

Abstract
Speckles are nuclear bodies that contain pre-mRNA splicing factors and polyadenylated RNA. Because nuclear poly(A) RNA consists of both mRNA transcripts and nucleus-restricted RNAs, we tested whether poly(A) RNA in speckles is dynamic or rather an immobile, perhaps structural, component. Fluorescein-labeled oligo(dT) was introduced into HeLa cells stably expressing a red fluorescent protein chimera of the splicing factor SC35 and allowed to hybridize. Fluorescence correlation spectroscopy (FCS) showed that the mobility of the tagged poly(A) RNA was virtually identical in both speckles and at random nucleoplasmic sites. This same result was observed in photoactivation-tracking studies in which caged fluorescein-labeled oligo(dT) was used as hybridization probe, and the rate of movement away from either a speckle or nucleoplasmic site was monitored using digital imaging microscopy after photoactivation. Furthermore, the tagged poly(A) RNA was observed to rapidly distribute throughout the entire nucleoplasm and other speckles, regardless of whether the tracking observations were initiated in a speckle or the nucleoplasm. Finally, in both FCS and photoactivation-tracking studies, a temperature reduction from 37 to 22°C had no discernible effect on the behavior of poly(A) RNA in either speckles or the nucleoplasm, strongly suggesting that its movement in and out of speckles does not require metabolic energy.
INTRODUCTION
Nuclear speckles are morphologically distinct regions of the nucleoplasm that contain pre-mRNA splicing components as well as poly(A) RNA (Carter et al., 1993; Zhang et al., 1994; Lamond and Spector, 2003). They are operationally defined by their immunostaining with a variety of pre-mRNA splicing factor antibodies, and they also show in situ hybridization signal using probes for poly(A). When these sites are viewed in the electron microscope, most of them are found to represent interchromatin granule clusters (Fakan et al., 1984, Deerinck et al., 1994). However, RNA polymerase II transcription sites are distributed throughout the nucleus, indicating that although some of the RNA present in interchromatin granules/speckles is nascent, transcription is not restricted to this compartment, and much of the poly(A) RNA there has likely been transcribed previously and/or at distant sites (Fakan and Nobis, 1978; Wansink et al., 1993, Zeng et al., 1997, Neugebauer and Roth, 1997, Cmarko et al., 1999; Guillot et al., 2004). Indeed, although transcripts that are being rapidly produced or that contain many introns are sometimes observed in speckles at the light microscopy level (Johnson et al., 2000; Shopland et al., 2003), the majority of studies over the years has indicated that most speckles are not primary sites of pre-mRNA splicing (for reviews, see Mattaj, 1994; Huang and Spector, 1996a; Neugebauer and Roth, 1997; Lamond and Spector, 2003). In two cases, a single species of pre-mRNA has been followed from its transcription site to the nuclear pore, and in neither case did the RNA seem to accumulate in nuclear subcompartments (Singh et al., 1999; Shav-Tal et al., 2004). Instead, it seems that splicing components typically move away from speckles to sites of gene expression where splicing occurs cotranscriptionally (Huang and Spector, 1996b; Misteli et al., 1997, 1998; Eils et al., 2000). These findings have therefore made it difficult to understand why poly(A) RNA is associated with speckles.
One idea has been that perhaps there is a metabolically stable, nucleus-restricted poly(A) RNA population that helps to organize and structurally define certain nuclear bodies such as speckles (Fakan et al., 1984; Huang et al., 1994; Lamond and Spector, 2003). This idea stems from observations that as much as 30% of the poly(A) RNA never leaves the nucleus in growing mammalian cells (Perry et al., 1974, Herman et al., 1976). At least some of this poly(A) RNA consists of noncoding sequences that seem to have a variety of nuclear functions (Morey and Avner, 2004). We earlier studied the movement of poly(A) RNA in the nucleus of live rat myoblasts using the technique of fluorescence correlation spectroscopy (FCS) (Politz et al., 1998) and found that although there was a class of poly(A) RNA that moved rather rapidly throughout the nucleoplasm, there also was a sizable fraction that moved much more slowly. It was therefore possible that this fraction represented molecules that were confined within nuclear bodies such as speckles and thus constrained in their motion.
In the present investigation, we developed methods to address the possible structural nature of the poly(A) RNA directly in the nuclear speckles of living cells. A stable cell line expressing a chimeric SC35 protein coupled to a monomeric red fluorescent protein (mRFP-SC35) was generated, and the chimeric protein was found to behave similarly to the native SC35 protein (Politz et al., 2003). Endogenous nuclear poly(A) RNA was tagged in these cells using fluorescently labeled oligo(dT) as a hybridization probe (Pederson, 1999; Politz, 1999; Politz et al., 1999). We used both FCS (Magde et al., 1972; Politz et al., 1998; Wachsmuth et al., 2000) and photoactivation-tracking techniques (Politz, 1999; Politz et al., 1999, 2003, 2004) to measure the mobility of the tagged poly(A) RNA inside speckles and in the nucleoplasm. Time-lapse digital imaging microscopy was also used to visually track the movement of photoactivated tagged poly(A) RNA as it moved between speckles and the nucleoplasm. We show that poly(A) RNA moves in and out of speckles with the characteristics of a diffusive process and that there is not a substantial immobile population of poly(A) RNA within the speckle as might be expected if it were serving as a structural scaffolding. In fact, the behavior of poly(A) RNA within and outside speckles was indistinguishable, and the rate of movement did not change when the temperature was lowered from 37 to 22°C. The results indicate that rather than speckles harboring an actively sequestered, kinetically distinct population, the poly(A) RNA that is associated with a speckle at any given time exchanges freely with the nucleoplasm and other speckles.
MATERIALS AND METHODS
mRFP-SC35 Stable Cell Line and Transfection
PCR was used to generate a restriction site at the stop codon of a human SC35 cDNA (Prasanth et al., 2003) for cloning into a vector encoding the mRFP (Campbell et al., 2002). A HeLa stable cell line containing the mRFP-SC35 was generated and maintained in DMEM (low glucose) with 10% fetal bovine serum (FBS) and 0.5 mg/ml G418 (Invitrogen, Carlsbad, CA).
β-Tropomyosin Mini-Gene Transfection and In Situ Hybridization
Electroporation was performed on trypsinized cells (240 V; 950 μF), which were then resuspended in 250 μl of growth medium and incubated with 4 μg of a rat β-tropomyosin mini-gene construct (Helfman et al., 1988) plus 20 μg of sheared salmon sperm DNA. Cells were then seeded onto acid-washed coverslips and processed for in situ hybridization 24 h posttransfection. The β-tropmyosin probe was labeled with Spectrum-Green-dUTP using a nick translation reagent kit (Vysis, Downer's Grove, IL), and in situ hybridization was performed essentially as described previously (Huang and Spector, 1996b). Cells were hybridized with 100 ng of probe in 20 μl of hybridization mixture (50% formamide, 2× SSC, 5% dextran sulfate, and 20 μg of yeast tRNA) at 37°C for 12–14 h. After posthybridization washes, DNA was stained with 4′-6-diamidino-2-phenylindole. Imaging acquisitions for in situ experiments were performed with a Zeiss Axioplan microscope and a 100× oil immersion objective. Images were processed using Openlab software (Improvision, Lexington, MA).
Immunoblot Analysis
Cell lysates were prepared from the mRFP-SC35 stable cell line and treated for 30 min at 37°C with 500 U/ml calf intestinal phosphatase (CIP; New England Biolabs, Beverly, MA), and, after gel electrophoresis, were blotted onto a nitrocellulose membrane and probed by an antibody that recognizes mRFP (Chemicon International, Temecula, CA).
