

Abstract
The activation of the cyclin-depdndent kinase Cdk1 at the transition from interphase to mitosis induces important changes in microtubule dynamics. Cdk1 phosphorylates a number of microtubule- or tubulin-binding proteins but, hitherto, tubulin itself has not been detected as a Cdk1 substrate. Here we show that Cdk1 phosphorylates β-tubulin both in vitro and in vivo. Phosphorylation occurs on Ser172 of β-tubulin, a site that is well conserved in evolution. Using a phosphopeptide antibody, we find that a fraction of the cell tubulin is phosphorylated during mitosis, and this tubulin phosphorylation is inhibited by the Cdk1 inhibitor roscovitine. In mitotic cells, phosphorylated tubulin is excluded from microtubules, being present in the soluble tubulin fraction. Consistent with this distribution in cells, the incorporation of Cdk1-phosphorylated tubulin into growing microtubules is impaired in vitro. Additionally, EGFP-β3-tubulinS172D/E mutants that mimic phosphorylated tubulin are unable to incorporate into microtubules when expressed in cells. Modeling shows that the presence of a phosphoserine at position 172 may impair both GTP binding to β-tubulin and interactions between tubulin dimers. These data indicate that phosphorylation of tubulin by Cdk1 could be involved in the regulation of microtubule dynamics during mitosis.
INTRODUCTION
In eukaryotic cycling cells, interphase microtubules form a dynamic network that is essential for cell polarity and intracellular traffic. When cells enter into mitosis, the interphase microtubule network rearranges into a mitotic spindle that is responsible for faithful chromosome segregation between daughter cells. Microtubule dynamics and turnover increase strikingly as cells progress from interphase to mitosis, with a microtubule half-life of 5–10 min in interphase and of 60–90 s in mitosis (Wittmann et al., 2001).
Microtubule dynamics in cells rely in part on the intrinsic properties of the microtubule building block, the α-β-tubulin dimer, and its ability to bind and hydrolyze a GTP nucleotide (Mitchison and Kirschner, 1984). The tubulin dimer is subject to special posttranslational modifications such as glutamylation, tyrosination, and acetylation (MacRae, 1997; Westermann and Weber, 2003). There is little evidence for a role of these modifications in the regulation of microtubule dynamics. Tubulin can also be phosphorylated by several kinases (Westermann and Weber, 2003). However, tubulin phosphorylation has not been connected with the cell-cycle-dependent regulation of microtubule dynamics.
Microtubule dynamics are also regulated by a number of microtubule effectors, including microtubule-associated proteins (MAPs), molecular motors such as kinesins, the Ras-like GTPase Ran-GTP, microtubule plus end-directed proteins and tubulin-binding proteins (Andersen, 1999, 2000; Carazo-Salas et al., 2001; Cassimeris, 2002; Heald and Nogales, 2002; Kinoshita et al., 2002; Galjart and Perez, 2003). These microtubule- or tubulin-associated proteins are themselves under the control of a balance of protein phosphatases and kinases.
The cyclin-dependent kinase Cdk1 (or Cdc2), associated with its cognate partner cyclin B, is a key enzyme for entry in mitosis (Nigg, 2001) and is essential for spindle morphogenesis. Cdk1 up-regulates microtubule dynamics when added in cell-free Xenopus extracts (Verde et al., 1990, 1992). Cdk1 inactivation is necessary for proper anaphase spindle dynamics and for cytokinesis (Wheatley et al., 1997). Large amounts of Cdk1 induce the depolymerization of interphase microtubules when injected into mammalian cells (Lamb et al., 1990) and the destabilization of microtubule arrays when added on lysed mammalian cells (Lieuvin et al., 1994).
Among Cdk1 substrates are a number of microtubule effectors (Ubersax et al., 2003). MAP4 has been shown to be phosphorylated by Cdk1 in vivo (Ookata et al., 1997), and other nonneuronal MAPs like E-MAP115 or XMAP215/TOG exhibit consensus sequences for this kinase (Masson and Kreis, 1995; Vasquez et al., 1999; Charrasse et al., 2000). Furthermore, MAP4 and XMAP215/TOG interact with cyclin B and this interaction could target Cdk1 to microtubules (Ookata et al., 1995; Charrasse et al., 2000). Phosphorylation of MAPs either dissociates them from the microtubule lattice (Masson and Kreis, 1995; Drewes et al., 1998) or reduces their ability to stabilize microtubules (Ookata et al., 1995). Other substrates of Cdk1 are microtubule motors, such as the kinesin-related proteins Eg5, Kid, or MKLP1 (Blangy et al., 1995; Sawin and Mitchison, 1995; Ohsugi et al., 2003; Mishima et al., 2004). These microtubule motors are clearly implicated in different steps of mitosis. Phosphorylation by Cdk1 has been shown to regulate their localization in the mitotic spindle (Blangy et al., 1995; Ohsugi et al., 2003; Mishima et al., 2004). Likewise, Op18/stathmin, a protein that sequesters tubulin dimers and destabilizes microtubules during interphase, is phosphorylated on two serine residues by Cdk1 at the onset of mitosis (Cassimeris, 2002). Phosphorylation turns off the microtubule destabilizing activity of Op18/stathmin.
In the present study, we show that, in addition to regulating microtubule effectors, Cdk1 can directly phosphorylate β-tubulin in vitro and in mitotic cells, and that this phosphorylation impairs tubulin incorporation into microtubules. We suggest that the phosphorylation of tubulin by Cdk1 may represent a means by which mitotic cells regulate microtubule dynamics.
MATERIALS AND METHODS
Antibodies
Anti-phospho-peptide P172 polyclonal antibody (Ab) was made by Eurogentech (Seraing, Belgium). Two rabbits were immunized with phosphopeptide Ac-VVPpSPKVSDTVVEC-CONH2, and serum was affinity-purified against phosphopeptide and then depleted against the same unphosphorylated peptide. Anti-α-tubulin AG monoclonal antibody (mAb) and anti-α-tubulin YL1/2 rat mAb were gifts of Laurence Lafanechère (CEA, Grenoble, France). Anti-β-tubulin TUB2.1 mAb was from Sigma (La Verpilliére, France), anti-GFP polyclonal Ab (A11122) from Molecular Probes, and anti-phosphorylated vimentin 4A4 mAb from MBL International (Woburn, MA). Cy3-conjugated anti-rabbit (Jackson ImmunoResearch Laboratories, West Grove, PA) and Alexa 488-conjugated anti-mouse (Molecular Probes) antibodies were used as secondary antibodies for immunofluorescence studies. For Western blots, peroxidase-conjugated goat anti-rabbit IgG and anti-mouse IgG secondary antibodies (Sigma) were used.
Cell Culture and Transfection
HeLa S3 cells were grown at 37°C in suspension cultures in MEM Eagle Joklik's formulation medium (Cambrex Bioscience, Walkersville, MD), supplemented with 10% horse serum (Invitrogen, Cergy, France) and 1% penicillin/streptomycin (Invitrogen). Adherent HeLa cells were grown in RPMI medium complemented with 10% fetal calf serum and 1% penicillin/streptomycin on 100-mm plastic dishes for cell extract preparation or on glass coverslips in 30-mm plastic dishes for transfection and immunofluorescence. Transient transfection was carried out with 1 μg of plasmid DNA using Lipofectamine plus reagent (Invitrogen) according to manufacturer's instructions. Cells were maintained in the presence of DNA for 30 h. For analysis of GFP constructs by Western blot, transfected cells were scrapped in Laemmli buffer. For immunofluorescence, cells were then either lysed in OPT buffer (80 mM Pipes, pH 6.7, 1 mM EGTA, 1 mM MgCl2, 0.5% Triton X-100, 10% glycerol) at 30°C for 2 min before fixation, or fixed directly for 10 min in methanol at –20°C. Cells were then treated for immunofluorescence and visualized either on a Zeiss Axioscop conventional microscope (Le Pecq, France), or on a Leica TCS-SP2 confocal microscope (Wetzlar, Germany).
