

Abstract
The regulated localization of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-type glutamate receptors (AMPARs) to synapses is an important component of synaptic signaling and plasticity. Regulated ubiquitination and endocytosis determine the synaptic levels of AMPARs, but it is unclear which factors conduct these processes. To identify genes that regulate AMPAR synaptic abundance, we screened for mutants that accumulate high synaptic levels of the AMPAR subunit GLR-1 in Caenorhabditis elegans. GLR-1 is localized to postsynaptic clusters, and mutants for the BTB-Kelch protein KEL-8 have increased GLR-1 levels at clusters, whereas the levels and localization of other synaptic proteins seem normal. KEL-8 is a neuronal protein and is localized to sites adjacent to GLR-1 postsynaptic clusters along the ventral cord neurites. KEL-8 is required for the ubiquitin-mediated turnover of GLR-1 subunits, and kel-8 mutants show an increased frequency of spontaneous reversals in locomotion, suggesting increased levels of GLR-1 are present at synapses. KEL-8 binds to CUL-3, a Cullin 3 ubiquitin ligase subunit that we also find mediates GLR-1 turnover. Our findings indicate that KEL-8 is a substrate receptor for Cullin 3 ubiquitin ligases that is required for the proteolysis of GLR-1 receptors and suggest a novel postmitotic role in neurons for Kelch/CUL3 ubiquitin ligases.
INTRODUCTION
Glutamate is the most abundant excitatory neurotransmitter in the brain, and glutamatergic synapses play a critical role in learning, memory, and developmental plasticity of the central nervous system (Meldrum, 2000). Ionotropic glutamate receptors (GluRs) receive and transduce glutamatergic signals on the postsynaptic face of synapses, where these multitransmembrane spanning proteins assemble into tetrameric glutamate-gated channels of differing subunit composition (Hollmann and Heinemann, 1994; Dingledine et al., 1999). The regulation of these receptors, the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) type in particular, is a critical mechanism by which neurons modulate synaptic strength (Bredt and Nicoll, 2003; Malenka, 2003; Malinow, 2003; Sheng and Hyoung Lee, 2003). In addition, GluRs play a role in several diseases of the nervous system, ranging from neurodegeneration to drug addiction and schizophrenia (Tzschentke, 2002; Mattson, 2003; Moghaddam, 2003; Aarts and Tymianski, 2004). To better understand how AMPA-type GluRs (AMPARs) function in the nervous system, it is essential to determine how the synaptic level and activity of AMPARs are regulated.
AMPAR synaptic levels are controlled by proteins that interact with signaling elements on the carboxy-terminal tail sequences that are exposed to the cytosol (Dong et al., 1997; Rongo et al., 1998; Song et al., 1998; Srivastava et al., 1998; Xia et al., 1999; Sans et al., 2001). Although some of the tail sequence signaling elements function to maintain high levels of AMPARs at the synapse, other signaling elements recruit factors that result in the ubiquitination, endocytosis, and proteolysis of AMPARs (Ehlers, 2000; Osten et al., 2000; Burbea et al., 2002; Esteban et al., 2003). AMPARs targeted for destruction are endocytosed in lateral regions adjacent to postsynaptic elements, although the machinery that degrades these membrane-bound proteins is less well understood (Racz et al., 2004).
To investigate these questions in a genetic system, we examined the GLR-1 AMPAR subunit in C. elegans. GLR-1 is expressed in interneurons, where it functions to mediate nose-touch mechanosensation and regulate the frequency of spontaneous reversals in locomotion (Hart et al., 1995; Maricq et al., 1995; Brockie et al., 2001). Mutants lacking GLR-1 are nose-touch defective and rarely reverse direction, whereas nematodes with elevated GLR-1 activity reverse direction at an increased frequency (Hart et al., 1995; Maricq et al., 1995; Zheng et al., 1999; Brockie et al., 2001; Mellem et al., 2002). Chimeric GLR-1 receptors tagged with the green fluorescent protein (GLR-1::GFP) are used to visualize glutamate receptors in living animals; these chimeric receptors are functional because they fully rescue glr-1 mutants (Rongo et al., 1998; Rongo and Kaplan, 1999). GLR-1::GFP is postsynaptically localized to clusters in the nerve ring (a region of proximal neurites that circumscribe the pharynx) and along the ventral nerve cord (a fascicle of distal neurites that runs anterior to posterior along the ventral midline of the body) (Rongo et al., 1998; Burbea et al., 2002). The synaptic abundance of GLR-1::GFP is regulated by the ubiquitination and subsequent endocytosis of the GLR-1 subunit (Burbea et al., 2002; Juo and Kaplan, 2004).
Ubiquitination is used to regulate numerous biological processes, and ubiquitin-mediated proteolysis is a critical component in protein turnover (Hegde, 2004). Ubiquitination is conducted in a stepwise manner by E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases, which recognize target proteins and catalyze the covalent attachment of ubiquitin to these target substrates. Ubiquitinated membrane proteins are substrates for endocytosis, although the exact mechanism by which such endocytosed membrane proteins are eventually degraded is unclear (Haglund et al., 2003; Hicke and Dunn, 2003). Improper ubiquitination in the nervous system has been implicated in Parkinson's, Huntington's, and Alzheimer's diseases as well as in other neurological disorders, further supporting the importance of ubiquitination and protein turnover in proper nervous system function (Ehlers, 2004).