Oligodeoxynucleotide (Oligo) Synthesis
Oligo(dT) and control oligo(dA) probes were synthesized by Integrated DNA Technologies (Coralville, IA) as 43mers, with a thymidine containing a C6-aminohexyl group present at every 10 bases [in both oligo(dT) and oligo(dA)]. The oligos were then labeled with either fluorescein (Molecular Probes, Eugene, OR) or caged fluorescein (Mitchison et al., 1994) as described previously (Politz et al., 1999, 2004). Both the aminohexyl linker arms and the spacing of the labeling groups along the oligo are thought to prevent RNase H degradation of the RNA target after the oligo is hybridized (Ueno et al., 1997).
Oligo Uptake and Live Cell Imaging
Cells were either plated into two-well dishes (Nalge Nunc, Naperville, IL) or onto 25-mm coverslips and transfected with oligo(dT) or oligo(dA) as described previously (Politz et al., 2004) except that the concentration of oligo was 0.125 μM. After a 1-h incubation without oligo in DMEM (with serum), cells on coverslips were mounted in a holder and then maintained at 37 or 22°C as described in DMEM buffered with 25 mM HEPES (10% FBS, no phenol red) (Politz et al., 2004). Cells containing fluorescein-labeled oligo(dT) growing in two-well dishes were imaged using a Quantix 57 charge-coupled device camera (Roper Scientific Photometrics, Tucson, AZ) coupled to a Leica DMIRB microscope equipped with a 100× objective (numerical aperture 1.4) as described previously (Politz et al., 2000). Signal intensity was quantitatively scaled using MetaMorph software. All uncaging, rapid acquisition digital microscopy, and image processing were performed using equipment and software previously described in detail (Politz et al., 1999, 2004). Some images were deconvolved using exhaustive photon reassignment (Carrington et al., 1995).
In Situ Reverse Transcription
Cells were allowed to take up oligo as described above (at an oligo concentration of 0.5 μM in the medium) and after a 1-h efflux period, fixed and subjected to in situ reverse transcription as described in detail previously (Politz et al., 1995; Politz and Singer, 1999), except that the anti-digoxigenin antibodies were linked to horseradish peroxidase (anti-digoxigenin-POD, Fab fragments; Roche Diagnostics, Indianapolis, IN), and the blocking step was performed using 1% normal sheep serum (The Jackson Laboratory, Bar Harbor, ME) in SSC. The colorimetric assay was carried out using a diaminobenzidine substrate according manufacturer's instructions (Roche Diagnostics), and bright field images of the resulting signal were captured using the Leica microscope system described above. Contrast was quantitatively scaled using MetaMorph software (Universal Imaging, Downingtown, PA).
Fluorescence Correlation Spectroscopy
FCS (Magde et al., 1972) was carried out using a previously described instrument (Wachsmuth et al., 2003). Cells were grown in eight-well dishes (Nalge Nunc) in RPMI medium (with 10% serum, no phenol red) and transfected as described above. The dishes were then mounted on the stage of the FCS microscope, and either a speckle or a site in the nucleoplasm was aligned in the laser path using a galvanometer scanner in point-addressable mode. Measurements were recorded at a laser intensity of ∼1–5 kW/cm2 (0.1–0.5-mW laser power at the focus). A 485-nm excitation filter (DF 22; Omega Optical, Brattleboro, VT) was placed in the laser light path to reduce the intensity of the mRFP for imaging of the fluorescein-labeled oligo. For experiments done at 37°C, a chamber around the microscope and stage was heated to maintain a temperature of 37°C in a humidified 5% CO2 atmosphere. Ten readings were taken at each site and averaged; readings that showed bleaching were discarded. Best fits for the autocorrelation curves were chosen and recorded using Quickfit (Press et al., 1992), and in some cases, the MaxEnt fitting program (Modos et al., 2004) was also used to help determine which fit was optimal.
Simulations
A 100-μm aperture placed in the UV illumination path during photoactivation produced an ∼1-μm uncaging spot at the focal plane. This spot is the convolution of the three-dimensional point-spread-function (3-D PSF) of the microscope with the two-dimensional (2-D) illumination aperture. In wide-field imaging systems, the captured images of fluorescence are the result of the convolution of the 3-D fluorescent oligo distribution with this 3-D PSF. To generate computer simulations of the uncaging and imaging processes, the 3-D PSF of the microscope system was measured using subresolution fluorescent beads (Carrington et al., 1995). Images spanning ± 10 μm of focus about the bead were acquired at 0.25-μm intervals, with a pixel size of 0.15 μm to match the experimental data. Custom software designed for modeling reaction and diffusion events inside cells in 3-D (Zou et al., 1999; Fogarty et al., 2000; ZhuGe et al., 2000) was then used to simulate both the uncaging and diffusion of the fluorescent oligos. The nucleus was modeled as a cylinder with its central axis along the z-axis (axis of focus) of the microscope (x,y = 15 μm; z = 10 μm; pixel size = 0.15 μm). The initial 3-D distribution of uncaged oligo was calculated by digitally convolving a computer generated image of a1-μm, 2-D aperture with the empirically measured 3-D PSF and was used as the starting distribution for the diffusion simulations. The 3-D uncaged spot was centered within the cylinder (as necessitated by the imposed symmetry) and focused 4 μm from the bottom. The edges of the nucleus were a barrier to diffusion. Diffusion was simulated using a single diffusion constant, D (units of square micrometers per second), for up to 30 s, and images were saved at times corresponding to the experimental data. The saved simulation images were converted from cylindrical coordinates to Cartesian coordinates (x,y,z) using interpolation to produce 3-D images having pixel sizes of 0.15 μm in x and y, and 0.25 μm in z. Each 3-D image time point was then “blurred” by convolving with the 3-D PSF and the infocus image was extracted, producing a temporal sequence of 2-D images of diffusion modeling the experimental data.
Simulations were conducted using a range of diffusion constants. Because the microscope system can be approximated as a linear system (Carrington et al., 1995), mixes of two or more different Ds were created by simulating each D separately. The resulting image sequences for each D were digitally added in the desired proportions. In the case of modeling oligo confined to a “speckle,” the initial uncaged distribution was masked using a 2-μm-diameter sphere center at the infocus position of the spot, and D was set to zero. For the simulation shown in Figure 6, D–F, the step convolving the simulation with the 3-D PSF was omitted.
Figure 6.

Simulations of a population of molecules moving away from a one micron diameter uncaging site with a diffusion coefficient of 0.3 μm2/s. (A) Simulated (see Materials and Methods) wide-field microscope image of the uncaged spot before any diffusion has occurred. The uncaged profile is reimaged with both in-focus (the aperture) and out-of-focus components. (B) Plot of intensities along a line through the center of the spot in A (circles), are well fit by a two component Gaussian model (black line) compared with a single Gaussian model (red line). The two Gaussian components capture the in-focus (narrower) and out-of-focus (broader) contributions (see Materials and Methods). (C) Plot of intensities along the same line over time, after allowing the initial uncaged distribution shown in A and B to diffuse in 3-D with D = 0.3 μm2/s. Colors (black, red, green, cyan, blue, magenta, violet, and orange) correspond to data from images at times 0.15, 0.6, 1.05, 1.5, 1.95 (2.4 and 2.85 not shown), 3.3, 3.75, and 8.25 s, respectively. The intensity line data from the images up to 3.75 s were jointly fit with the equation for a single (Gaussian) diffusion component convolved with a two Gaussian component initial uncaged distribution (solid lines). The diffusion coefficient from the fit was D = 0.38 μm2/s. (D) Same as in A except before being reimaged (blurred) by the PSF of the microscope. This yields only the infocus portion of the 3-D uncaged spot. (E) Plot of intensities along a line through the center of D (circles) are well fit by a one-component gaussian model (black line). (F) Same as C except the lines were through D, and the equation fit to the data is a single diffusion component convolved with a single Gaussian component for the initial distribution. From the fit, D = 0.34 μm2/s. (G) From the same simulation as in A, after diffusing for 8.25 s (orange circles in C). (H) Same as in G except for this simulation, 5% of the uncaged molecules at the uncaging spot were assumed to be immobile. (I) Same as C, but the line was drawn through the center of the uncaging site on H. The presence of the 5% fixed molecules within a 2 μm speckle is quite evident by 8 s (orange circles, compare with C). Bars (A, C, F and G), 4 μm.