Native and Recombinant Tubulin Purification
HeLa cell tubulin and bovine brain tubulin were purified as described (Caudron et al., 2000; Fourest-Lieuvin, 2005). Recombinant mouse α2- and β5-tubulins were produced from pET3a plasmids provided by Ronald Melki (LEBS, Gif-sur-Yvette, France). As adapted from Melki et al. (1997), recombinant tubulins were overexpressed in milligram quantities in Escherichia coli BL21(DE3), isolated as inclusion bodies from lysed cells using Bugbuster protein extraction reagent (Novagen, Darmstadt, Germany), dissolved in 20 mM Tris, pH 7.5, 10 mM dithiothreitol, 7.5 M urea, dialyzed against 20 mM Tris, pH 7.5, 0.1 mM dithiothreitol, and stored at –80°C until use.
Mutagenesis
Point mutations were performed on mouse β5-tubulin-pET3a (same as above) and EGFP-mouse β3-tubulin constructs, using the QuickChange kit from Stratagene (La Jolla, CA). The EGFP-mouse β3-tubulin construct was a generous gift from Jürgen Wehland and coworkers (Gesellschaft für Biotechnologische Forschung, Braunschweig, Germany).
Phosphorylation Reactions
HeLa cell tubulin (20 pmol), bovine brain tubulin (20 pmol), recombinant α- or β-tubulin (10 pmol), or maltose-binding protein-casein kinase 2 (CK2) fusion protein (10 pmol, a gift from Claude Cochet, Grenoble, France) were incubated for 15–30 min at 30°C with 5 μCi of γ32P-ATP (Perkin-Elmer Life Sciences, Wellesley, MA; reference BLU502A), 100 μM ATP, 5 mM MgCl2, and 40 U (2 μL) of human Cdk1-cyclin B (New England Biolabs, Beverly, MA) in 20 μL of MEM buffer (100 mM Mes, pH 6.7, 1 mM EGTA, 1 mM MgCl2, 0.1 mM dithiothreitol). Controls were performed with 2 μl of kinase storage buffer instead of kinase. Phosphorylation reactions were stopped with Laemmli buffer supplemented with 10 mM EDTA, and samples were processed for SDS-PAGE and autoradiography or analysis with a phosphorimager (Molecular Imager FX; Bio-Rad, Hercules, CA). Relative amounts of incorporated radioactivity were quantified using Quantity One software (Bio-Rad). For phosphorylation reactions on peptides, peptides mapping the human β1-tubulin sequence, RRMNTFSVVPSPKVSDTVVEP (pep172) and RRMNTFSVVPpSPKVSDTVVEP (pep172-P), were synthesized by Eurogentec. Note that two Arg residues were added at the N-terminus of each peptide to allow their binding onto P81 phosphocellulose (see below). Each peptide (150 μM) was phosphorylated in same conditions as above. Reactions were stopped and amounts of radioactivity incorporated in peptides were quantified by peptide binding onto P81 phosphocellulose paper as described (Filhol et al., 1991).
Assembly Assays after Tubulin Phosphorylation
To allow proper tubulin assembly, we used a source of γ32P-ATP without any additives (Perkin-Elmer Cetus; reference BLU/NEG/002A). Bovine brain tubulin (35 μM) was phosphorylated for 20 min at 30°C with 20 μCi γ32P-ATP, 100 μM ATP, 5 mM MgCl2, 10 mM β-glycerophosphate, 0.5 μM microcystine, 200 U (10 μL) Cdk1-cyclin B in 100 μL of PEM buffer (100 mM Pipes, pH 6.7, 1 mM EGTA, 1 mM MgCl2). Controls were performed with 2 μl of kinase storage buffer instead of kinase. Afterward, samples were incubated for 10 min on ice, to depolymerize any tubulin structure that could have assembled during the phosphorylation reaction. GTP (1 mM) was added and each mixture was allowed to polymerize for 30 min at 35°C. For experiments with taxol, taxol was added progressively to a final concentration of 35 μM during the polymerization step. Samples were then layered on PEM-60% (vol/vol) glycerol cushions (with 10 mM β-glycerophosphate and 0.5 μM microcystine) at 35°C and ultracentrifuged at 200,000 × g for 20 min at 35°C. Supernatants (100 μL), containing nonpolymerized tubulin, were removed from the top of the glycerol cushions and kept for SDS-PAGE. Cushions were discarded, and pellets were washed once with PEM buffer at 35°C before being incubated for 15 min at 4°C in 100 μL of PEM buffer with 10 mM β-glycerophosphate and 0.5 μM microcystine (and supplemented with 5 mM CaCl2 and 50 mM KCl for experiments with taxol) to depolymerize microtubules. Pellets were then cleared by ultracentrifugation at 200,000 × g for 10 min at 4°C and were processed for SDS-PAGE.
Tubulin Extraction from Synchronized Cells
Drugs used were purchased from Sigma, and stock solutions were in dimethyl sulfoxide (DMSO). HeLa S3 or HCT116 cells were synchronized to G1/S phase or to M phase by a 19-h treatment with 5 μg/ml aphidicolin or 0.3 μM nocodazole, respectively. At this concentration of nocodazole, cells were blocked in mitosis with no net microtubule disassembly (Jordan et al., 1992). Synchronization was controlled by flow cytometry analysis. For nocodazole-release experiments, mitotic cells were washed twice with phosphate-buffered saline (PBS) and released into normal medium for 30 or 60 min, until most of them had reached metaphase (Sauer et al., 2005). For experiments with roscovitine and nocodazole, cells were treated for 21 h with 50 μM roscovitine or with DMSO alone and then treated for 3 h with 0.3 μM nocodazole, as adapted from Meijer et al. (1997). To perform tubulin extraction, 108 cells were washed in PBS and lysed with 1 ml of cold OPT buffer supplemented with 10 mM NaF, 10 mM β-glycerophosphate, 0.5 μM microcystine, 1 μM pepstatin, and 400 μM phenylmethylsulfonyl fluoride (PMSF). After a 15-min incubation on ice with shearing through a pipette tip, lysed cells were ultracentrifuged at 200,000 × g for 10 min at 4°C, and supernatant (i.e., cell extract) was stored at –80°C. To perform soluble and insoluble tubulin fractions, the protocol was as above except that cells were extracted with 1 ml of OPT at 37°C for 2 min (soluble fraction) and then washed two times with 40 ml of OPT buffer at 37°C, before being incubated 15 min on ice with 1 ml of cold OPT buffer (insoluble fraction). Eighteen milligrams of cell extract proteins were incubated for 1 h at 4°C with 300 μL YL1/2-coupled Sepharose 4B previously washed with PEM supplemented with 10 mM NaF and β-glycerophosphate. Bound tubulin was eluted with peptide VGVDSVEGEGEEEGEEY at 0.1 mg/ml in same buffer, and fractions were analyzed by SDS-PAGE and Western blotting using P172 Ab. For analysis with anti-phosphorylated vimentin 4A4 mAb, 106 treated cells were washed once with PBS, lysed in 100 μl of 1% SDS, and sonicated before SDS-PAGE and Western blotting.