Ubiquitination specificity is conferred by E3 ligases, including the diverse superfamily of Cullin-RING ligases (reviewed in Willems et al., 2004; Petroski and Deshaies, 2005). Cullin-RING ligases are complexes containing a RING protein, which recruits E2-conjugating enzymes, a Cullin scaffold protein, a substrate receptor, and sometimes an adaptor between the Cullin and substrate receptor. There are seven different subfamilies of Cullin-RING ligases (named CDL1–7 for Cullin-dependent ligase), and each subfamily can assemble with numerous substrate receptors. Each substrate receptor contains a domain for interacting with a specific substrate and a subfamily-specific domain for interacting with a specific Cullin subfamily member. For example, CDL1 (or SCF) is comprised of CUL1 bound to an F-box-containing substrate receptor, CDL2 is comprised of CUL2 bound to a SOCS/BC-box-containing substrate receptor, and CDL3 is comprised of CUL3 bound to a BTB-domain-containing substrate receptor. Cullin-RING ligases have been primarily studied for their role in mitosis and cell division, particularly for CDL1. Recently, CDL3s were found to regulate the meiosis/mitosis transition in C. elegans (Pintard et al., 2003; Xu et al., 2003). CDL3s assemble with BTB proteins, which are substrate receptors that directly bind to Cul3 without the aid of an adaptor (Deshaies, 1999; Joazeiro and Weissman, 2000; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003). There are many known BTB proteins, suggesting the assembly of a diverse array of CDL3s, each with unique substrate specificity (Willems et al., 2004; Petroski and Deshaies, 2005). The biological function of these proteins is largely unknown, particularly for postmitotic Cullin-RING ligases.
To identify the factors that regulate the turnover of AMPARs, we screened for mutants with increased synaptic abundance of GLR-1. Here, we describe mutants for the gene kel-8 (kelch-repeat containing protein 8), which have increased GLR-1 levels in neurites. Mutants for kel-8 also show an increased frequency of spontaneous reversals of locomotion, consistent with increased GLR-1 synaptic levels. By contrast, we found that the levels and localization of other synaptic proteins seem normal in kel-8 mutants. KEL-8 is a member of the BTB-Kelch superfamily of proteins and contains six Kelch repeats and a BTB domain. Kelch repeats are protein–protein interaction domains, and Kelch proteins interact with a variety of other proteins, including actin (Adams et al., 2000). We show that KEL-8 is expressed and required in GLR-1-expressing neurons and is localized to sites adjacent to GLR-1 postsynaptic clusters along the ventral cord neurites. We also show that KEL-8 is required for efficient ubiquitin-mediated turnover of GLR-1 subunits. Moreover, KEL-8 binds to CUL-3, a Cullin3-type scaffold for E3 ubiquitin ligases, and expression of a dominant negative CUL-3 results in GLR-1 accumulation. Our findings suggest that KEL-8 is a substrate receptor for CDL3 that regulates GLR-1 turnover and indicate a novel biological role for Cullin-RING ligases: the regulation of glutamate receptor localization and signaling in postmitotic neurons.
MATERIALS AND METHODS
Strains
Standard methods were used to maintain C. elegans (Wood, 1988). Animals were grown at 20°C on standard NGM plates seeded with OP50 Escherichia coli. Some strains were provided by the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN). Strains used in this study include nuIs25[glr-1::gfp], odIs1[snb-1::gfp], odIs16[glr-1::rfp], odIs22[lin-10::gfp], nuIs68[unc-43::gfp], nuIs89[MUb, ttx-3::gfp], unc-11(e47), dpy-11(e224), unc-34(e315ts), rfl-1(or198ts), and CB4856 (Hawaiian strain).
Isolation and Mapping of the kel-8 Mutant
P0 nuIs25 nematodes were ethyl methanesulfonate mutagenized using standard procedures (Brenner, 1974). F2 animals from individual plates were sampled (n = 30–50) by mounting on 2% agarose pads containing levamisol. Animals were scored by fluorescence microscopy for defects in GLR-1::GFP localization. Mutants were recovered either directly from the slide or by isolating siblings from the parental F1 plate. The mutant containing the kel-8(od38) mutation also contained a second unlinked mutation with an uncoordinated behavioral phenotype and its own GLR-1 localization phenotype. This second mutation was crossed away from kel-8(od38) after five rounds of backcrossing and will be described elsewhere.
The kel-8(od38) mutation was mapped between dpy-11 and unc-34 on the left arm of LGV. A dpy-11 kel-8 unc-34 recombinant chromosome was constructed and placed in trans over the CB4856 LGV chromosome to allow three-factor single-nucleotide polymorphism (SNP) mapping. Multiple recombinants placed kel-8 between SNP pkP5103 (map position –12.41) and SNP C29G2.1 (map position –13.37), a region of ∼200 kb. This region is spanned by six cosmids, which were independently injected into kel-8 mutants. Cosmid W02G9 (coinjected with rol-6dm) rescued the kel-8 mutant phenotype with respect to GLR-1 in five of six Rol extrachromosomal lines. By contrast, we observed no rescue from five other cosmids (C24B9, 0 of 2 lines; R05D8, 0 of 2 lines; C29G2, 0 of 2 lines; and W02H2, 0 of 5 lines) containing sequences adjacent to W02G9 in the genome but lacking the kel-8 locus. We sequenced the most promising candidate on W02G9 and identified a premature stop codon mutation in W02G9.2 from multiple independent PCR reactions using kel-8 genomic DNA as template. Sequences from full-length cDNAs yk1096f10 and yk1360a11 (a gift from Y. Kohara, National Institute of Genetics, Mishima, Japan), which include an SL1 splice leader marking the 5′ end of transcription, were used to determine the gene structure.