The spatial profile of the UV uncaging spot intensity at the focal plane, before diffusion, can be well fit with a single Gaussian:
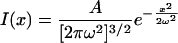 |
(1) |
where ω is the e–1 half-width of the spot. The equation for the spatial profile produced by diffusion of this initial spot is
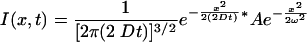 |
(2) |
where D is the diffusion constant, t is time, ω is as for Eq. 1, and * denotes convolution. The convolution of two Gaussians is another Gaussian:
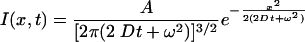 |
(3) |
We can well fit the spatial profile of the image of the initial uncaged fluorescence, before diffusion, with the sum of two Gaussian spatial profiles, roughly corresponding to the infocus and out-of-focus components of the 3-D fluorescence distribution:
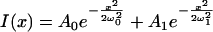 |
(4) |
and the corresponding diffusion profile:
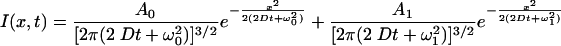 |
(5) |
The 2-D spatial and temporal intensity profile of both simulated images and experimental images were fit to either Eq. 3 or Eq. 5, using custom software implementing a multiparameter least-squares curve fit approach, to derive estimates of both D and ωi.
RESULTS
Characterization of Oligo(dT) Uptake by a Stable HeLa Cell Line Expressing mRFP-SC35
A stable HeLa cell line expressing the splicing factor SC35 fused to monomeric red fluorescent protein (mRFP-SC35) was generated using standard procedures (see Materials and Methods). Greater than 95% of the cell population showed expression of the red SC35 protein, which was observed to be most concentrated at multiple nucleoplasmic sites (Figure 1A). Signal was also observed in nucleoplasmic regions outside the speckles. A similar distribution pattern has also been observed in stable cell lines expressing the splicing protein SF2/ASF linked to green fluorescent protein (GFP) (Phair and Misteli, 2000). The transformed cell population maintained nearly 100% expression of mRFP-SC35 over several generations.
Figure 1.

Characteristics of HeLa cell line stably expressing mRFP-SC35. (A) Raw and restored (exhaustive photon reassignment; see Materials and Methods) midplanes of an optical stack showing a live HeLa cell stably expressing mRFP-SC35. Bars, 4.2 μm. (B) Immunoblot of total cellular proteins probed with RFP antibody before (–) and after (+) treatment with CIP in wild-type HeLa cells (left lane) and the stable SC35 HeLa cell line (middle and right lanes). (C) Fluorescent in situ hybridization to fixed SC35 stable HeLa cell line transiently transfected with a plasmid that expresses β-tropomyosin mRNA at high levels (see Materials and Methods). β-Tropomyosin mRNA was detected with a Spectrum-Green-labeled probe (green) and overlap with mRFP-SC35 (red) is shown as yellow. Bar, 5 μm.
We examined the phosphorylation status of the chimeric mRFP-SC35 protein and found it to be present in its active, phosphorylated form in the stably transfected cell line (Figure 1B), demonstrating that this mRFP fusion protein was behaving similarly to its endogenous SC35 counterpart (Misteli and Spector, 1996). To further test the degree to which the behavior of the mRFP-SC35 fusion protein resembled that of the native SC35 protein, we investigated its dynamic association with a specific polymerase II transcription site. As shown in Figure 1C, the mRFP-SC35 chimeric protein was recruited to the sites of active transcription of a transfected β-tropomyosin gene, similar to the behavior of native SC35 (Huang and Spector, 1991; Jimenez-Garcia and Spector, 1993; Huang and Spector, 1996b; Misteli et al., 1997; Misteli and Spector, 1999). The cells also seemed morphologically normal and grew with a doubling time similar to the parental cell line. Therefore, by all these criteria the mRFP-SC35 fusion protein seemed to be mirroring the behavior of the native SC35 protein and therefore was judged to be a valid marker for speckles in these cells.
Introduction of fluorescein-labeled oligo(dT) into this stably transfected HeLa cell line (see Materials and Methods) was optimized to give detectable signal in greater than 50% of the cells, with minimal effect on cell viability (as judged by degree of spreading and population size 24 h later; Politz 1999; Politz et al., 2004). As previously found in L6 myoblasts (Politz et al., 1995, 1999), oligo(dT) signal was distributed throughout the nucleus and within speckles but did not seem concentrated in speckles (Figure 2, A and B). An in situ reverse transcription assay (Eberwine et al., 1992; Politz and Singer, 1999; Politz et al., 1999) was used to detect hybridization of the oligo to endogenous RNA. In this assay, only oligo(dT) that is hybridized to poly(A) can act as a primer for reverse transcription. As can be seen in Figure 2C, reverse transcription products were observed in both the nucleus and the cytoplasm of cells that took up oligo(dT), as expected for hybridization to poly(A) RNA. Cells that had taken up the control oligo(dA) showed no signal (Figure 2D), indicating that the oligo(dA) does not hybridize appreciably to RNA in the cell. (HeLa cell nuclear RNA contains some oligo(U) tracts, but these are present at a much lower concentration than poly(A); Kish and Pederson, 1977).
Figure 2.

In vivo signal distribution and in situ reverse transcription in the mRFP-SC35 HeLa cell line after uptake of fl-oligo(dT) or fl-oligo(dA). (A and B) HeLa cells stably transfected with SC35-mRFP were allowed to take up fluorescently labeled oligo(dT) and the distribution of signal was visualized as described in Materials and Methods. (A) SC35-mRFP. (B) fl-oligo(dT). Bars, 3 μm. (C and D) After uptake of either fl-oligo(dT) or fl-oligo(dA), cells were fixed and subjected to in situ reverse transcription as described in Materials and Methods. Incorporation of biotin-labeled deoxynucleotides was detected using anti-biotin antibody coupled to horseradish peroxidase as described in text. (C) Bright field image of cells containing oligo(dT). (D) Bright field image of cells containing oligo(dA). Bars, 9.4 μm.
FCS Used to Measure Mobility of Poly(A) RNA in Speckles and the Nucleoplasm
We first used the technique of FCS (Magde et al., 1972; Krichevsky and Bonnet, 2002; Kim and Schwille, 2003) to compare the mobility of poly(A) RNA in speckles and the nucleoplasm. This method quantitatively measures the amount of fluorescence in a very small volume and rapidly monitors the fluctuation of that signal intensity over time. The more the signal fluctuates, the more rapidly the molecules are moving in and out of the interrogated volume. This is manifest as an increased rate of decay in the autocorrelation function. Using curve-fitting algorithms, the number of differently diffusing components within the sample area can be determined, and their respective diffusion coefficients were estimated (Politz et al., 1998; Wachsmuth et al., 2000). The confocal assay volume of FCS is less than a femtoliter; in our experiments, the radial diameter of the confocal volume was 0.44 μm and the z-axis height was ∼1.6 μm when exciting fluorescein (and 0.5 and 2 μm, respectively, when exciting mRFP). These dimensions are similar to or smaller than that of a speckle; deconvolved images of the stable SC35 cell line used to estimate speckle size gave radial diameters of ∼0.5–2.5 μm and z-axis heights of ∼1.5–2.5 μm (our unpublished data). Therefore, FCS measurements could be taken inside a speckle without substantial excitation of the surrounding nucleoplasm.