Incubation of Blotted Recombinant Tubulins with Mitotic Cell Extracts
Equal amounts of wild-type (WT) and S172A recombinant β-tubulins were processed for SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were saturated overnight at 37°C in PBS plus 5% Tween-20 and 2% gelatin and were washed twice with kinase buffer (see below) before incubation with extracts. Mitotic cell extracts with kinase activity were performed as adapted from Matsumoto-Taniura et al. (1996): Nocodazole-arrested cells were washed twice with PBS and were then homogenized in a Dounce grinder with two volumes of kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM dithiothreitol, 0.01% Brij 35, 0.5 μM microcystine, 1 μM pepstatin, and 400 μM PMSF). Extracts were ultracentrifuged at 250,000 × g for 15 min at 4°C and were supplemented with 1 mM ATP and 5 mM MgCl2 just before incubation for 30 min at 37°C with tubulin-blotted and -saturated membranes. Membranes were then washed 20 min in PBS-0.5 M NaCl, 20 min in stripping buffer (0.75% glycine, 0.1% Nodinet-P40, 1% SDS, pH 2.2) and washed three times in PBS-0.1% Tween. All washings were performed in the presence of 10 mM NaF and 10 mM β-glycerophosphate. Membranes were finally immunoblotted with P172 Ab.
Evaluation of the Proportion of Phosphorylated Tubulin in Mitotic Cell Extracts
Purified bovine brain tubulin was phosphorylated in vitro with Cdk1 in the presence of γ32P-ATP as described in Phosphorylation Reactions (see above). The stoichiometry of transferred ATP per mole of β-tubulin was calculated after SDS-PAGE using a phosphorimager and Quantity One software (BioRad). In parallel, aliquots from the same phosphorylation reaction were blotted together with tubulin from mitotic cell extracts. The blot was stained with Ponceau red and then incubated with the P172 Ab. The quantity of cell extract tubulin was evaluated from the Ponceau red staining by comparison with the known amounts of bovine brain tubulin, using Quantity One software. Likewise, the P172 signal was quantified. With the in vitro-phosphorylated tubulin, it was possible to link the ratio [P172 staining/amount of tubulin] to a stoichiometry of phosphorylation. Therefore, it was possible to calculate the approximate stoichiometry of phosphorylation in mitotic cells from the ratio [P172 staining/amount of tubulin] for mitotic cell extract tubulin.
RESULTS
In Vitro Phosphorylation of β-Tubulin by Cdk1
We have previously shown that Cdk1 induces the destabilization of microtubule networks when added on lysed mammalian cells in culture (Lieuvin et al., 1994). Subsequently, we have designed a method for the isolation, in biochemical quantities, of proteins associated with HeLa cell microtubules (Fourest-Lieuvin, 2005). In a survey of Cdk1 substrates in these cell extracts, we identified a band comigrating with tubulin (unpublished data).
To test directly whether Cdk1 could phosphorylate tubulin, we incubated purified HeLa cell tubulin with or without the human Cdk1-cyclin B1 complex (referred below as Cdk1), in the presence of γ32P-ATP. Results showed a phosphorylation of β-tubulin (Figure 1A). In the same manner, bovine brain β-tubulin was phosphorylated by Cdk1 (see for example, Figure 6A). An additional phosphorylated peptide was observed above β-tubulin (Figure 1A), not comigrating with α-tubulin but apparently corresponding to cyclin B1, which is a known substrate for Cdk1 (Labbé et al., 1989). To test directly whether β-tubulin, and not α-tubulin, was a Cdk1 substrate, we incubated purified recombinant β- and α-tubulins with Cdk1. As a positive control, we included a recombinant form of protein kinase CK2, which is a well-established substrate for Cdk1 in vitro and in vivo (Mulner-Lorillon et al., 1990; Litchfield et al., 1992; Bosc et al., 1995). Both CK2 and β-tubulin were phosphorylated by Cdk1 at similar levels, whereas α-tubulin was not phosphorylated (Figure 1B, right panel). These results indicate that β-tubulin, not α-tubulin, is a bona fide Cdk1 substrate in vitro.
Figure 1.
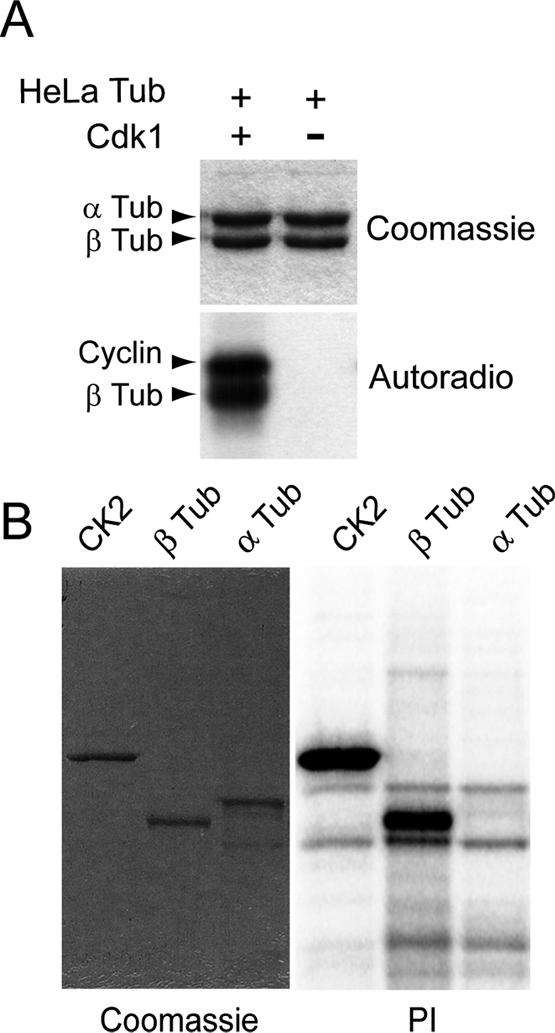
Cdk1 phosphorylates native and recombinant β-tubulin in vitro. (A) Purified HeLa cell tubulin was incubated in the presence of γ32P-ATP either with (+) or without (–) Cdk1-cyclin B. Samples were then processed for SDS-PAGE (top) and analyzed by autoradiography (bottom). (B) Maltose-binding protein-CK2 fusion protein, recombinant β-tubulin, and recombinant α-tubulin were incubated with Cdk1-cyclin B in the presence of γ32P-ATP. Samples were processed for SDS-PAGE (left) and analyzed with a phosphorimager (PI, right).
Figure 6.

Tubulin phosphorylated in vitro by Cdk1 incorporates very poorly into microtubules. (A) Bovine brain tubulin was incubated in the presence of γ32P-ATP with (lanes 3 and 4) or without (lanes 1 and 2) Cdk1-cyclin B. Afterward, tubulin was allowed to polymerize and centrifuged to separate microtubule pellet (P) from unpolymerized tubulin (S). Equal volumes of each sample were processed for SDS-PAGE and were either Coomassie stained and analyzed with a phosphorimager (PI) or blotted with P172 Ab (bottom). Ponceau red staining of the blot is shown. (B) Same experiment as in A, except that the polymerization step was done in the presence of taxol. (C) Semiquantification of the proportions of tubulin (as visualized by Ponceau red staining) and of phospho-tubulin (visualized by P172 Ab staining) in supernatants (S) and pellets (P) of experiments done with or without taxol during the polymerization step. Results are given as percentages either of total tubulin (S+P, top panel) or of total phospho-tubulin (S+P, bottom panel).