Transgenes and Germline Transformation
To observe the localization of different synaptic proteins, we used several previously published integrated transgenes: nuIs25[glr-1::gfp], odIs1[snb-1::gfp], odIs16[glr-1::rfp], odIs22[lin-10::gfp], nuIs68[unc-43::gfp], and nuIs89[MUb, ttx-3::gfp] (Rongo et al., 1998; Rongo and Kaplan, 1999; Burbea et al., 2002; Shim et al., 2004). Other transgenic strains were isolated by microinjecting various plasmids (typically at 50 ng/ml) using rol-6dm (a gift from C. Mello, University of Massachusetts Medical School, Worcester, MA) as a marker. The following transgenes were introduced into the germline and followed as extrachromosomal arrays. The kel-8::gfp transgene was generated by subcloning 5 kb of upstream genomic sequence and the entire kel-8 open reading frame from genomic cosmid W02G9 into the GFP vector pPD95.75 (a gift from A. Fire, Stanford University School of Medicine, Palo Alto, CA) so that the GFP sequences were fused in frame at the carboxy terminus of KEL-8. The Pglr-1::kel-8::yfp transgene was generated by subcloning the kel-8 cDNA from yk362h1 (a gift from Y. Kohara) into pV6 (a gift from V. Maricq, University of Utah, Salt Lake, UT), which contains the glr-1 promoter. Yellow fluorescent protein (YFP) sequences were then inserted in frame at the kel-8 carboxy terminus. The kel-8(1-219)::gfp transgene was generated by subcloning 5 kb of upstream genomic sequence and the first five exons through amino acid 219 into pPD95.75 so that GFP was fused in frame at the carboxy terminal end of the KEL-8 BTB domain. The Pglr-1::kel-8 transgene was generated by subcloning kel-8 cDNA sequences from yk362h1 into pV6. The Pglr-1::cul-3, Pglr-1::cul-3(1–500), and Pglr-1::cul-3(501–777) transgenes were generated by subcloning cul-3 cDNA sequences from pGST-CUL-3 (a gift from L. Xu and W. Harper, Harvard Medical School, Boston, MA) into pV6.
Fluorescent Microscopy
GFP-, cyan fluorescent protein (CFP)-, and YFP-tagged fluorescent proteins were visualized in nematodes by mounting L4 and young adults on 2% agarose pads with 10 mM levamisole at room temperature. Fluorescent images were observed using a Zeiss Axioplan II and either a 100× or 63× (1.4 numerical aperture PlanApo for both) objective and imaged with an ORCA charge-coupled device (CCD) camera (Hamamatsu, Bridgewater, NJ) using Image-Pro version 4.1 (Media Cybernetics, Silver Spring, MD) and VayTek version 6.2 software (VayTek, Fairfield, IA). Exposure times were chosen to fill the 12-bit dynamic range without saturation, and out-of-focus light was removed with a constrained iterative deconvolution algorithm (VayTek).
To quantify the fluorescently tagged proteins, images of nematodes were captured by CCD as described above using a constant gain and exposure time (filling the 12 bit dynamic range) for all samples. Background fluorescence from the coverslip and from nonspecific tissue autofluorescence was removed by subtracting an image filtered with a low pass Gaussian filter. Cluster outlines were calculated for fluorescent signals that were 2 SDs above the unlocalized baseline using a macro written for Image-Pro. We found this algorithm agreed with puncta assessed by eye. Cluster size was measured as the maximum radius for each outlined cluster. Cluster number was calculated by counting the average number of clusters per 10 μm of dendrite length.
Behavioral Assays
Nose-touch sensory responses were assayed as described previously (Hart et al., 1995). Each animal was tested on food for reversal of locomotion after a forward collision with a hair. Each animal was tested 10 times, and 20 or more animals were tested for each genotype. The reversal frequency of individual animals was assayed as described previously (Zheng et al., 1999). Single young adult hermaphrodites were placed on NGM plates in the absence of food. The animals were allowed to adjust to the plates for 5 min, and the number of spontaneous reversals for each animal was counted over a 5-min period. Twenty animals were tested for each genotype, and the reported scores reflect the mean number of reversals per minute.
Anti-KEL-8 Antibody
GST-KEL-8(1-100) protein was produced in BL21 E. coli by subcloning kel-8 cDNA sequences encoding the first 100 amino acids into pGEX-KG (Guan and Dixon, 1991). GST-KEL-8(1-100) protein was expressed and purified using glutathione-Sepharose (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) and used to immunize rabbits using Freunds (Pocono Rabbit Farms, Canadensis, PA). The resulting antisera recognized both glutathione S-transferase (GST) and KEL-8 proteins.
Immunoprecipitations and GST Pull Downs
KEL-8 protein was produced in COS-7 cells by subcloning kel-8 cDNA sequences into pCMV-Tag1 (Stratagene, La Jolla, CA). GST, GST::CUL-3(H2), and GST::CUL-3 proteins were produced in COS-7 cells using previously described plasmids (Xu et al., 2003). COS-7 cells were cultured as described previously (Firestein et al., 1999) and transiently cotransfected with FLAG and GST-tagged proteins using LipofectAMINE Plus (Invitrogen, Carlsbad, CA). Typically 2.5 μg of each plasmid was transfected at a 1:1 ratio. After 48 h, transfected cells were lysed in 1 ml of cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, and 0.25% sodium deoxycholate) with protease inhibitors (Roche Diagnostics, Indianapolis, IN). Lysates were cleared of debris and used either for coimmunoprecipitation with anti-FLAG-conjugated protein A-agarose (Sigma, St. Louis, MO) or for GST pull-down assays using glutathione-Sepharose beads (GE Healthcare). Beads were washed in RIPA buffer and bound proteins were eluted in SDS loading buffer and analyzed by SDS-PAGE. Precipitated proteins were detected by Western blotting using polyclonal anti-GST (Sigma) and anti-KEL-8 primary antibodies (this study), horseradish peroxidase-conjugated secondary antibodies (Upstate Biotechnology, Lake Placid, NY), and enhanced chemiluminescence reagents (GE Healthcare). Similar results were observed in six independent transfection experiments.