We used the galvanometer scanner of the fluorescence fluctuation microscope in the point-addressable mode to direct a laser beam into a confocal volume inside the nucleus of live SC35-expressing HeLa cells containing fluorescent oligo(dT). The beam was focused either within a speckle or into an area of the nucleoplasm devoid of a speckle. Fluorescence fluctuations in each interrogated volume were then measured over time and recorded as an autocorrelation curve. Figure 3A shows representative autocorrelation curves obtained within (blue) and outside (red) a speckle, with the best fit curves (smooth lines) shown for each in the same color. It can be seen that very similar autocorrelation curves were obtained from both speckle and nucleoplasmic sites, indicating that the oligo(dT) had similar molecular dynamics at both sites. Additionally, the inverse particle number (the y-intercept), which is proportional to the total concentration of oligo(dT), is very similar in both cases. The best fit curves most often represented two or three components of differing mobilities. The relative fraction of each of these components is shown in Figure 3B (dT on and dT off).
Figure 3.

Mobility of oligo(dT) and oligo(dA) on and off speckles in mRFP-SC35 HeLa cells measured using FCS. (A) Autocorrelation curves of cells containing oligo(dT), based on FCS measured either within a speckle (blue) or in the nucleoplasm (off a speckle, red). (B) Fraction of oligo(dT) and oligo(dA) present in different mobility classes measured within speckles (on) and in the nucleoplasm (off). Each kinetic component is indicated by black, red, or open bars in the histogram. The 10- to 100-ms component (red) was undetectable in oligo(dA) containing-cells. (C) Fraction of mRFP-SC35 protein present in different mobility classes measured within speckles (on) and in the nucleoplasm (off). (D) Average fraction of oligo(dT) present in different mobility classes measured using FCS within speckles (on) and in the nucleoplasm (off) at 22 and 37°C.
One of these kinetic components was not observed when measurements were made in cells containing the nonhybridizing control probe, oligo(dA), and is therefore highly likely to represent oligo(dT) hybridized to poly(A) RNA (Figure 3B, compare red bars in dT columns to the zero-level red bars in the dA columns). No significant difference in the mobility of this fraction was observed when measurements were made either within a speckle or in the nucleoplasm (Figure 3B, compare red fraction in dT off to dT on). It took the oligo(dT):poly(A) RNA hybrids an average of 22 ± 5 ms (D = 0.65 ± 0.19 μm2/s) to traverse the confocal volume within a speckle and 20 ± 4 ms (D = 0.70 ± 0.19 μm2/s) to traverse the same distance in the nucleoplasm. Thus, we observed no significant difference in mobility of the poly(A) RNA populations inside or outside a speckle.
The fastest moving component present in the oligo(dT)-containing cells (Figure 3B, dT black bars) closely corresponded to the most prevalent (∼80% of signal) mobility fraction obtained when measurements were made in control oligo(dA)-containing cells (Figure 3B, dA black bars) and therefore probably represents unhybridized oligo(dT) in the nucleus (average dT on, 1.1 ± 0.2 ms; dT off, 1.1 ± 0.3 ms; dA on, 1.8 ± 0.2 ms; and dA off, 2.0 ± 0.3 ms).
Interestingly, when the mobility of the mRFP-SC35 protein itself was measured in speckles, the same two mobility fractions were observed (Figure 3C), although the faster moving fraction was a somewhat higher percentage of the total. A similar distribution of SC35 mobility classes was observed when measurements were taken outside a speckle (Figure 3C). Therefore, the behavior of the mRFP-SC35 protein as judged by FCS was similar to the behavior of oligo(dT), both with respect to the presence of two mobility categories, presumably one bound and the other free, and in the fact that there was no difference in the mobility of these fractions on or off speckles.
A low abundance, very slow third component was also identified using FCS in some oligo(dT)-containing cells (Figure 3B, dT white bars). This minor, low-mobility fraction was not thought to contain hybridized oligo(dT) because it was also present in some control oligo(dA)-containing cells (Figure 3B, dA white bars). The nature of this slow fraction of oligo(dT) and oligo(dA) is not understood at present, but it should be noted that a slow component with similar characteristics is also detected in some cells in which the intranuclear mobility of GFP is measured (Wachsmuth et al., 2000; Baudendistel and Langowski, unpublished observations). Another observation, perhaps related, was that in some FCS measurements an initial bleaching of signal occurred. We were not able to characterize this very slow moving or immobile fraction in detail, but it was detected in both speckles and in the nucleoplasm at approximately equal frequencies.
We also used FCS to measure the mobility of the hybridized oligo(dT) in cells at 22°C compared with 37°C. Our previous studies had shown no difference in poly(A) RNA mobility in L6 myoblasts as a function of temperature (Politz et al., 1999) or under conditions that inhibited ATP production (Politz et al., 1998). However, in these previous studies it was not possible to distinguish speckles from other areas within the nucleoplasm. Using the stable mRFP-SC35 HeLa cell line, where oligo(dT) mobility within and outside speckles could easily be compared at 22 and 37°C, no significant difference was observed in the shape of the autocorrelation curves obtained at each temperature (our unpublished data), and the best fit mobility distributions showed no significant difference in the behavior of the poly(A) RNA at either temperature (Figure 3D, red bars). As described above, at 37°C it took poly(A) RNA an average of 22 ± 5 ms to traverse a confocal volume within a speckle and 20 ± 4 ms to traverse the same distance in the nucleoplasm proper. When the measurements were made at 22°C instead, it took poly(A) RNA an average of 27 ± 6 ms to traverse the confocal volume within a speckle and 28 ± 8 ms in the nucleoplasm. Additional details of the FCS measurements are provided in the Supplemental Material.
Visualization and Tracking of Poly(A) RNA by Photoactivation in Speckles and Nucleoplasm
To visually track the movement of poly(A) RNA in and out of speckles and within the nucleoplasm, we next introduced oligo(dT) labeled with photoactivatable fluorescein (Mitchison et al., 1994; Politz, 1999; Politz et al., 1999) to the SC35-expressing HeLa cell line. The coverslips containing the growing cells were mounted in a temperature-controlled chamber on an inverted microscope, and the oligo present in a particular speckle was photoactivated using a laser directed through a pinhole (Politz et al., 1999, 2004). The uncaging spot within the nucleus was typically 1.0–1.5 μm in diameter (see Figure 6 for image), similar to or smaller than the speckles. The movement of the photoactivated signal away from the speckle was tracked over time by capturing sequential digital images (Figure 4). The signal was observed to move out in all directions from the uncaging site into the surrounding nucleoplasm but did not enter nucleoli. Signal moved to remote speckles in a manner indistinguishable from the movement through the nucleoplasm, but with no accumulation of signal in other speckles (see red circled speckles in Figure 4). There was no evidence of a concentration of signal moving in a directed way toward another speckle or toward the nuclear envelope. Instead, the pattern of signal dispersion reproducibly approximated a Gaussian distribution.
Figure 4.

Movement of oligo(dT):poly(A) RNA hybrids away from a speckle after photoactivation. mRFP-SC35 HeLa cells were allowed to take up caged-fl oligo(dT) and then the probe was photoactivated using 360-nm light directed at a speckle in a live cell nucleus as described in Materials and Methods. The uncaging site is marked with a white circle in the “caged” panel, and two speckles are circled in red in both the SC35 panel and in the final panel. High-speed time-lapse digital image microscopy was used to capture 2D images of the signal as it moved away from the uncaging site. (The brighter dots visualized here sometimes occur in cells that have taken up oligodeoxynucleotides and do not correspond to hybridization sites (Politz and Pederson, unpublished data; also see Lorenz et al., 1998). Bar, 2.6 μm.