Determination of the Cdk1 Phosphorylation Site in β-Tubulin Sequence
Analysis of β-tubulin sequences of diverse origins revealed the presence of two putative sites for phosphorylation by Cdk1. One site, S172PK, was a good candidate for phosphorylation by Cdk1 (Holmes and Solomon, 1996). This site is conserved among β-tubulin genes of different species, from the yeast Saccharomyces cerevisiae to humans (Figure 2A). It is also conserved between human β-tubulin isoforms. The other putative site, T219P, is very short and less conserved (Figure 2A).
Figure 2.

Cdk1 phosphorylates β-tubulin on the serine 172 residue. (A) The sequence of human β1-tubulin (accession number PO7436 in NCBI protein database) from residues 161–190 and 204–233 was aligned with β-tubulin sequences of other organisms, using the software ClustalW (Infobiogen, Evry, France). Conserved residues are shown on a gray background, S172PK and T219P sites are in bold, and T5 loop is underlined. (B) Each of two synthetic peptides, matching human β1-tubulin sequence from residue 164 to residue 182, were incubated with Cdk1-cyclin B and γ32P-ATP. One peptide, pep172-P, was synthesized with a phosphate on Ser172. As a control, a mixture of the two peptides was incubated with γ32P-ATP but without Cdk1. After incubation, incorporated radioactivity in each peptide was counted. (C) Wild-type (WT) or point-mutated recombinant β-tubulins were incubated in the presence of γ32P-ATP with (lanes 5–8) or without (lanes 1–4) Cdk1-cyclin B. Mutants tested were as follows: Ser172 to Ala (S172A) β-tubulin (lanes 2 and 6), Thr219 to Ala (T219A) β-tubulin (lanes 3 and 7), and S172A/T219A double-mutated β-tubulin (lanes 4 and 8). After incubation, samples were processed for SDS-PAGE (top) and analyzed with a phosphorimager (bottom).
To test whether the S172PK site could indeed be phosphorylated by Cdk1, we tested two synthetic peptides containing this site, pep172 and pep172-P. These peptides matched the human β1-tubulin sequence and were indistinguishable, except that a phosphate was incorporated at the Ser 172 residue of pep172-P. When incubated with Cdk1 and γ32P-ATP, pep172 incorporated large amounts of radioactivity, whereas pep172-P did not (Figure 2B). This experiment indicates that the S172PK site, and no other amino-acid in pep172, can be phosphorylated by Cdk1.
To test whether the S172PK site was the preferred phosphorylation site in β-tubulin, we performed point mutations in recombinant β-tubulin. S172A and T219A point mutants and a S172A/T219A double mutant were assayed for phosphorylation by Cdk1 along with WT β-tubulin (Figure 2C). The S172A point mutation strongly inhibited the phosphorylation of β-tubulin by Cdk1 (Figure 2C, lane 6), whereas the T219A point mutation did not (Figure 2C, lane 7). Phosphorylation was also inhibited in the case of the double mutant (Figure 2C, lane 8). These results indicate that the S172PK site is the main Cdk1 phosphorylation site in recombinant β-tubulin.
Anti-Phospho-Peptide P172 Antibody
To study β-tubulin phosphorylation in cells, an anti-phospho-peptide polyclonal Ab directed against the phosphorylated S172PK site was raised (P172 Ab; see Materials and Methods). We first tested whether the P172 Ab was specific for phosphorylated tubulin, compared with unphosphorylated tubulin, using both recombinant β-tubulin and bovine brain tubulin. Western blot analysis showed that P172 Ab reacted with β-tubulin phosphorylated by Cdk1 (Figure 3A, lanes 1 and 3), not with unphosphorylated tubulin (Figure 3A, lanes 2 and 4). To test whether the P172 Ab was specific of the phosphorylated Ser172 residue on β-tubulin, we tested the antibody on recombinant β-tubulin point mutants incubated or not with Cdk1. Results showed that only mutants with phosphorylated Ser172 residue reacted with the antibody (Figure 3B, lanes 5 and 7). We conclude that the P172 Ab reacts specifically with phosphorylated Ser 172, when tubulin is exposed to active Cdk1.
Figure 3.

The anti-phospho-peptide P172 Ab is specific of β-tubulin phosphorylated by Cdk1 in vitro. (A) Recombinant β-tubulin (lanes 1 and 2) and native bovine brain tubulin (lanes 3 and 4) were incubated in the presence of nonradioactive ATP, either with (lanes 1 and 3) or without (lanes 2 and 4) Cdk1-cyclin B. Samples were then processed for Western blotting with P172 Ab (bottom). Ponceau red staining is shown. (B) Wild-type (WT) or point-mutant recombinant β-tubulins were incubated in the presence of ATP with (lanes 5–8) or without (lanes 1–4) Cdk1-cyclin B. β-tubulin mutants tested were as in Figure 2. After incubation, samples were processed for Western blotting with P172 Ab.
Phosphorylation on Ser 172 of β-Tubulin in Mitotic Cells
To examine the β-tubulin phosphorylation status in interphase or mitotic cells, we used cell extracts from aphidicolin- or nocodazole-arrested HeLa S3 cells, respectively. The phospho-specific P172 Ab was blotted on affinity-purified tubulin from these cell extracts. Tubulin from nocodazole-arrested cells was distinctly labeled with P172 Ab, whereas tubulin extracted from cells arrested in G1/S using aphidicolin yielded a barely detectable signal (Figure 4A, bottom panel). The low concentration of nocodazole used in these experiments has been shown to block microtubule dynamics (Jordan et al., 1992). To test whether anomalies in spindle dynamics could interfere with tubulin phosphorylation, nocodazole-arrested cells were released into fresh medium for 30 and 60 min to allow recovery of spindle functionality before extraction (Sauer et al., 2005). The extent of tubulin phosphorylation was comparable in nocodazole-released cell extracts as in nocodazole-arrested cell extracts (Figure 4A). Similar results were obtained for adherent HeLa or HCT116 cells (unpublished data). When cells were pretreated with roscovitine, an inhibitor of Cdk1 (Meijer et al., 1997), before exposure to nocodazole, the P172 Ab signal was strongly diminished on immunoblots (Figure 4B, middle panel). These results were consistent with β-tubulin being phosphorylated by Cdk1 in mitotic cells, but did not preclude the possibility that β-tubulin was phosphorylated by another kinase at a site cross-reacting with the P172 Ab. To test this possibility, equal amounts of WT and S172A purified recombinant β-tubulins were transferred onto nitrocellulose membranes, which were incubated with mitotic cell extracts at 37°C in the presence of ATP and MgCl2 to activate mitotic kinases. Membranes were then washed and immunoblotted with P172 Ab (Figure 4C). WT β-tubulin reacted strongly with P172 Ab after phosphorylation by mitotic extracts, whereas S172A β-tubulin did not. This showed that P172 Ab was specific of the phosphorylated Ser172 in β-tubulin, even when tubulin was exposed to a panel of mitotic kinases. Taken together, all these experiments demonstrate that β-tubulin is phosphorylated on Ser172 in mitotic cells, presumably by Cdk1 itself.
Figure 4.