Yeast two-hybrid experiments were performed by placing kel-8 cDNA sequences into the pDEST22 prey vector (Invitrogen). The resulting pDESTAD::KEL-8 plasmid was cotransformed into yeast strain AH109 with pDESTDB::CUL-3 or pDESTDB, an empty bait vector (Xu et al., 2003). Transformed yeast were recovered on –Leu –Trp dropout plates and were streaked on either –Leu–Trp–His or –Leu–Trp–Ade dropout plates to test for interactions.
RESULTS
GLR-1 Accumulates in kel-8 Mutants
Chimeric GLR-1 receptors tagged with the green fluorescent protein (GLR-1::GFP) are localized to synaptic clusters at neuron–neuron synapses within the C. elegans neuropil in living animals (Figure 1A; Rongo et al., 1998; Rongo and Kaplan, 1999; Burbea et al., 2002). From a direct visual screen for mutants with defects in GLR-1::GFP localization, we identified an allele, od38, of a gene that we have named kel-8 based on its sequence (Figure 2; see below). Mutants for kel-8(od38) lack small synaptic clusters of GLR-1::GFP and instead accumulate large accretions of GLR-1::GFP (Figure 1B). The size of GLR-1::GFP accretions in kel-8 mutants ranges from 2 to 10 times larger compared with GLR-1::GFP clusters in wild-type animals (Figure 1, B and I). There is a 40% decrease in the number of accretions per length of neurite in kel-8 mutants compared with the number of clusters in wild-type animals (Figure 1J), although the drop in number observed in kel-8 mutants is a likely consequence of the large accretion size. These results suggest that KEL-8 regulates the abundance of GLR-1 receptors in ventral cord neurites.
Figure 1.

KEL-8 regulates GLR-1 abundance in dendrites. GLR-1::GFP (A and B), SNB-1::GFP (C and D), UNC-43::GFP (E and F), and LIN-10::GFP (G and H) fluorescence was observed along the ventral cord dendrites of wild-type nematodes (A, C, E, and G) or kel-8 mutants (B, D, F, and H). Whereas wild-type animals have small clusters of GLR-1::GFP (100%; n = 20), most kel-8 mutants have large (>2-μm) clusters of GLR-1::GFP (95%; n = 20). Bar, 5 μm. The mean cluster area (I) and the mean number of clusters per 10 μm (J) of ventral cord length is plotted for adult nematodes of the given genotype and expressing the transgene indicated beneath the graph. White bars indicate wild-type animals, whereas black bars indicate kel-8 mutants. AU, arbitrary units. Error bars are SEM for all graphs. ***p < 0.0001 and **p < 0.001 compared with wild type expressing the corresponding transgene by Student's t test. n = 15–20 animals for each genotype–transgene combination.
Figure 2.

KEL-8 encodes a member of the Kelch Superfamily. (A) The predicted intron/exon gene structure of kel-8 based on cDNA sequence is shown at top. Black boxes indicate coding sequences, whereas white boxes indicate untranslated regions. At bottom is the predicted protein domain structure, including the BTB, BACK, and six Kelch repeats. Amino acid identities and similarities to human Kelch-like 8 (KHL8) for each domain are shown. The molecular nature of the od38 mutation is indicated. (B) Phylogenetic tree for KEL-8 and various Kelch proteins in the human and C. elegans genomes. KEL-8 is most similar to human Kelch-like 8. (C) Amino acid alignment of KEL-8, human KHL8, and Drosophila KELCH. Black highlighting indicates identities, and gray highlighting indicates similarities. Overlines indicate specific protein domains. (D) Fluorescence from GLR-1::GFP in the ventral cord bundle of kel-8 mutants. (E) kel-8 mutants rescued with genomic cosmid W02G9 containing the kel-8 locus. (F) kel-8 mutants rescued cell autonomously with a Pglr-1::kel-8 transgene containing wild-type kel-8 cDNA fused to the glr-1 promoter. Bar, 5 μm.
The change in GLR-1 ventral cord accumulation in kel-8 mutants could reflect a general defect in protein trafficking, or in synapse formation per se. To test this possibility, we examined the localization of three other synaptic proteins: SNB-1 (synaptobrevin), UNC-43 (CaMKII), and LIN-10 (Mint2). We previously generated transgenes that express GFP-tagged versions of these proteins using the glr-1 promoter (Rongo et al., 1998; Rongo and Kaplan, 1999; Shim et al., 2004). We introduced these transgenes into wild-type and kel-8 mutant animals to observe the subcellular localization of their protein products in the ventral cord neurites. SNB-1::GFP is localized to presynaptic terminals (Rongo et al., 1998; Nonet, 1999), and we found no significant change in the size and number of SNB-1::GFP-labeled terminals in kel-8 mutants compared with wild type (Figure 1, C, D, I, and J). UNC-43::GFP and LIN-10::GFP colocalize with GLR-1 at postsynaptic elements in the ventral cord (Rongo and Kaplan, 1999; Glodowski et al., 2005; Umemura et al., 2005), and we found no significant difference in the size or number of either UNC-43::GFP (Figure 1, E, F, I, and J) or LIN-10::GFP (Figure 1, G–J) clusters in kel-8 mutants compared with wild type. These results indicate that the accumulation of GLR-1 in kel-8 mutants is not because of gross defects in trafficking or synapse formation in the GLR-1-expressing neurons.
KEL-8 Encodes a BTB-Kelch-like Protein
We identified the kel-8 gene as predicted coding region W02G9.2 by genetic mapping and transformation rescue (Figure 2, A, D, and E; see Materials and Methods for details). The kel-8 gene encodes a 690-amino acid protein in the BTB-Kelch superfamily that is predicted to contain a BTB domain, a BACK domain, and six Kelch repeats (Bork and Doolittle, 1994; Ahmad et al., 1998; Adams et al., 2000; Stogios and Prive, 2004). Multiple complete cDNA clones of kel-8 were obtained from the C. elegans expressed sequence tag project. Multiple subtypes of BTB-Kelch-like proteins have been identified in vertebrates based on their sequence similarity to Drosophila KELCH. Of the vertebrate subtypes, we found that the KEL-8 gene product shows the highest similarity to Kelch-like 8 (Figure 2B). Two other BTB-Kelch-like proteins, KEL-1 and SPE-26, have been identified in the C. elegans genome, and a BLAST search of the C. elegans genome revealed five additional genes encoding BTB-Kelch-related proteins. Based on the similarity to Kelch-like 8, we named the W02G9.2 gene kel-8 for kelch-repeat containing protein 8 (Figure 2C).
