [PubMed] [Google Scholar]
- 5.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, Legall JR, Morris A, Spragg R. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–824. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 6.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, Lamy M, LeGall JR, Morris A, Spragg R. Report of the American-European consensus conference on ARDS: definitions, mechanisms, relevant outcomes and clinical trial coordination. The Consensus Committee. Intensive Care Med. 1994;20:225–232. doi: 10.1007/BF01704707. [DOI] [PubMed] [Google Scholar]
- 7.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 8.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 9.Buckley S, Barsky L, Driscoll B, Weinberg K, Anderson KD, Warburton D. Apoptosis and DNA damage in type 2 alveolar epithelial cells cultured from hyperoxic rats. Am J Physiol Lung Cell Mol Physiol. 1998;274:L714–L720. doi: 10.1152/ajplung.1998.274.5.L714. [DOI] [PubMed] [Google Scholar]
- 10.Budinger GR, Chandel NS, Donnelly HK, Eisenbart J, Oberoi M, Jain M. Active transforming growth factor-beta1 activates the procollagen I promoter in patients with acute lung injury. Intensive Care Med. 2005;31:121–128. doi: 10.1007/s00134-004-2503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98:2396–2402. doi: 10.1182/blood.v98.8.2396. [DOI] [PubMed] [Google Scholar]
- 12.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B, Polunovsky V, White J, Blazar B, Nakhleh R, Jessurun J, Peterson M, Bitterman P. Mesenchymal cells isolated after acute lung injury manifest an enhanced proliferative phenotype. J Clin Invest. 1992;90:1778–1785. doi: 10.1172/JCI116052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med. 1997;156:840–845. doi: 10.1164/ajrccm.156.3.9701124. [DOI] [PubMed] [Google Scholar]
- 15.Clark JG, Milberg JA, Steinberg KP, Hudson LD. Type III procollagen peptide in the adult respiratory distress syndrome. Association of increased peptide levels in bronchoalveolar lavage fluid with increased risk for death. Ann Intern Med. 1995;122:17–23. doi: 10.7326/0003-4819-122-1-199501010-00003. [DOI] [PubMed] [Google Scholar]
- 16.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 17.Deheinzelin D, Jatene FB, Saldiva PH, Brentani RR. Upregulation of collagen messenger RNA expression occurs immediately after lung damage. Chest. 1997;112:1184–1188. doi: 10.1378/chest.112.5.1184. [DOI] [PubMed] [Google Scholar]
- 18.Del Peso L, Gonzalez VM, Hernandez R, Barr FG, Nunez G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene. 1999;18:7328–7333. doi: 10.1038/sj.onc.1203159. [DOI] [PubMed] [Google Scholar]
- 19.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 20.Dhainaut JF, Charpentier J, Chiche JD. Transforming growth factor-beta: a mediator of cell regulation in acute respiratory distress syndrome. Crit Care Med. 2003;31:S258–264. doi: 10.1097/01.CCM.0000057901.92381.75. [DOI] [PubMed] [Google Scholar]
- 21.Dubaybo BA, Rubeiz GJ, Fligiel SE. Dynamic changes in the functional characteristics of the interstitial fibroblast during lung repair. Exp Lung Res. 1992;18:461–477. doi: 10.3109/01902149209064340. [DOI] [PubMed] [Google Scholar]
- 22.Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28:499–503. doi: 10.1165/rcmb.2002-0092OC. [DOI] [PubMed] [Google Scholar]
- 23.Fireman E, Ben Efraim S, Messer G, Dabush S, Greif J, Topilsky M. Cell-free supernatants of sarcoid alveolar macrophages suppress proliferation of sarcoid alveolar fibroblasts. Clin Immunol Immunopathol. 1991;59:368–378. doi: 10.1016/0090-1229(91)90032-6. [DOI] [PubMed] [Google Scholar]
- 24.Fireman E, Shahar I, Shoval S, Messer G, Dvash S, Grief J. Morphological and biochemical properties of alveolar fibroblasts in interstitial lung diseases. Lung. 2001;179:105–117. doi: 10.1007/s004080000051. [DOI] [PubMed] [Google Scholar]
- 25.Fukuda Y, Ishizaki M, Masuda Y, Kimura G, Kawanami O, Masugi Y. The role of intraalveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol. 1987;126:171–182. [PMC free article] [PubMed] [Google Scholar]
- 26.Hagimoto N, Kuwano K, Inoshima I, Yoshimi M, Nakamura N, Fujita M, Maeyama T, Hara N. TGF-beta 1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J Immunol. 2002;168:6470–6478. doi: 10.4049/jimmunol.168.12.6470. [DOI] [PubMed] [Google Scholar]
- 27.Harbeck RJ. Immunophenotyping of bronchoalveolar lavage lymphocytes. Clin Diagn Lab Immunol. 1998;5:271–277. doi: 10.1128/cdli.5.3.271-277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hertz MI, Woodward ME, Gross CR, Swart M, Marcy TW, Bitterman PB. Safety of bronchoalveolar lavage in the critically ill, mechanically ventilated patient. Crit Care Med. 1991;19:1526–1532. doi: 10.1097/00003246-199112000-00015. [DOI] [PubMed] [Google Scholar]