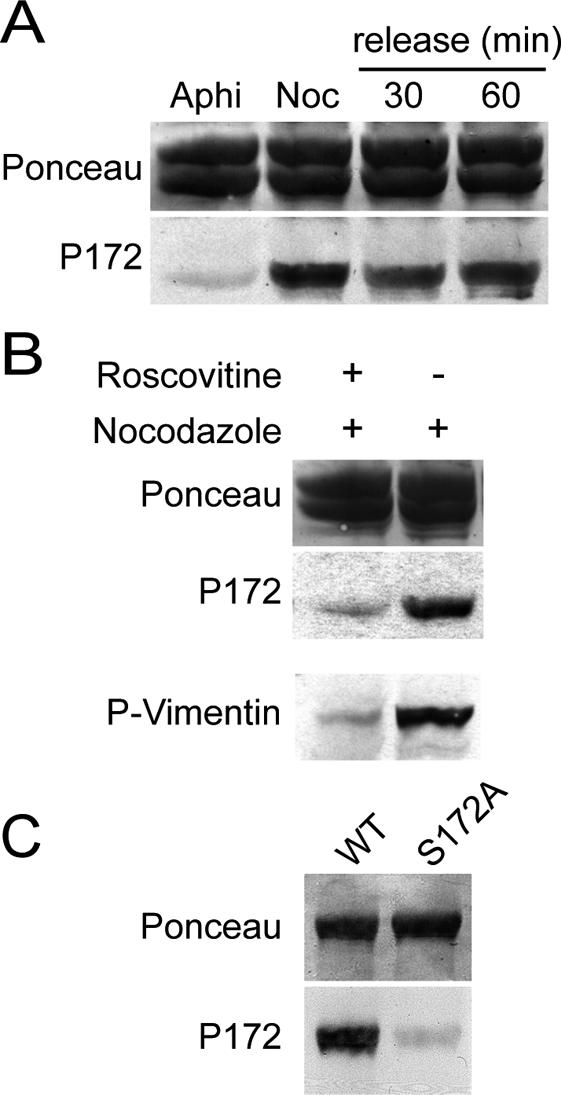
β-tubulin is phosphorylated on Ser172 in M-phase cell extracts. (A) HeLa S3 cells were arrested in G1/S using aphidicolin (Aphi), or in M-phase using nocodazole (Noc), or released for 30 or 60 min from the nocodazole block. Tubulin was isolated from these cells and processed for Western blotting with P172 Ab (bottom panel). Ponceau red staining of the blot is shown (top panel). (B) HeLa S3 cells were treated or not with roscovitine before nocodazole exposure. Tubulin was isolated from these cells and processed for Western blotting with P172 Ab (middle panel). Ponceau red staining of the blot is shown (top panel). As a control, total extracts from the same cells were blotted with 4A4 mAb, which specifically recognizes a Cdk1-phosphorylated vimentin site (bottom panel; Meijer et al., 1997). (C) Equal quantities of WT and S172A recombinant β-tubulins were transferred onto nitrocellulose after SDS-PAGE. A Ponceau staining was performed to control transfer (top panel). The nitrocellulose membrane was incubated at 37°C with mitotic cell extracts supplemented with ATP and MgCl2. The membrane was then thoroughly washed before immunoblotting with P172 Ab (bottom panel).
We then evaluated the proportion of phosphorylated tubulin versus total tubulin in mitotic cell extracts (see Materials and Methods). This evaluation indicated that phosphorylated tubulin represented ∼0.2–0.5% of the total tubulin in M-phase extracts.
Localization of Phosphorylated Tubulin in Cells
We used both immunofluorescence microscopy and cell fractionation to localize phosphorylated β-tubulin. In immunofluorescence studies, adherent HeLa cells were either labeled with P172 Ab alone (unpublished data) or sequentially double-labeled with P172 Ab and anti-β-tubulin TUB2.1 mAb (Figure 5, A and B). The P172 Ab stained mitotic cells, from metaphase to telophase, but not interphase cells (Figure 5A). Microtubule structures such as spindles or midbodies in mitotic cells were not labeled, as assessed by confocal microscopy analysis (Figure 5B). The P172 labeling was the same with single stain, in every cell type tested (Rat 2, MDCK, and HCT116 cells), whatever the method of fixation used. The P172 Ab may recognize other phospho-proteins than tubulin in whole mitotic cells, but, conservatively, our results demonstrate an absence of detectable incorporation of phosphorylated tubulin in microtubules in cells.
Figure 5.

P172 Ab stains mitotic cells, but phosphorylated tubulin is not detected in the microtubules of mitotic cells. (A) Adherent HeLa cells were double-stained with P172 Ab (left) and with anti-β-tubulin TUB 2.1 mAb (middle). DNA was stained with Hoechst 33258 (right). Pictures were taken on a conventional fluorescence microscope. (B) Adherent HeLa cells were labeled as in A and analyzed on a confocal microscope. Stacked images were merged (right). Bars, 20 μm. (C) HeLa S3 cells were arrested in M-phase with nocodazole, and the free tubulin fraction and the polymerized tubulin fraction (corresponding to microtubules in cells) were extracted. These two fractions were analyzed by Western blotting with P172 Ab (bottom). Ponceau red staining of the blot is shown (top). (D) Semiquantification of the phospho-tubulin in the free and polymerized tubulin fractions. For each tubulin fraction, quantities of phospho-tubulin (P172 staining relative to Ponceau staining) were evaluated and results are given as percentages of total phospho-tubulin (free + polymerized).
To substantiate this conclusion, we analyzed the soluble (corresponding to the free tubulin) and the insoluble (corresponding to microtubules) tubulin fractions from nocodazole-arrested HeLa S3 cells, by Western blotting with P172 Ab. Results showed that free tubulin was much more labeled by P172 Ab than polymerized tubulin (Figure 5C). A semiquantification of the P172 signal showed that ∼77% of the phospho-tubulin was in the free tubulin pool (Figure 5D).
Taken together, these results indicate that the bulk of phosphorylated tubulin remains unpolymerized in mitotic cells.
Assembly Properties of Phosphorylated Tubulin
To test the effect of phosphorylation on tubulin polymerization directly, we performed in vitro experiments. Bovine brain tubulin was phosphorylated or not with Cdk1 in the presence of γ32P-ATP and then allowed to polymerize (see Materials and Methods). After polymerization, samples were centrifuged to separate microtubule pellets from unpolymerized tubulin. Supernatants and pellets were analyzed both with a phosphorimager (PI) and by Western blotting with P172 Ab (Figure 6A). Results showed that the bulk (58%) of total tubulin was in pellets (Figure 6A, lane 4 of Coomassie and Ponceau panels, and Figure 6C, top panel, –taxol histograms). In contrast, the majority (81%) of phospho-tubulin was present in the supernatant and thus was unpolymerized (Figure 6A, lane 3 of PI and P172 panels, and Figure 6C, bottom panel, –taxol histograms).
The poor incorporation of phosphorylated tubulin in microtubules could reflect either an impairment of tubulin polymerization by phosphorylation or the presence of a large proportion of denatured tubulin among phospho-tubulin molecules. To test these possibilities, taxol was added in tubulin samples during the polymerization reaction, to increase total tubulin polymerization. Taxol addition induced an increase in the quantity of phosphorylated tubulin in the microtubule pellet (Figure 6B, P172 panel), which amounted to ∼63% of total phospho-tubulin (Figure 6C, bottom panel, + taxol histograms). These experiments indicate that the bulk of phospho-tubulin is not denatured but has an impaired polymerization capacity.
Behavior of EGFP-β-TubulinS172A and EGFP-β-TubulinS172D/E Mutants in Cells
To test whether, in cells as in vitro, phosphorylation of tubulin on Ser172 is sufficient to impair tubulin polymerization, we examined the incorporation, in cell microtubules, of various EGFP-tubulin mutants. These mutants mimicked either unphosphorylated tubulin (S172A mutation) or phosphorylated tubulin (S172E or S172D mutations). The WT and mutant constructs were transfected into adherent HeLa cells. Each of the constructs was expressed full-length as assessed in a control experiment (Supplementary Data, Figure S1A). After transfection, cells were either fixed directly (Figure 7A) or lysed before fixation to improve the visualization of microtubules (Figure 7B). Whole cells and lysed cells were either stained only with anti-GFP Ab (unpublished data), or double-stained with anti-GFP Ab and with anti-α-tubulin mAb (Figure 7).