The oligo(dT) moved away from the speckle much more slowly than a control nonhybridizing oligo(dA). Over half of the oligo(dA) had left the uncaging site by 2 s, whereas ∼90% of the oligo(dT) remained and then moved away more slowly (Figure 5A). This is the expected result because, as observed in the FCS experiments, the oligo(dT) that is hybridized to poly(A) RNA moves much more slowly than does free oligo in the nucleus. The signal intensity along a line drawn through the nucleus and the center of the uncaging site generally described a Gaussian distribution (typical example in Figure 5B, broken curves), although the curves were never completely smooth. The small fluctuations in signal distribution along the curve represent regions where the signal did not spread in a completely uniform way and thus may reflect the presence of obstacles in the intranuclear landscape.
Figure 5.

Characterization of signal movement away from photoactivation site. (A) Probe was uncaged in speckles in cells containing either oligo(dT) (red curve) or oligo(dA) (black curve) and the average signal/pixel remaining at the uncaging site (bleach adjusted) was calculated for each time point and plotted. The bar on each time point represents the SE of the mean. (B) The Gaussian distributions of the signal intensity across a line across the nucleus and through the center of the uncaging site were digitally recorded at successive times after photoactivation (broken curves, top to bottom, 65, 450, 900, 1350, 1800, 3150, and 3500 ms), and a global algorithm was used to determine the best fit diffusion coefficient for the curves and time simultaneously (see Figure 6; see Materials and Methods). A representative distribution and the simulated fit (smooth lines) for an uncaging on a speckle is shown here; the diffusion coefficient estimated from this uncaging was 0.346 μm2/s. (C) The mean square displacement (at e–2) versus time plotted for the same uncaging site as shown in B. The line through the points is based on a linear least squares regression analysis (R2 = 0.92) and predicts a diffusion coefficient of 0.7 μm2/s. (D) Average signal remaining at uncaging sites over time after uncaging on (red curve) or off (blue curve) a speckle in cells containing oligo(dT). Error bars for each point have been omitted so that the two curves can be seen clearly. For each point the SE of the mean was less than or equal to ±3%. (E) Same as in B except the uncaging site was nucleoplasmic (off a speckle). The diffusion coefficient calculated from this global fit is 0.285 μm2/s. (F) The mean square displacement (at e–2) versus time plotted for the same uncaging site as shown in E. (D = 0.6 μm2/s; R2 = 0.98). (G) Average fraction of signal remaining at uncaging site after photoactivation of caged-fl oligo(dT) in speckles at 37°C (pink) and 22°C (blue). The bar on each point is the SE of the mean. (H) Same as B except at 22°C. The diffusion coefficient estimated from this global fit is 0.351 μm2/s.
When the rate of movement away from the uncaging site was plotted as a function of the width (at e–2) of the Gaussian distribution at increasing times (Cardullo et al., 1991), the mean square displacement of the signal was found to be linearly proportional to time (Figure 5C), as expected of a diffusive process. The average estimated diffusion coefficient using this method was 0.67 ± 0.05 μm2/s, which was very similar to that estimated earlier in the nucleoplasm of L6 cells using the same method (Politz et al., 1999). Because this method of estimating a diffusion coefficient is dependent on the position at which the width of the Gaussian is measured, and also does not account for any potential optical blurring, we also used a more refined algorithm to estimate the diffusion coefficient. This algorithm fit the relative shapes of the entire Gaussian distributions over multiple time points with a two-component model (Figure 5B, smooth lines) to more accurately estimate a diffusion coefficient (Fogarty et al., 2000; Figure 6). Using this method, an average diffusion coefficient of 0.32 ± 0.04 μm2/s was estimated.
When a site in the nucleoplasm outside the speckle was uncaged, the signal was observed to behave in a virtually identical manner to signal uncaged in a speckle: it still left the site at a similar rate (Figure 5, D and E), and the square of the displacement again varied linearly with time (Figure 5F). The average diffusion coefficient was estimated to be 0.39 ± 0.04 μm2/s using the global algorithm (Figure 5E, smooth lines show fit), very similar to the diffusion coefficient estimated within speckles. This result is consistent with the FCS experiments described above in which no significant difference was observed in the mobility of poly(A) RNA within speckles or in the nucleoplasm.
We next followed the movement of poly(A) RNA after photoactivation inside a speckle at 22°C, rather than at 37°C. No change in the characteristics of movement of the tagged poly(A) RNA was observed (Figure 5, G and H), and the average diffusion coefficient of 0.27 ± 0.08 μm2/s estimated from the global fit algorithm was not significantly different from that estimated at 37°C (0.32 ± 0.04 μm2/s; Figure 5B). No difference was observed in the rate of movement at the two temperatures in the nucleoplasm either (our unpublished data), as has been reported previously for poly(A) RNA in L6 myoblasts (Politz et al., 1999; Politz and Pederson, 2000). Therefore, using either FCS or photoactivation techniques, poly(A) RNA showed similar dynamics in the nucleoplasm and the speckle, and no evidence was obtained for the presence of a distinct slow-moving poly(A) RNA population within the speckle.
Although the results presented up to now indicated that most poly(A) in speckles was dynamically exchanging with the nucleoplasm, we wanted to address the degree to which our methods could have detected a minor population of immobile RNA in the speckle because this was the key hypothesis being tested. Additionally, we wanted to determine whether uncaged signal above and below the plane of focus was affecting our diffusion coefficient estimates. We therefore carried out a series of quantitative uncaging simulations. Figure 6 shows simulations (see Materials and Methods) of a population of molecules moving away from a 1-μm-diameter uncaging site with a diffusion coefficient of 0.3 μm2/s within a nucleus that is 15 μs in diameter under our standard widefield imaging conditions (blurred, Figure 6, A and B) versus a simulated ideal case where no optical blurring of the signal takes place (i.e., contributions from uncaged molecules above and below the plane of focus were omitted from the simulation; Figure 6, D and E). When the global fit algorithm was used to fit the Gaussian curves for the blurred simulation at each time point (Figure 6C, smooth lines), the best fit was obtained when a two-component Gaussian spatial profile was assumed. The second component thus very likely represents the blurred, or out-of-focus light, because it is not present in the fits to the nonblurred image (Figure 6, E and F). It therefore can be concluded that the contribution of the out-of-focus signal above and below the plane of focus in our experiments inflates our estimate for the diffusion coefficient of poly(A) RNA by at most a factor of 2, and we can account for this using the two component global fit algorithm. As mentioned, we used this two-component global fit algorithm to calculate the Ds for all the uncaging data described above, so that the contribution of the optical blurring was removed.
We noted that the global fits to the real data Gaussian distributions shown in Figure 5 do not fit the early time points as well as the global fits to the simulations shown in Figure 6 (compare Figure 5C to 6C). We think this is because of the presence of free oligo near the uncaging site at these early times points. Indeed, the FCS studies described above predict that about half of the oligo(dT) in the nucleus in unhybridized, but this fraction cannot be tracked in the time frame of the uncaging studies because free oligo would move away from the site within the first few time points (seconds). This interpretation is supported by the fact that if the global fits are done only to the first two time points after uncaging, a faster diffusion coefficient is obtained.
Finally, we built differing levels of a putative immobile RNA population into the simulation as fixed parameters to determine what fraction of immobile molecules would be detectable in the photoactivation-tracking experiments. These simulations revealed that an immobile RNA population comprising as little as 5% of the total signal within the uncaging site would have been easily visualized in both the tracking images (compare Figure 6G with H) and in the Gaussian plots of the intensity distribution used to estimate the diffusion coefficient (compare Figure 6I with B).