- 30.Hill JD, Ratliff JL, Parrott JC, Lamy M, Fallat RJ, Koeniger E, Yaeger EM, Whitmer G. Pulmonary pathology in acute respiratory insufficiency: lung biopsy as a diagnostic tool. J Thorac Cardiovasc Surg. 1976;71:64–71. [PubMed] [Google Scholar]
- 31.Hirohata S, Yanagida T, Nagai T, Sawada T, Nakamura H, Yoshino S, Tomita T, Ochi T. Induction of fibroblast-like cells from CD34(+) progenitor cells of the bone marrow in rheumatoid arthritis. J Leukoc Biol. 2001;70:413–421. [PubMed] [Google Scholar]
- 32.Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT) Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 35.Konttinen YT, Nykanen P, Nordstrom D, Saari H, Sandelin J, Santavirta S, Kouri T. DNA synthesis in prolyl 4-hydroxylase positive fibroblasts in situ in synovial tissue. An autoradiography-immunoperoxidase double labeling study. J Rheumatol. 1989;16:339–345. [PubMed] [Google Scholar]
- 36.Larsen K, Tufvesson E, Malmstrom J, Morgelin M, Wildt M, Andersson A, Lindstrom A, Malmstrom A, Lofdahl CG, Marko-Varga G, Bjermer L, Westergren-Thorsson G. Presence of activated mobile fibroblasts in bronchoalveolar lavage from patients with mild asthma. Am J Respir Crit Care Med. 2004;170:1049–1056. doi: 10.1164/rccm.200404-507OC. [DOI] [PubMed] [Google Scholar]
- 37.Liebler JM, Qu Z, Buckner B, Powers MR, Rosenbaum JT. Fibroproliferation and mast cells in the acute respiratory distress syndrome. Thorax. 1998;53:823–829. doi: 10.1136/thx.53.10.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludwicka A, Ohba T, Trojanowska M, Yamakage A, Strange C, Smith EA, Leroy EC, Sutherland S, Silver RM. Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J Rheumatol. 1995;22:1876–1883. [PubMed] [Google Scholar]
- 39.Marshall RP, Bellingan G, Webb S, Puddicombe A, Goldsack N, McAnulty RJ, Laurent GJ. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med. 2000;162:1783–1788. doi: 10.1164/ajrccm.162.5.2001061. [DOI] [PubMed] [Google Scholar]
- 40.Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107:196–200. doi: 10.1378/chest.107.1.196. [DOI] [PubMed] [Google Scholar]
- 41.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 42.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 43.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol. 2005;166:675–684. doi: 10.1016/S0002-9440(10)62289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moulin V, Larochelle S, Langlois C, Thibault I, Lopez-Valle CA, Roy M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J Cell Physiol. 2004;198:350–358. doi: 10.1002/jcp.10415. [DOI] [PubMed] [Google Scholar]
- 45.Pache JC, Christakos PG, Gannon DE, Mitchell JJ, Low RB, Leslie KO. Myofibroblasts in diffuse alveolar damage of the lung. Mod Pathol. 1998;11:1064–1070. [PubMed] [Google Scholar]
- 46.Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med. 2004;141:460–470. doi: 10.7326/0003-4819-141-6-200409210-00012. [DOI] [PubMed] [Google Scholar]
- 48.Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polunovsky VA, Chen B, Henke C, Snover D, Wendt C, Ingbar DH, Bitterman PB. Role of mesenchymal cell death in lung remodeling after injury. J Clin Invest. 1993;92:388–397. doi: 10.1172/JCI116578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 51.Reynolds HY. Use of bronchoalveolar lavage in humans–past necessity and future imperative. Lung. 2000;178:271–293. doi: 10.1007/s004080000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitt-Graff A, Desmouliere A, Gabbiani G. Heterogeneity of myofibroblast phenotypic features: an example of fibroblastic cell plasticity. Virchows Arch. 1994;425:3–24. doi: 10.1007/BF00193944. [DOI] [PubMed] [Google Scholar]
- 53.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150:113–122. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- 54.Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, Standiford TJ, Thannickal VJ. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 56.Yao HW, Xie QM, Chen JQ, Deng YM, Tang HF. TGF-beta1 induces alveolar epithelial to mesenchymal transition in vitro. Life Sci. 2004;76:29–37. doi: 10.1016/j.lfs.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 57.Zapol WM, Trelstad RL, Coffey JW, Tsai I, Salvador RA. Pulmonary fibrosis in severe acute respiratory failure. Am Rev Respir Dis. 1979;119:547–554. doi: 10.1164/arrd.1979.119.4.547. [DOI] [PubMed] [Google Scholar]