Figure 7.

Transfected EGFP-β-tubulinS172D remains soluble, whereas EGFP-β-tubulinWT and EGFP-β-tubulinS172A colocalize with microtubules. Adherent HeLa cells were transfected with EGFP-β-tubulinWT, EGFP-β-tubulinS172A, or EGFP-β-tubulinS172D. Cells were either fixed directly (A) or lysed before fixation (B). Whole cells and lysed cells were double-stained with GFP Ab and α-tubulin AG mAb. In the case of EGFP-β-tubulinS172D (B, right panel), the GFP staining had been overexposed to visualize lysed transfected cells. The data presented here are representative of an analysis of 100 transfected cells from three independent experiments. Bars, 20 μm.
When cells were not lysed before fixation, a GFP staining was observed in the cytoplasm of transfected cells, for every construct tested. However, it was very difficult to distinguish microtubules against the fluorescent background, probably because HeLa cells are not very flat (Figure 7A). When cells were lysed before fixation, we observed a colocalization of GFP staining with the α-tubulin staining in the case of cells transfected with EGFP-β-tubulinWT (Figure 7B, left panels). Concerning EGFP-β-tubulinS172A, a colocalization of the GFP staining with microtubules, although less extensive than in the case of the WT protein, was also evident (Figure 7B, middle panels). In contrast, when cells were transfected with either EGFP-β-tubulinS172E (unpublished data) or EGFP-β-tubulinS172D (Figure 7B, right panels), no GFP staining was observed on microtubules after cell lysis. Hence, EGFP-β-tubulinS172E and EGFP-β-tubulinS172D proteins, which mimic phosphorylated tubulin, appeared unable to incorporate into microtubules. In control experiments, EGFP-β-tubulinS172D protein coimmunoprecipitated with α-tubulin, indicating that mutant β-tubulin was present as tubulin dimers in cells (Supplementary Data, Figure S1B). Taken together, these data are consistent with an impairment of the polymerization capacity of tubulin by phosphate addition on Ser172.
Localization of the Cdk1 Phosphorylation Site in the Tubulin 3-D Structure
In an attempt to explain why phosphorylation of β-tubulin by Cdk1 may inhibit polymerization, we looked at the position of Ser172 in the 3-D structure of the tubulin. This serine residue lies in the T5 loop of β-tubulin, which is near the ribose of the GDP or GTP nucleotide (Figure 8A). Additionally, this loop is at the interface between two tubulin dimers within a protofilament (Lowe et al., 2001). We also modeled a phosphoserine at position 172 in the tubulin structure using the graphics package TURBO-FRODO (Roussel and Cambillau, 1991), to examine the possible incidence of a phosphate group on the binding of the GDP/GTP nucleotide. As shown in our model (Figure 8B), the presence of a bulky negatively charged phosphate group could induce modifications in the nucleotide-binding area that may impair GDP/GTP binding and turnover, thereby impairing tubulin assembly.
Figure 8.

Ser172 is located near the nucleotide in β-tubulin, and phosphorylated Ser172 might interfere with GTP/GDP binding and turnover. (A) Localization of the Ser172 residue in the 3-D structure of the tubulin heterodimer (accession number PDB#1JFF, Lowe et al., 2001). Note that Ser172 is numbered Ser174 in PDB#1JFF. The serine residue is in red, the GDP nucleotide in β-tubulin is in green, and the GTP nucleotide in α-tubulin is in yellow. (B) Modeling of a phosphoserine (P-Ser) at the position 172, using the graphics package TURBO-FRODO. Some of the possible interferences between the phosphate group and the ribose of GDP are depicted with blue dotted lines.
DISCUSSION
In this study, we find that Cdk1 can phosphorylate β-tubulin. We provide evidence that tubulin phosphorylation by Cdk1 occurs in mitotic mammalian cells and impairs tubulin incorporation into microtubules both in cells and in vitro. It is known that protein phosphatases and kinases affect the overall dynamic properties of cellular microtubules during interphase as well as mitosis (Verde et al., 1990; Lieuvin et al., 1994; Yoshida et al., 2003; Schaar et al., 2004; Trinczek et al., 2004). However, the literature stipulates that kinases and phosphatases act through the phosphorylation/dephosphorylation of effector proteins associated with microtubules or with tubulin. In this context, our findings are new, showing a direct interplay between a kinase and tubulin, not mediated by an associated effector.
We find that phosphorylation by Cdk1 mainly occurs on the Ser172 residue of β-tubulin. In the 3-D structure of β-tubulin, the Ser172 residue is located in the T5 loop, which binds the ribose of GDP or GTP, at the exchangeable nucleotide binding site of tubulin (Hesse et al., 1987; Lowe et al., 2001). As shown in our model (Figure 8B), addition of a phosphate group to Ser172 can interfere with nucleotide binding. Nucleotide binding and exchange are important for tubulin assembly, and interference with nucleotide affinity or binding kinetics could logically be involved in the observed inhibition of tubulin assembly capacity by Ser172 phosphorylation. Additionally, residues in the T5 loop are involved in contacts between dimers along protofilaments (Lowe et al., 2001) and modification of these contacts in phosphorylated tubulin might also perturb tubulin assembly. A comprehensive study of the mechanism(s) through which phosphorylation affects tubulin assembly will require biochemical amounts of isolated phosphorylated tubulin.
Our finding that phosphorylation inhibits tubulin polymerization is consistent with previous observations of microtubule disassembly in cells injected with large amounts of Cdk1 or in lysed cells incubated with Cdk1 (Lamb et al., 1990; Lieuvin et al., 1994). However, we find that <1% of total tubulin is phosphorylated in mitotic cells, and this raises questions concerning the mechanisms through which such a low stoichiometry of tubulin phosphorylation could influence microtubule dynamics.
One possibility is that phosphorylated tubulin interacts with microtubule plus ends in cells. In a mitotic cell, considering that polymerized tubulin represents ∼50% of total tubulin, and based on an average microtubule length of 5 μm and on a tubulin dimer size of 8 nm (Caudron et al., 2002), the ratio of the number of microtubule plus ends to the number of microtubule dimers is ∼1/16,000. For a stoichiometry of phosphorylated tubulin/total tubulin of 0.2% (20 phosphorylated dimers vs. 10,000 dimers), there is a 30-fold excess of phosphorylated tubulin dimers compared with microtubule plus ends. Additionally, Cdk1 may be specifically targeted to microtubule plus ends. For instance, in the yeast S. cerevisiae, the Cdk1–mitotic cyclin Clb4 complex has been found to be localized to the plus ends of astral microtubules by the protein Kar9, which is related to the mammalian tumor suppressor adenomatous polyposis coli (APC; Maekawa et al., 2003; Maekawa and Schiebel, 2004). In Δclb4 yeast cells, astral microtubule dynamics are altered, showing a moderate increase of growth and shrinkage rates (Maekawa and Schiebel, 2004). Maybe Cdk1 localization mechanisms exist in higher eukaryotic cells. Indeed, in mammalian cells, a subpopulation of Cdk1 is associated with spindles (Bailly et al., 1989; Riabowol et al., 1989; Rattner et al., 1990; Andreassen and Margolis, 1994). The Cdc14 phosphatase also associates with spindles and is essential for microtubule bundling and stabilization during anaphase and exit from mitosis (Cho et al., 2005; Higuchi and Uhlmann, 2005). Therefore, it would be of great interest to examine whether phosphorylated tubulin is a substrate for Cdc14.