DISCUSSION
In this work, we have found that the mobility of poly(A) RNA in HeLa cells is essentially the same both in speckles and in the nucleoplasm. Therefore, although this RNA population includes not only processed mRNA destined for cytoplasmic export but also a substantial fraction of nucleus-retained poly(A) RNA molecules (Perry et al., 1974, Herman et al., 1976; Huang et al., 1994; Morey and Avner, 2004) that might have been thought to be involved in maintaining nuclear substructure in some way, we did not find any poly(A) RNA to be specifically tethered within speckles. Our results do not rule out the presence of a very small slow-moving or immobile class of poly(A) RNA in the nucleus, but if it exists, our results indicate that it is <5% of the total poly(A) RNA and is present in similar amounts in both speckles and the nucleoplasm. Therefore, it is unlikely that poly(A) RNA serves as an immobile scaffolding to form the speckle; rather, it can move freely in and out of speckles in all directions and visit other speckles.
These findings are consistent with numerous studies on the mobilities of nuclear proteins that have demonstrated rapid exchange between the nucleoplasm and speckles (Kruhlak et al., 2000; Phair and Misteli, 2000; Molenaar et al., 2004), the nucleolus (Phair and Misteli, 2000; Snaar et al., 2000; Chen and Huang, 2001), and Cajal bodies (Snaar et al., 2000; Handwerger et al., 2003; Dundr et al., 2004). We also show here that the SC35 protein is extremely dynamic in both within and outside speckles. It is therefore becoming increasingly clear that nuclear proteins that were once thought to perhaps serve static structural roles because they seemed concentrated in a certain structure, actually move in and out of these structures very rapidly (Misteli, 2001; Bubulya and Spector, 2004). Our results demonstrate that poly(A) RNA behaves in a similar manner and exchanges rapidly between speckles and the nucleoplasm.
We looked for temperature-dependent behavior of the poly(A) RNA movement in speckles, but we observed no difference in the rate of movement of poly(A) RNA at 22 versus 37°C within speckles or in the nucleoplasm. If movement into or out of speckles were dependent on enzymatic activity (perhaps requiring a motor protein driven by ATP hydrolysis, for example), one would expect to see a three- to fivefold slowing of the rate at the lower temperature because enzymatic reactions have Q10 s (change in reaction rate with every 10 K change in temperature) between 2 and 3. Furthermore, if poly(A) were being directed specifically to the nuclear pores by a metabolic energy-requiring process, one would expect to see a change in the distribution of the poly(A) RNA as it moved out into the nucleoplasm from the speckle at the different temperatures. However, we did not see this. Thus, the simplest interpretation is that poly(A) RNA can move freely between speckles and the nucleoplasm of HeLa cells without a direct input of metabolic energy.
Calapez et al. (2002) reported an ∼1.5- to 2-fold (estimated from Figure 8 in Calapez et al., 2002) reduction in the rate of movement (which gives a threefold reduction in diffusion coefficient) of two different GFP-labeled proteins bound to poly(A) RNA at 37 versus 22°C and suggested that this implies metabolic energy-dependent mobility. Similarly, Molenaar et al. (2004) have reported an ∼1.5-fold (estimated from their Figure 7B) decrease in the rate of recovery after photobleaching of a tagged poly(A) RNA at 37 versus 22°C and suggested on this basis that metabolic energy is necessary for poly(A) RNA mobility. However, reductions of this magnitude are not easily explained by rate decreases in enzymatic reactions because, as explained above, one would expect to see a much larger effect upon a 15°C temperature shift (see also Shav-Tal et al., 2004 for a discussion of this point). In addition, a recent study of mRNA movement in the nucleus of mammalian cells has somewhat clarified the situation by revealing that ATP is required for the resumption of movement when RNA becomes corralled within tight confinements, rather than the nucleotide being involved in the movement itself (Vargas et al., 2005).
Molenaar et al. (2004) also reported that several poly(A) binding proteins moved in and out of speckles at rates similar to those reported here for both bound SC35 and poly(A) RNA, and by others for RNA-binding proteins (Phair and Misteli, 2000; Misteli, 2001; Calapez et al., 2002), but they reported a 5- to 10-fold slower diffusion coefficient for poly(A) RNA itself. It is unclear, however, whether the 2′-O-methyl modified oligo(U) probe used by Molenaar et al. (2004) was actually hybridized to poly(A). Hybridization was tested in only one way: U2OS cells were treated with the cytotoxic drug cordycepin for 16 h, and after this treatment time, it was observed that newly injected 2′-O-methyl oligo(U) was more mobile within the nucleus and no longer accumulated in speckles. Because cordycepin interferes with polyadenylation, it was concluded that the probe must have been hybridized in the untreated cells. However, cordycepin has not been used in previous studies on mammalian cell nuclear RNA metabolism for more than 1–3 h (Siev et al., 1969; Penman et al., 1970; Darnell et al., 1971; Mendecki et al., 1972) because of severe toxicity effects. Moreover, it has been clearly established that cordycepin inhibits the production of cytoplasmic mRNA in HeLa cells within 25–40 min (Penman et al., 1970). The mRNA population of HeLa cells consists of at least two kinetic components; 30–40% has an average half-life of 2–7 h, and the rest has an average half-life of 24 h (Singer and Penman, 1973; Puckett and Darnell, 1977). A 16-h treatment with cordycepin would therefore reduce the shorter lived mRNA population to 6–25% of its original concentration; cells treated in this way may not reflect the behavior of normal cells.
We observed one more interesting property of poly(A) RNA in the experiments described here: poly(A) RNA can visit more than one speckle as it travels throughout the nucleoplasm. This implies that a visit to a speckle does not change the characteristics of the poly(A) RNA in such a way that it is unable to pass through other speckles. A similar phenomenon was observed in live cell tracking of 28S rRNA, which was observed to move between nucleoli (Politz et al., 2003). Certain nuclear proteins have also been observed to move between nuclear compartments, such as fibrillarin between nucleoli and SF2/ASF between speckles (Kruhlak et al., 2000; Phair and Misteli, 2000), but these proteins are not destined for transport to the cytoplasm, as is the aforementioned 28S rRNA and polyadenylated mRNA. It therefore seems that even macromolecules that must be transported to the cytoplasm can roam freely not only throughout the nucleoplasm but also enter and exit similar nuclear compartments with no direct requirement of metabolic energy.
Supplementary Material
Acknowledgments
We thank Supriya Prasanth for help in Western blot analysis. This work was supported by National Institutes of Health Grants GM-60551 to J. P., R. T. and T. P.; GM-42694 to D. S.; and DK-32520 to K. F.; National Science Foundation Grants DB19200027 and DB19724611 to the University of Massachusetts Biomedical Imaging Facility; and a grant to J. L. from the Volkswagen Foundation as part of the program “Physics, Chemistry, and Biology with Single Molecules.”
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–10–0952) on December 21, 2005.
Abbreviations used: FCS, fluorescence correlation spectroscopy; mRFP-SC35, monomeric red fluorescent protein fused to SC35 protein; oligo, oligodeoxynucleotide.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Bubulya, P. A., and Spector, D. L. (2004). “On the move”ments of nuclear components in living cells. Exp. Cell Res. 296, 4–11. [DOI] [PubMed] [Google Scholar]
- Calapez, A., Pereira, H. M., Calado, A., Braga, J., Rino, J., Carvalho, C., Tavanez, J. P., Wahle, E., Rosa, A. C., and Carmo-Fonseca, M. (2002). The intranuclear mobility of messenger RNA binding proteins is ATP dependent and temperature sensitive. J. Cell Biol. 159, 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 11, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardullo, R. A., Mungovan, R. M., and Wolf, D. E. (1991). Imaging membrane organization and dynamics. In: Biophysical and Biochemical Aspects of Fluorescence Spectroscopy, ed. T. D. Dewey, New York: Plenum Publishing, 231–260.