To determine the molecular nature of the kel-8(od38) allele, we sequenced genomic DNA from kel-8 mutants. The mutation alters the conceptual translation of KEL-8 protein from arginine to an Opal stop codon at amino acid 102, resulting in a protein lacking all functional domains (Figure 2A). Thus, kel-8(od38) is a likely null allele in the kel-8 gene.
KEL-8 Negatively Regulates GLR-1 Function
KEL-8 negatively regulates GLR-1 abundance, which could result in increased levels of GLR-1 on the membrane surface of kel-8 mutants. The levels of GLR-1 on the postsynaptic membrane can be monitored through changes in behavior (Burbea et al., 2002; Juo and Kaplan, 2004). C. elegans spend the majority of their time moving forward; however, this forward locomotion is occasionally halted by spontaneous reversals in the direction of movement, and GLR-1 signaling positively regulates these spontaneous reversals (Zheng et al., 1999; Mellem et al., 2002). C. elegans backward locomotion can also be induced by stimulating the mechanosensory neuron ASH, which makes glutamatergic connections to the GLR-1-expressing interneurons (White et al., 1986; Kaplan and Horvitz, 1993). Mutants with reduced GLR-1 signaling have a lower frequency of spontaneous reversal and are nose-touch insensitive, whereas mutants with increased GLR-1 signaling or higher levels of cell surface GLR-1 have a higher frequency of spontaneous reversal (Hart et al., 1995; Maricq et al., 1995). Wild-type animals reverse direction in response to nose-touch with a frequency of ∼83% (20 animals, 10 trials per animal), whereas glr-1 mutants only reverse direction in response to nose-touch with a frequency of ∼7% (Figure 3A). We found that kel-8 mutants responded to nose-touch with a frequency of ∼82%. We also examined spontaneous reversal frequency. Wild-type animals spontaneously reversed ∼2.6 times per minute (20 animals, 5 min trial per animal), whereas glr-1 mutants only spontaneously reverse direction ∼1.4 times per minute (Figure 3B). We found that kel-8 mutants spontaneously reversed direction ∼4.1 times per minute, a frequency that was statistically greater than that for wild-type animals (Figure 3B). To determine whether the increased reversal frequency is because of increased GLR-1 in kel-8 mutants, we examined glr-1; kel-8 double mutants. The double mutants behave similarly to glr-1 single mutants, demonstrating that glr-1 suppresses the behavioral defects of kel-8 and that the increased reversal frequency in kel-8 mutants requires GLR-1 function. Our results suggest that the increase in GLR-1::GFP abundance in kel-8 mutants correlates with an increase in GLR-1-mediated locomotion behavior and is consistent with increased synaptic strength.
Figure 3.
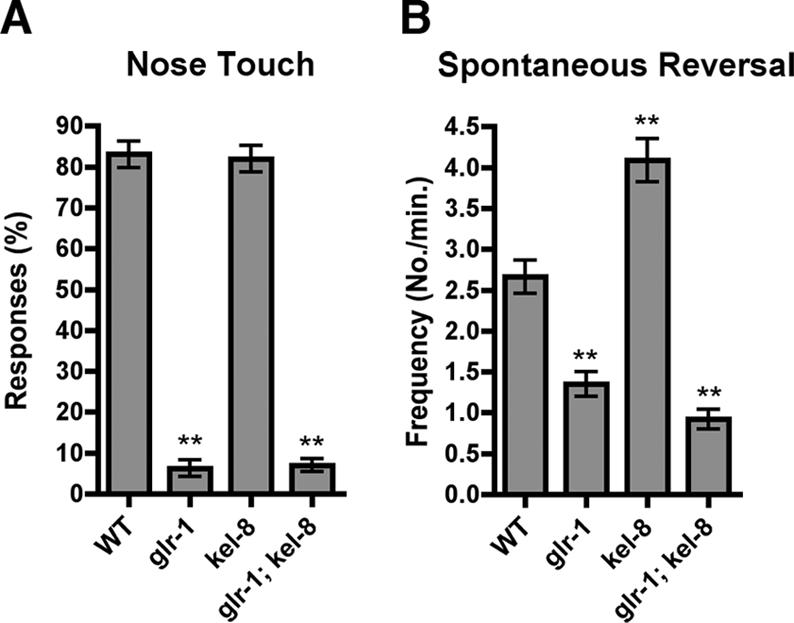
KEL-8 negatively regulates GLR-1 function. (A) The mean nose-touch sensitivity (percentage of 10 total trials per animal in which the animal reversed direction upon forward collision with an eyelash) is plotted for the given genotype. WT, wild-type animals. (B) The mean spontaneous reversal frequency (number of reversals per minute over a 5-min period) is plotted for the given genotype. Error bars are SEM for both graphs. **p < 0.01 for comparisons to wild type using analysis of variance (ANOVA) and Dunnett's multiple comparison test. n = 20 animals for each genotype.