Phosphorylated tubulin at microtubule ends could conceivably affect microtubule dynamics. In previous studies, Verde et al. (1990) reported that Cdk1 had no effect on pure tubulin polymerization on isolated centrosomes. A requirement for APC or for other factors capable of targeting Cdk1 at microtubule plus ends may account for this negative result.
Because the S172PK site and the T5 loop are both very well conserved in evolution and are present in the yeast S. cerevisiae (Figure 2A), yeast genetics should provide powerful tools to investigate the effects of tubulin Ser172 phosphorylation on the cell cycle and on microtubule dynamics.
Supplementary Material
Acknowledgments
We thank Bernard Eddé and Mathilde Louwagie for their help in preliminary experiments and Ronald Melki, Claude Cochet, and Jürgen Wehland for providing plasmids and purified proteins. We are most grateful to Richard H. Wade for helpful discussions and to Bernard Eddé and Robert L. Margolis for critical reading of this manuscript. I.G.-S. was supported by the European Union Quality of Life Integrated Project SPINE (Structural Proteomics in Europe; contract no. QLG2-CT-2002-00988), and L.P. by the Fondation pour la Recherche Médicale. This work was supported by a grant from La Ligue Nationale Contre le Cancer to D.J.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–07–0621) on December 21, 2005.
Abbreviations used: Cdk1, cyclin-dependent kinase 1; CK2, casein kinase 2; EGFP, enhanced green fluorescent protein; MAP, microtubule-associated protein; PI, phosphorimager; WT, wild-type.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Andersen, S. S. (1999). Balanced regulation of microtubule dynamics during the cell cycle: a contemporary view. Bioessays 21, 53–60. [DOI] [PubMed] [Google Scholar]
- Andersen, S. S. (2000). Spindle assembly and the art of regulating microtubule dynamics by MAPs and Stathmin/Op18. Trends Cell Biol. 10, 261–267. [DOI] [PubMed] [Google Scholar]
- Andreassen, P. R., and Margolis, R. L. (1994). Microtubule dependency of p34cdc2 inactivation and mitotic exit in mammalian cells. J. Cell Biol. 127, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly, E., Doree, M., Nurse, P., and Bornens, M. (1989). p34cdc2 is located in both nucleus and cytoplasm; part is centrosomally associated at G2/M and enters vesicles at anaphase. EMBO J. 8, 3985–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy, A., Lane, H. A., d'Herin, P., Harper, M., Kress, M., and Nigg, E. A. (1995). Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83, 1159–1169. [DOI] [PubMed] [Google Scholar]
- Bosc, D. G., Slominski, E., Sichler, C., and Litchfield, D. W. (1995). Phosphorylation of casein kinase II by p34cdc2. Identification of phosphorylation sites using phosphorylation site mutants in vitro. J. Biol. Chem. 270, 25872–25878. [DOI] [PubMed] [Google Scholar]
- Carazo-Salas, R. E., Gruss, O. J., Mattaj, I. W., and Karsenti, E. (2001). Ran-GTP coordinates regulation of microtubule nucleation and dynamics during mitotic-spindle assembly. Nat. Cell Biol. 3, 228–234. [DOI] [PubMed] [Google Scholar]
- Cassimeris, L. (2002). The oncoprotein 18/stathmin family of microtubule destabilizers. Curr. Opin. Cell Biol. 14, 18–24. [DOI] [PubMed] [Google Scholar]
- Caudron, N., Arnal, I., Buhler, E., Job, D., and Valiron, O. (2002). Microtubule nucleation from stable tubulin oligomers. J. Biol. Chem. 277, 50973–50979. [DOI] [PubMed] [Google Scholar]
- Caudron, N., Valiron, O., Usson, Y., Valiron, P., and Job, D. (2000). A reassessment of the factors affecting microtubule assembly and disassembly in vitro. J. Mol. Biol. 297, 211–220. [DOI] [PubMed] [Google Scholar]
- Charrasse, S., Lorca, T., Doree, M., and Larroque, C. (2000). The Xenopus XMAP215 and its human homologue TOG proteins interact with cyclin B1 to target p34cdc2 to microtubules during mitosis. Exp. Cell Res. 254, 249–256. [DOI] [PubMed] [Google Scholar]
- Cho, H. P., Liu, Y., Gomez, M., Dunlap, J., Tyers, M., and Wang, Y. (2005). The dual-specificity phosphatase CDC14B bundles and stabilizes microtubules. Mol. Cell. Biol. 25, 4541–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes, G., Ebneth, A., and Mandelkow, E. M. (1998). MAPs, MARKs and microtubule dynamics. Trends Biochem. Sci. 23, 307–311. [DOI] [PubMed] [Google Scholar]
- Filhol, O., Cochet, C., Wedegaertner, P., Gill, G. N., and Chambaz, E. M. (1991). Coexpression of both α- and beta subunits is required for assembly of regulated casein kinase II. Biochemistry 30, 11133–11140. [DOI] [PubMed] [Google Scholar]
- Fourest-Lieuvin, A. (2005). Purification of tubulin from limited volumes of cultured cells. Protein Expr. Purif. 45, 183–190. [DOI] [PubMed] [Google Scholar]
- Galjart, N., and Perez, F. (2003). A plus-end raft to control microtubule dynamics and function. Curr. Opin. Cell Biol. 15, 48–53. [DOI] [PubMed] [Google Scholar]
- Heald, R., and Nogales, E. (2002). Microtubule dynamics. J. Cell Sci. 115, 3–4. [DOI] [PubMed] [Google Scholar]
- Hesse, J., Thierauf, M., and Ponstingl, H. (1987). Tubulin sequence region beta 155–174 is involved in binding exchangeable guanosine triphosphate. J. Biol. Chem. 262, 15472–15475. [PubMed] [Google Scholar]
- Higuchi, T., and Uhlmann, F. (2005). Stabilization of microtubule dynamics at anaphase onset promotes chromosome segregation. Nature 433, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, J. K., and Solomon, M. J. (1996). A predictive scale for evaluating cyclin-dependent kinase substrates. A comparison of p34cdc2 and p33cdk2. J. Biol. Chem. 271, 25240–25246. [DOI] [PubMed] [Google Scholar]
- Jordan, M. A., Thrower, D., and Wilson, L. (1992). Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 102(Pt 3), 401–416. [DOI] [PubMed] [Google Scholar]
- Kinoshita, K., Habermann, B., and Hyman, A. A. (2002). XMAP 215, a key component of the dynamic microtubule cytoskeleton. Trends Cell Biol. 12, 267–273. [DOI] [PubMed] [Google Scholar]
- Labbé, J. C., Capony, J. P., Caput, D., Cavadore, J. C., Derancourt, J., Kaghad, M., Lelias, J. M., Picard, A., and Doree, M. (1989). MPF from starfish oocytes at first meiotic metaphase is a heterodimer containing one molecule of cdc2 and one molecule of cyclin B. EMBO J. 8, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, N. J., Fernandez, A., Watrin, A., Labbe, J. C., and Cavadore, J. C. (1990). Microinjection of p34cdc2 kinase induces marked changes in cell shape, cytoskeletal organization, and chromatin structure in mammalian fibroblasts. Cell 60, 151–165. [DOI] [PubMed] [Google Scholar]