- Carrington, W. A., Lynch, R. M., Moore, E. D., Isenberg, G., Fogarty, K. E., and Fay, F. S. (1995). Superresolution three-dimensional images of fluorescence in cells with minimal light exposure. Science 268, 1483–1487. [DOI] [PubMed] [Google Scholar]
- Carter, K. C., Bowman, D., Carrington, W., Fogarty, K., McNeil, J. A., Fay, F. S., and Lawrence, J. B. (1993). A three-dimensional view of precursor messenger RNA metabolism within the mammalian nucleus. Science 259, 1330–1335. [DOI] [PubMed] [Google Scholar]
- Chen, D., and Huang, S. (2001). Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J. Cell Biol. 153, 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmarko, D., Verschure, P. J., Martin, T. E., Dahmus, M. E., Krause, S., Fu, X. D., van Driel, R., and Fakan, S. (1999). Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell 10, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell, J. E., Philipson, L., Wall, R., and Adesnik, M. (1971). Polyadenylic acid sequences: role in conversion of nuclear RNA into messenger RNA. Science 174, 507–510. [DOI] [PubMed] [Google Scholar]
- Deerinck, T. J., Martone, M. E., Lev-Ram, V., Green, D. P., Tsien, R. Y., Spector, D. L., Huang, S., and Ellisman, M. H. (1994). Fluorescence photooxidation with eosin: a method for high resolution immunolocalization and in situ hybridization detection for light and electron microscopy. J. Cell Biol. 126, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dundr, M., Hebert, M. D., Karpova, T. S., Stanek, D., Xu, H., Shpargel, K. B., Meier, U. T., Neugebauer, K. M., Matera, A. G., and Misteli, T. (2004). In vivo kinetics of Cajal body components. J. Cell Biol. 164, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine, J., Spencer, C., Miyashiro, K., Mackler, S., and Finnell, R. (1992). Complementary DNA synthesis in situ: methods and applications. Methods Enzymol. 216, 80–100. [DOI] [PubMed] [Google Scholar]
- Eils, R., Gerlich, D., Tvarusko, W., Spector, D. L., and Misteli, T. (2000). Quantitative imaging of pre-mRNA splicing factors in living cells. Mol. Biol. Cell 11, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakan, S., and Nobis, P. (1978). Ultrastructural localization of transcription sites and of RNA distribution during the cell cycle of synchronized CHO cells. Exp. Cell Res. 113, 327–337. [DOI] [PubMed] [Google Scholar]
- Fakan, S., Leser, G., and Martin, T. E. (1984). Ultrastructural distribution of nuclear ribonucleoproteins as visualized by immunocytochemistry on thin sections. J. Cell Biol. 98, 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty, K. E., Kidd, J. F., Tuft, R. A., and Thorn, P. (2000). Mechanisms underlying InsP3-evoked global Ca2+ signals in mouse pancreatic acinar cells. J. Physiol. 526, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot, P. V., Xie, S. Q., Hollinshead, M., and Pombo, A. (2004). Fixation-induced redistribution of hyperphosphorylated RNA polymerase II in the nucleus of human cells. Exp. Cell Res. 295, 460–468. [DOI] [PubMed] [Google Scholar]
- Handwerger, K. E., Murphy, C., and Gall, J. G. (2003). Steady-state dynamics of Cajal body components in the Xenopus germinal vesicle. J. Cell Biol. 160, 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfman, D. M., Ricci, W. M., and Finn, L. A. (1988). Alternative splicing of tropomyosin pre-mRNAs in vitro and in vivo. Genes Dev. 2, 1627–1638. [DOI] [PubMed] [Google Scholar]
- Herman, R. C., Williams, J. G., and Penman, S. (1976). Message and nonmessage sequences adjacent to poly(A) in steady state heterogeneous nuclear RNA of HeLa cells. Cell 7, 429–437. [DOI] [PubMed] [Google Scholar]
- Huang, S., and Spector, D. L. (1991). Nascent pre-mRNA transcripts are associated with nuclear regions enriched in splicing factors. Genes Dev. 5, 2288–2302. [DOI] [PubMed] [Google Scholar]
- Huang, S., Deerinck, T. J., Ellisman, M. H., and Spector, D. L. (1994). In vivo analysis of the stability and transport of nuclear poly(A)+ RNA. J. Cell Biol. 126, 877–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., and Spector, D. L. (1996a). Dynamic organization of pre-mRNA splicing factors. J. Cell Biochem. 62, 191–197. [DOI] [PubMed] [Google Scholar]
- Huang, S., and Spector, D. L. (1996b). Intron-dependent recruitment of pre-mRNA splicing factors to sites of transcription. J. Cell Biol. 133, 719–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Garcia, L. F., and Spector, D. L. (1993). In vivo evidence that transcription and splicing are coordinated by a recruiting mechanism. Cell 73, 47–59. [DOI] [PubMed] [Google Scholar]
- Johnson, C., Primorac, D., McKinstry, M., McNeil, J., Rowe, D., and Lawrence, J. B. (2000). Tracking COL1A1 RNA in osteogenesis imperfecta: splice-defective transcripts initiate transport from the gene but are retained within the SC35 domain. J. Cell Biol. 150, 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. A., and Schwille, P. (2003). Intracellular applications of fluorescence correlation spectroscopy: prospects for neuroscience. Curr. Opin. Neurobiol. 13, 583–590. [DOI] [PubMed] [Google Scholar]
- Kish, V. M., and Pederson, T. (1977). Heterogeneous nuclear RNA secondary structure: oligo(U) sequences base-paired with poly(A) and their possible role as binding sites for heterogeneous nuclear RNA-specific proteins. Proc. Natl. Acad. Sci. USA 74, 1426–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky, O., and Bonnet, G. (2002). Fluorescence-correlation spectroscopy: the technique and its applications. Rep. Prog. Phys. 65, 251–297. [Google Scholar]
- Kruhlak, M. J., Lever, M. A., Fischle, W., Verdin, E., Bazett-Jones, D. P., and Hendzel, M. J. (2000). Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J. Cell Biol. 150, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond, A. I., and Spector, D. L. (2003). Nuclear speckles: a model for nuclear organelles. Nat. Rev. 4, 605–612. [DOI] [PubMed] [Google Scholar]
- Lorenz, P., Baker, B. F., Bennett, C. F., and Spector, D. L. (1998). Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell 9, 1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magde, D., Elson, E. L., and Webb, W. W. (1972). Thermodynamic fluctuations in a reacting system: measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 29, 705–708. [Google Scholar]
- Mattaj, I. (1994). Splicing in space. Nature 372, 727–728. [DOI] [PubMed] [Google Scholar]
- Mendecki, J., Lee, S. Y., and Brawerman, G. (1972). Characteristics of the polyadenylic acid segment associated with messenger ribonucleic acid in mouse sarcoma 180 ascites cells. Biochemistry 11, 792–798. [DOI] [PubMed] [Google Scholar]
- Misteli, T. (2001). Protein dynamics: implications for nuclear architecture and gene expression. Science 291, 843–847. [DOI] [PubMed] [Google Scholar]
- Misteli, T., Caceres, J. F., Clement, J. Q., Krainer, A. R., Wilkinson, M. F., and Spector, D. L. (1998). Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol. 143, 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., Caceres, J. F., and Spector, D. L. (1997). The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527. [DOI] [PubMed] [Google Scholar]
- Misteli, T., and Spector, D. L. (1996). Serine/threonine phosphatase 1 modulates the subnuclear distribution of pre-mRNA splicing factors. Mol. Biol. Cell 7, 1559–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., and Spector, D. L. (1999). RNA Polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol. Cell 3, 697–705. [DOI] [PubMed] [Google Scholar]
- Mitchison, T. J., Sawin, K. E., and Theriot, J. A. (1994). Caged fluorescent probes for monitoring cytoskeleton dynamics. In: Cell Biology: A Laboratory Handbook, Vol. 3, ed. J. E. Celis, New York: Academic Press, 65–74. [Google Scholar]