KEL-8 Is Expressed in Neurons
Mutants for kel-8 have no obvious morphological or developmental defects and have apparently regular, coordinated locomotion. We reasoned that the relatively specific phenotype of kel-8 mutants might be because of limited expression of the gene. We generated a transcriptional reporter by fusing 5.0 kb of upstream sequences and the entire genomic kel-8 coding sequences in frame to GFP sequences. The resulting kel-8::gfp transgene was introduced into nematodes, where it expressed KEL-8::GFP protein in a subset of neurons (Figure 4A). We also introduced the kel-8::gfp transgene into nematodes expressing GLR-1::RFP (monomeric red fluorescent protein [RFP]; Campbell et al., 2002) from a previously described transgene (Figure 4, B and C; Glodowski et al., 2005). We found that KEL-8::GFP and GLR-1::RFP were expressed in the same subset of interneurons, including AVA, AVB, AVD, and AVE (Figure 4, G–I) as well as PVC (Figure 4, J–L), suggesting that KEL-8 functions in the same cells as GLR-1. To confirm this possibility, we made a transgene, Pglr-1::kel-8, containing kel-8 cDNA sequences under the control of the glr-1 promoter. We introduced Pglr-1::kel-8 into kel-8 mutants and found that mutant nematodes carrying the Pglr-1::kel-8 extrachromosomal array (4 independent lines, 15–25 animals examined per line) were rescued for the GLR-1 localization phenotype (Figure 2F), whereas their untransformed siblings were not (Figure 2D; our unpublished data), indicating that KEL-8 functions cell autonomously.
Figure 4.

KEL-8 is expressed in neurons and localized to clusters in the ventral cord dendrites. KEL-8::GFP (A, D, G, and J) and GLR-1::RFP (B, E, H, and K) fluorescence were observed throughout the entire animal (A–C), along ventral cord neurites (D–F), along lateral head ganglia (G–I), and in the lumbar ganglia of the tail (J–L). KEL-8::GFP is observed in the nerve ring (NR), lateral and ventral ganglia (L&VG), the ventral nerve cord (VNC), and the lumbar ganglia (LG) of the tail (merged image in C). Intestinal autofluorescence (IA), which is nonspecific and does not indicate expression from either transgene, is observed throughout the midbody. Along the ventral cord, KEL-8::GFP is localized to clusters adjacent to clusters of GLR-1::RFP along the ventral cord (merged image in F). KEL-8::GFP and GLR-1::RFP are expressed in the same lateral and lumbar cells bodies (merged images in I and L, respectively; cell identities are indicated). (M and N) GLR-1::CFP fluorescence was observed in kel-8 mutants (M) or kel-8 mutants (N) rescued cell autonomously with a Pglr-1::kel-8::yfp transgene containing wild-type kel-8 cDNA fused in frame to YFP. The KEL-8::YFP chimeric protein functions to rescue the kel-8 mutant phenotype in these neurons. Bars, 20 μm (A–C), 5 μm (D–L), and 10 μm (M and N).
We observed that KEL-8::GFP was localized to clusters along the ventral cord neurites (Figure 4D). Coexpression of KEL-8::GFP and GLR-1::RFP resulted in KEL-8::GFP clusters that were localized adjacent to GLR-1::RFP clusters. We also generated a transgene, Pglr-1::kel-8::yfp, that expresses (via the glr-1 promoter) KEL-8 protein fused at its carboxy terminus to YFP. We introduced Pglr-1::kel-8::yfp as an extra-chromosomal transgene into kel-8 mutant nematodes that express an integrated glr-1::cfp transgene. Although kel-8 mutants accumulate large accretions of GLR-1::CFP in the absence of exogenous KEL-8 protein (Figure 4M), sibling kel-8 mutants that express KEL-8::YFP from a Pglr-1::kel-8::yfp transgene have properly-localized GLR-1::CFP (4 independent lines, 15–20 animals examined per line; Figure 4N). These results indicate that the KEL-8 fusion protein is functional.
Drosophila KELCH homodimerizes in vivo via the BTB domain, and BTB dimerization is sufficient for endogenous KELCH protein to localize an exogenous KELCH BTB domain to the actin-rich ring canals (Robinson and Cooley, 1997). To determine whether the BTB domain is playing a similar role in KEL-8, we made a transgene, kel-8(1-219)::gfp, that expresses the amino terminus of KEL-8 (amino acids 1–219, which contains the BTB domain) in frame with GFP under the kel-8 promoter. We introduced kel-8(1-219)::gfp into kel-8 mutants but could not detect KEL-8(1-219)::GFP protein (our unpublished data). We then introduced wild-type endogenous kel-8(+) and observed the same expression and punctate localization pattern for KEL-8(1-219)::GFP protein in ventral cord neurites (our unpublished data) that we observed for full-length KEL-8::GFP (4 of 4 transgenic lines). These results indicate that full-length endogenous KEL-8 stabilizes and localizes the exogenous BTB domain protein and suggest that the KEL-8 BTB domain dimerizes in vivo.
KEL-8 Is Required for Ubiquitin-mediated Degradation of GLR-1
KEL-8 negatively regulates GLR-1 abundance, and one mechanism by which this could occur is ubiquitination. Because of a limiting cellular concentration of monoubiquitin, overexpression of Myc epitope-tagged ubiquitin (MUb) by a nuIs89 transgene has been shown to negatively regulate GLR-1 abundance in neurites (Papa and Hochstrasser, 1993; Hegde et al., 1997; Swaminathan et al., 1999; Burbea et al., 2002). Because kel-8 mutants accumulate GLR-1 (Figure 5E), KEL-8 could be needed for ubiquitin-mediated turnover of GLR-1. To test this idea, we introduced nuIs89 into kel-8 mutants and found that mutations in kel-8 partially block the turnover of GLR-1 because of overexpressed ubiquitin (Figure 5F). Our quantification of GLR-1 cluster size (area) and number per length of ventral cord support these conclusions (Figure 5I). Overexpression of ubiquitin in wild-type animals results in fewer GLR-1 clusters, although cluster size remains unchanged. By contrast, overexpression of ubiquitin in kel-8 mutants does not result in fewer GLR-1 clusters. However, whereas the size of GLR-1 clusters in nuIs89[MUb]; kel-8 nematodes is larger than in nuIs89[MUb] nematodes, it is not as large as in kel-8 single mutants. These results demonstrate that KEL-8 is required for part of the ubiquitin-mediated degradation of GLR-1. They also suggest that overexpression of ubiquitin can partially decrease GLR-1 levels by a second mechanism that is independent of KEL-8.