- Lieuvin, A., Labbé, J. C., Dorée, M., and Job, D. (1994). Intrinsic microtubule stability in interphase cells. J. Cell Biol. 124, 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litchfield, D. W., Luscher, B., Lozeman, F. J., Eisenman, R. N., and Krebs, E. G. (1992). Phosphorylation of casein kinase II by p34cdc2 in vitro and at mitosis. J. Biol. Chem. 267, 13943–13951. [PubMed] [Google Scholar]
- Lowe, J., Li, H., Downing, K. H., and Nogales, E. (2001). Refined structure of α β-tubulin at 3.5 A resolution. J. Mol. Biol. 313, 1045–1057. [DOI] [PubMed] [Google Scholar]
- MacRae, T. H. (1997). Tubulin post-translational modifications—enzymes and their mechanisms of action. Eur. J. Biochem. 244, 265–278. [DOI] [PubMed] [Google Scholar]
- Maekawa, H., and Schiebel, E. (2004). Cdk1-Clb4 controls the interaction of astral microtubule plus ends with subdomains of the daughter cell cortex. Genes Dev. 18, 1709–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa, H., Usui, T., Knop, M., and Schiebel, E. (2003). Yeast Cdk1 translocates to the plus end of cytoplasmic microtubules to regulate bud cortex interactions. EMBO J. 22, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson, D., and Kreis, T. E. (1995). Binding of E-MAP-115 to microtubules is regulated by cell cycle-dependent phosphorylation. J. Cell Biol. 131, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto-Taniura, N., Pirollet, F., Monroe, R., Gerace, L., and Westendorf, J. M. (1996). Identification of novel M phase phosphoproteins by expression cloning. Mol. Biol. Cell 7, 1455–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer, L., Borgne, A., Mulner, O., Chong, J. P., Blow, J. J., Inagaki, N., Inagaki, M., Delcros, J. G., and Moulinoux, J. P. (1997). Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 243, 527–536. [DOI] [PubMed] [Google Scholar]
- Melki, R., Batelier, G., Soulie, S., and Williams, R. C., Jr. (1997). Cytoplasmic chaperonin containing TCP-1, structural and functional characterization. Biochemistry 36, 5817–5826. [DOI] [PubMed] [Google Scholar]
- Mishima, M., Pavicic, V., Gruneberg, U., Nigg, E. A., and Glotzer, M. (2004). Cell cycle regulation of central spindle assembly. Nature 430, 908. [DOI] [PubMed] [Google Scholar]
- Mitchison, T., and Kirschner, M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. [DOI] [PubMed] [Google Scholar]
- Mulner-Lorillon, O., Cormier, P., Labbe, J. C., Doree, M., Poulhe, R., Osborne, H., and Belle, R. (1990). M-phase-specific cdc2 protein kinase phosphorylates the beta subunit of casein kinase II and increases casein kinase II activity. Eur. J. Biochem. 193, 529–534. [DOI] [PubMed] [Google Scholar]
- Nigg, E. A. (2001). Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell. Biol. 2, 21–32. [DOI] [PubMed] [Google Scholar]
- Ohsugi, M., Tokai-Nishizumi, N., Shiroguchi, K., Toyoshima, Y. Y., Inoue, J., and Yamamoto, T. (2003). Cdc2-mediated phosphorylation of Kid controls its distribution to spindle and chromosomes. EMBO J. 22, 2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookata, K., Hisanaga, S., Bulinski, J. C., Murofushi, H., Aizawa, H., Itoh, T. J., Hotani, H., Okumura, E., Tachibana, K., and Kishimoto, T. (1995). Cyclin B interaction with microtubule-associated protein 4 (MAP4) targets p34cdc2 kinase to microtubules and is a potential regulator of M-phase microtubule dynamics. J. Cell Biol. 128, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookata, K., Hisanaga, S., Sugita, M., Okuyama, A., Murofushi, H., Kitazawa, H., Chari, S., Bulinski, J. C., and Kishimoto, T. (1997). MAP4 is the in vivo substrate for CDC2 kinase in HeLa cells: identification of an M-phase specific and a cell cycle-independent phosphorylation site in MAP4. Biochemistry 36, 15873–15883. [DOI] [PubMed] [Google Scholar]
- Rattner, J. B., Lew, J., and Wang, J. H. (1990). p34cdc2 kinase is localized to distinct domains within the mitotic apparatus. Cell Motil. Cytoskelet. 17, 227–235. [DOI] [PubMed] [Google Scholar]
- Riabowol, K., Draetta, G., Brizuela, L., Vandre, D., and Beach, D. (1989). The cdc2 kinase is a nuclear protein that is essential for mitosis in mammalian cells. Cell 57, 393–401. [DOI] [PubMed] [Google Scholar]
- Roussel, A., and Cambillau, C. (1991). The TURBO-FRODO graphics package. In: Silicon Graphics Geometry Partners Directory, Mountain View, CA: Silicon Graphics, 86.
- Sauer, G., Korner, R., Hanisch, A., Ries, A., Nigg, E. A., and Sillje, H. H. (2005). Proteome analysis of the human mitotic spindle. Mol. Cell Proteomics 4, 35–43. [DOI] [PubMed] [Google Scholar]
- Sawin, K. E., and Mitchison, T. J. (1995). Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc. Natl. Acad. Sci. USA 92, 4289–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar, B. T., Kinoshita, K., and McConnell, S. K. (2004). Doublecortin microtubule affinity is regulated by a balance of kinase and phosphatase activity at the leading edge of migrating neurons. Neuron 41, 203–213. [DOI] [PubMed] [Google Scholar]
- Trinczek, B., Brajenovic, M., Ebneth, A., and Drewes, G. (2004). MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J. Biol. Chem. 279, 5915–5923. [DOI] [PubMed] [Google Scholar]
- Ubersax, J. A., Woodbury, E. L., Quang, P. N., Paraz, M., Blethrow, J. D., Shah, K., Shokat, K. M., and Morgan, D. O. (2003). Targets of the cyclin-dependent kinase Cdk1. Nature 425, 859–864. [DOI] [PubMed] [Google Scholar]
- Vasquez, R. J., Gard, D. L., and Cassimeris, L. (1999). Phosphorylation by CDK1 regulates XMAP215 function in vitro. Cell Motil. Cytoskelet. 43, 310–321. [DOI] [PubMed] [Google Scholar]
- Verde, F., Dogterom, M., Stelzer, E., Karsenti, E., and Leibler, S. (1992). Control of microtubule dynamics and length by cyclin A- and cyclin B-dependent kinases in Xenopus egg extracts. J. Cell Biol. 118, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde, F., Labbé, J. C., Dorée, M., and Karsenti, E. (1990). Regulation of microtubule dynamics by cdc2 protein kinase in cell-free extracts of Xenopus eggs. Nature 343, 233–238. [DOI] [PubMed] [Google Scholar]
- Westermann, S., and Weber, K. (2003). Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell. Biol. 4, 938–947. [DOI] [PubMed] [Google Scholar]
- Wheatley, S. P., Hinchcliffe, E. H., Glotzer, M., Hyman, A. A., Sluder, G., and Wang, Y. (1997). CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J. Cell Biol. 138, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, T., Hyman, A., and Desai, A. (2001). The spindle: a dynamic assembly of microtubules and motors. Nat. Cell Biol. 3, E28–E34. [DOI] [PubMed] [Google Scholar]
- Yoshida, N., Haga, K., and Haga, T. (2003). Identification of sites of phosphorylation by G-protein-coupled receptor kinase 2 in β-tubulin. Eur. J. Biochem. 270, 1154–1163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.