- Modos, K., Galantai, R., Bardos-Nagy, I., Wachsmuth, M., Toth, K., Fidy, J., and Langowski, J. (2004). Maximum-entropy decomposition of fluorescence correlation spectroscopy data: application to liposome-human serum albumin association. Eur. Biophys. J. 33, 59–67. [DOI] [PubMed] [Google Scholar]
- Molenaar, C., Abdulle, A., Gena, A., Tanke, H. J., and Dirks, R. W. (2004). Poly(A) RNAs roam the cell nucleus and pass through speckle domains in transcriptionally active and inactive cells. J. Cell Biol. 165, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morey, C., and Avner, P. (2004). Employment opportunities for non-coding RNAs. FEBS Lett. 567, 27–34. [DOI] [PubMed] [Google Scholar]
- Neugebauer, K. M., and Roth, M. B. (1997). Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev. 11, 3279–3285. [DOI] [PubMed] [Google Scholar]
- Pederson, T. (1999). Movement and localization of RNA in the cell nucleus. FASEB J. 2, S238–242. [DOI] [PubMed] [Google Scholar]
- Penman, M., Rosbash, M., and Penman, S. (1970). Messenger and heterogeneous nuclear RNA in HeLa cells: differential inhibition by cordycepin. Proc. Natl. Acad. Sci. USA 67, 1878–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. P., Kelley, D. E., and LaTorre, J. (1974). Synthesis and turnover of nuclear and cytoplasmic polyadenylic acid in mouse L cells. J. Mol. Biol. 82, 315–331. [DOI] [PubMed] [Google Scholar]
- Phair, R. D., and Misteli, T. (2000). High mobility proteins in the mammalian cell nucleus. Nature 404, 604–609. [DOI] [PubMed] [Google Scholar]
- Politz, J. C. (1999). Use of caged fluorochromes to track macromolecular movement in living cells. Trends Cell Biol. 9, 284–287. [DOI] [PubMed] [Google Scholar]
- Politz, J. C., Browne, E. S., Wolf, D. E., and Pederson, T. (1998). Intranuclear diffusion and hybridization state of oligonucleotides measured by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 95, 6043–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politz, J. C., and Pederson, T. (2000). Movement of mRNA from transcription site to nuclear pores. J. Struct. Biol. 129, 252–257. [DOI] [PubMed] [Google Scholar]
- Politz, J. C., and Singer, R. H. (1999). In situ reverse transcription for the detection of hybridization between oligonucleotides and their intracellular targets. Methods 18, 281–285. [DOI] [PubMed] [Google Scholar]
- Politz, J. C., Taneja, K. L., and Singer, R. H. (1995). Characterization of hybridization between synthetic oligodeoxynucleotides and RNA in living cells. Nucleic Acids Res. 23, 4946–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politz, J.C.R., Tuft, R. A., and Pederson, T. (2003). Diffusion-based transport of nascent ribosomes in the nucleus. Mol. Biol. Cell 14, 4805–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politz, J.C.R., Tuft, R. A., and Pederson, T. (2004). Photoactivation-based labeling and in vivo tracking of RNA molecules in the nucleus. In: Live Cell Imaging: A Laboratory Manual, ed. D. L. Spector and R. D. Goldman, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 177–185.
- Politz, J. C., Tuft, R. A., Pederson, T., and Singer, R. H. (1999). Movement of nuclear poly(A) RNA throughout the interchromatin space in living cells. Curr. Biol. 9, 285–291. [DOI] [PubMed] [Google Scholar]
- Politz, J. C., Yarovoi, S., Kilroy, S. M., Gowda, K., Zwieb, C., and Pederson, T. (2000). Signal recognition particle components in the nucleolus. Proc. Natl. Acad. Sci. USA 97, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth, K. V., Sacco-Bubulya, P. A., Prasanth, S. G., and Spector, D. L. (2003). Sequential entry of components of the gene expression machinery into daughter nuclei. [erratum appears in Mol. Biol. Cell (2003) 14 following 1743]. Mol. Biol. Cell 14, 1043–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., Teukolsky, S. A., Vetterling, W. T., and Flannery, B. P. (1992). Numerical Recipes in C: The Art of Scientific Computing, Cambridge: Cambridge University Press.
- Puckett, L., and Darnell, J. E. (1977). Essential factors in the kinetic analysis of RNA synthesis in HeLa cells. J. Cell Physiol. 90, 521–534. [DOI] [PubMed] [Google Scholar]
- Shav-Tal, Y., Darzacq, X., Shenoy, S. M., Fusco, D., Janicki, S. M., Spector, D. L., and Singer, R. H. (2004). Dynamics of single mRNPs in nuclei of living cells. Science 304, 1797–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland, L. S., Johnson, C. V., Byron, M., McNeil, J., and Lawrence, J. B. (2003). Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighborhoods. J. Cell Biol. 162, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siev, M., Weinberg, R., and Penman, S. (1969). The selective interruption of nucleolar RNA synthesis in HeLa cells by cordycepin. J. Cell Biol. 41, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, R. H., and Penman, S. (1973). Messenger RNA in HeLa cells: kinetics of formation and decay. J. Mol. Biol. 78, 321–334. [DOI] [PubMed] [Google Scholar]
- Singh, O. P., Björkroth, B., Masich, S., Wieslander, L., and Daneholt, B. (1999). The intranuclear movement of Balbiani ring premessenger ribonucleoprotein particles. Exp. Cell Res. 251, 135–146. [DOI] [PubMed] [Google Scholar]
- Snaar, S., Wiesmeijer, K., Jochemsen, A. G., Tanke, H. J., and Dirks, R. W. (2000). Mutational analysis of fibrillarin and its mobility in living human cells. J. Cell Biol. 151, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, Y., Kumagai, I., Haginoya, N., and Matsuda, A. (1997). Effects of 5-(N-aminohexyl)carbamoyl-2′deoxyuridine on endonuclease stability and the ability of oligodeoxynucleotide to activate RNase H. Nucleic Acids Res. 25, 3777–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas, D. Y., Raj, A., Marras, S.A.E., Kramer, F. R., and Tyagi, S. (2005). Mechanism of mRNA transport in the nucleus. Proc. Natl. Acad. Sci. USA 102, 17008–17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachsmuth, M., Waldeck, W., and Langowski, J. (2000). Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially resolved fluorescence correlation spectroscopy. J. Mol. Biol. 298, 677–689. [DOI] [PubMed] [Google Scholar]
- Wachsmuth, M., Weidemann, T., Muller, G., Hoffmann-Rohrer, U. W., Knoch, T. A., Waldeck, W., and Langowski, J. (2003). Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 84, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansink, D. G., Schul, W., van der Kraan, I., van Steensel, B., van Driel, R., and de Jong, L. (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol. 122, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, C., Kim, E., Warren, S. L., and Berget, S. M. (1997). Dynamic relocation of transcription and splicing factors dependent upon transcriptional activity. EMBO J. 16, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G., Taneja, K. L., Singer, R. H., and Green, M. R. (1994). Localization of pre-mRNA splicing in mammalian nuclei. Nature 372, 809–812. [DOI] [PubMed] [Google Scholar]
- ZhuGe, R., Fogarty, K. E., Tuft, R. A., Lifshitz, L. M., Sayer, K., and Walsh, J. V. (2000). A novel method for direct measurement of Ca2+ spark signal mass reveals dynamics of signaling between ryanodine receptors and Ca2+-activated K+ channels in smooth muscle. J. Gen. Physiol. 116, 845–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, H., Lifshitz, L. M., Tuft, R. A., Fogarty, K. E., and Singer, J. J. (1999). Imaging Ca2+ entering the cytoplasm through a single opening of a plasma membrane cation channel. J. Gen. Physiol. 114, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.