Figure 5.

KEL-8 is required for ubiquitin-mediated turnover of GLR-1. (A–G) GLR-1::GFP fluorescence was observed along ventral cord dendrites of wild-type nematodes (A), wild-type nematodes carrying a transgene that overexpresses MUb (B), unc-11 mutants (C), unc-11 mutants overexpressing MUb (D), kel-8 mutants (E), kel-8 mutants overexpressing MUb (F), and unc-11 kel-8 double mutants (G). Overexpression of ubiquitin results in decreased GLR-1 levels, whereas unc-11 and kel-8 mutations result in increased levels of GLR-1. Moreover, kel-8 mutations, like unc-11 mutations, partially block the downregulation of GLR-1 by overexpression of ubiquitin. Double mutants for kel-8 and unc-11 accumulate GLR-1 in a similar manner to kel-8 mutants. Bar, 5 μm. (H) Quantification of the GLR-1 cluster area and number (per 10 μm of neurite length) comparing unc-11 and kel-8 single and double mutants. (I) Quantification of the GLR-1 cluster area and number comparing kel-8 in the presence or absence of overexpressed ubiquitin (MUb transgene). Error bars are SEM for all graphs. *p < 0.05 and ***p < 0.001 for intergenotype comparisons using ANOVA and Bonferroni multiple comparison test. n = 20 animals for each genotype.
Interestingly, mutations in the AP180 clathrin adaptor protein UNC-11 result in an increase of GLR-1 abundance (Burbea et al., 2002; Figure 5C), suggesting that clathrin-mediated endocytosis is required for the turnover of GLR-1 protein. Moreover, mutations in unc-11, like in kel-8, can partially prevent the turnover of GLR-1 because of overexpressed MUb (Burbea et al., 2002; Figure 5D). Thus, KEL-8 and UNC-11 could act together or could act independently in parallel processes to regulate GLR-1 abundance. If KEL-8 and UNC-11 work in parallel processes, then GLR-1 localization defects in kel-8 unc-11 double mutants should be dramatically more severe than in either single mutant. Instead, unc-11 kel-8 double mutants contained GLR-1 accretions that were only slightly larger compared with those found in kel-8 or unc-11 single mutants (Figure 5, G and H), suggesting that a mutation in one of these genes can partially occlude the effect of mutations in the other. Our results are most consistent with UNC-11 and KEL-8 working together in the same linear pathway to regulate GLR-1 abundance and suggest that KEL-8 is required for the ubiquitin-mediated turnover of GLR-1.
KEL-8 Interacts with the CUL-3 Subunit of Cullin 3 Ubiquitin Ligases
Several BTB proteins have been shown to be substrate receptors for Cullin 3 ubiquitin ligases (CDL3s) (Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003; Kobayashi et al., 2004; Zhang et al., 2004). Because KEL-8 contains a BTB domain, it might interact directly with CUL-3, the C. elegans Cullin 3 orthologue. To test whether these proteins interact, we used a transient expression system in COS-7 cells. We cotransfected either GST::CUL-3 protein or GST alone with FLAG-tagged KEL-8 protein (FLAG::KEL-8) in COS-7 cells. We detected GST, GST::CUL-3, and FLAG::KEL-8 on the same blot using an antibody raised against GST::KEL-8, which recognizes both GST and KEL-8 epitopes. Using glutathione agarose beads, we could pull down KEL-8 from COS-7 lysates cotransfected with GST::CUL-3 but not GST alone (Figure 6A). In addition, we immunoprecipitated GST::CUL-3 but not GST alone from cotransfected COS-7 lysates using anti-FLAG antibodies (Figure 6B). Two α-helices in the CUL-3 amino terminus make important contacts with the BTB domain of MEL-26, and a mutation in these helices reduces the interaction (Zheng et al., 2002; Xu et al., 2003). We engineered the L49A/E52A (called H2) mutation into GST::CUL-3 and tested whether the mutant protein could pull down FLAG::KEL-8 when cotransfected. We found that the amount of FLAG::KEL-8 pulled down by the GST::CUL-3(H2) mutant protein was significantly reduced compared with GST::CUL-3 wild-type protein (Figure 6A). In addition, less mutant GST::CUL-3(H2) protein was coimmunoprecipitated with FLAG::KEL-8 by anti-FLAG antibodies compared with wild-type GST::CUL-3 (Figure 6B). Finally, KEL-8 and CUL-3 can directly interact in a yeast two-hybrid system (Figure 6C). These results suggest that KEL-8 interacts with CUL-3 in analogous manner to other BTB–Cullin interactions.
Figure 6.

KEL-8 interacts with CUL-3 Cullin. (A) GST::CUL-3 and FLAG::KEL-8 were cotransfected into COS-7 cells, which were solubilized in RIPA buffer. GST affinity chromatography was performed, and bound proteins were detected by Western blotting (WB) with an antisera that recognizes both GST and KEL-8. GST::CUL-3 but not GST alone pulls down KEL-8 protein. GST::CUL-3(H2), which reduces the affinity of CUL-3 for other BTB proteins, pulls down reduced amounts of KEL-8. (B) Immunoprecipitations were performed with anti-FLAG antisera, and coimmunoprecipitated proteins were detected by Western blotting with an anti-GST antisera. FLAG::KEL-8 coprecipitates GST::CUL-3 but not GST alone. Reduced amounts of GST::CUL-3(H2) (only detectable with significantly longer chemiluminescence exposures than the blot shown) are coprecipitated compared with wild-type GST::CUL-3. For both A and B, arrowheads indicate the specific pulled down proteins. Brackets above the gels indicate the contransfected constructs. “Load” indicates 2.5% of the original lysate. Similar results were observed in six separate transfection experiments. (C) AH109 yeast expressing CUL-3 fused to Gal4 DNA binding domain (DB CUL-3) and KEL-8 fused to an activation domain (AD KEL-8) are able to support growth on two independent yeast two-hybrid reporter genes (HIS3 and ADE2). The DB CUL-3 and AD KEL-8 constructs, present individually in AH109 and streaked out in separate quadrants, are unable to support growth. (D–H) GLR-1::GFP clusters were visualized in wild-type nematodes (D), nematodes expressing full-length CUL-3 protein (E), nematodes expressing amino-terminal residues 1–500 (F), nematodes expressing carboxy-terminal residues 501–777 (G), and rfl-1 mutants (H). Expression of either CUL-3(1-500) or CUL-3(501-777), or a mutation in the Nedd8-activating enzyme RFL-1 results in the accumulation of GLR-1. Bar, 5 μm.
Known mutants for cul-3 do not exist and RNA interference of cul-3 results in embryonic lethality because of failed mitosis (Kurz et al., 2002). As an alternative approach for testing CUL-3 function in vivo, we generated dominant negative versions of CUL-3. Cullins have a modular structure, with an amino-terminal domain that interacts with F-box and BTB domain proteins and a carboxy-terminal domain that interacts with RING domain proteins and E2 ubiquitin-conjugating enzymes. Partial CUL-3 proteins can act as dominant negatives (Zhang et al., 2005). Thus, to interfere with endogenous CUL-3 function, we generated Pglr-1::cul-3(1-500) and Pglr-1::cul-3(501-777) transgenes that express the amino terminus and carboxy terminus of CUL-3, respectively, using the glr-1 promoter. We also generated a Pglr-1::cul-3(full length) transgene that expresses full-length CUL-3 protein as a control. We introduced the transgenes into nematodes separately and found that expression of full-length CUL-3 had little effect on GLR-1 clusters (4 independent lines, 15–25 animals examined per line; Figure 6, D and E). By contrast, expression of either the CUL-3 amino terminus (2 independent lines, 15–25 animals per line; Figure 6F) or carboxy terminus (2 independent lines, 15–25 animals per line; Figure 6G) resulted in accumulation of GLR-1, similar to kel-8 mutants.
As an independent test for the role of CUL-3 in KEL-8 function, we reasoned that neddylation should be required for GLR-1 degradation. Neddylation is the attachment of the ubiquitin-like NEDD8 specifically to cullin family members and is needed for the assembly and function of Cullin-RING ligases (Willems et al., 2004; Petroski and Deshaies, 2005). We took advantage of the temperature-sensitive rfl-1(or198ts) mutant, which is inviable at higher temperatures because of the decreased activity of the RFL-1 Nedd8-activating enzyme (Kurz et al., 2002). We introduced GLR-1::GFP into rfl-1(or198ts) and found that GLR-1::GFP accumulates in viable rfl-1 mutants at 15°C (Figure 6H). Together, our results suggest that KEL-8 and CUL-3 form a cullin complex, CDL3KEL-8, that is essential for proper degradation of GLR-1 receptors.
DISCUSSION
In this study, we used a genetic approach to identify kel-8, a gene required to regulate GLR-1 synaptic abundance. First, we showed that KEL-8 is a member of the Kelch superfamily of proteins that regulates the levels of GLR-1 but not other synaptic proteins. Second, we demonstrated that KEL-8 is expressed and required in neurons and is localized in clusters that are adjacent to GLR-1-containing postsynaptic elements. Finally, we found that KEL-8 binds to the CUL-3 subunit of the CDL3 ubiquitin ligase and is required for the ubiquitin-mediated turnover of GLR-1. Based upon these results, we propose that KEL-8 serves as a substrate receptor for CDL3 and that this interaction with the ubiquitin–proteasome apparatus is critical for regulating glutamate receptor abundance and synaptic signaling.
KEL-8 Regulates GLR-1 Synaptic Levels
Regulated turnover of GLR-1 subunits was first observed in unc-11/clathrin adaptor AP180 mutants, where endocytosis is blocked (Burbea et al., 2002). Ubiquitination of several critical lysines in the carboxy-terminal tail of GLR-1 results in the endocytosis and degradation of GLR-1. The ubiquitination and turnover of GLR-1 can be facilitated by elevating the levels of free ubiquitin using the nuIs89[MUb] transgene, and mutations in unc-11, components of the anaphase-promoting complex (APC), and kel-8 block some of the effects of elevated ubiquitin levels, suggesting that these proteins are needed for ubiquitin-mediated turnover of GLR-1 (Burbea et al., 2002; Juo and Kaplan, 2004). The effect of overexpressing ubiquitin from the nuIs89[MUb] transgene cannot be entirely mediated through KEL-8, because we would expect that nuIs89[MUb]; kel-8 nematodes would have a similar number and size of GLR-1 clusters to kel-8 single mutants. Whereas nuIs89[MUb]; kel-8 animals have a similar number of GLR-1 clusters relative to kel-8 single mutants, by contrast, the GLR-1 clusters of nuIs89[MUb]; kel-8 animals are of an intermediate size compared with either nuIs89[MUb] animals or kel-8 mutants alone. One explanation is that overexpression of ubiquitin might independently affect different facets (e.g., cluster size versus number) of GLR-1 synaptic localization and that KEL-8 mediates this effect on a specific facet: cluster number. Alternatively, all facets of GLR-1 localization might be determined solely by GLR-1 abundance, such that the large size of GLR-1 clusters in kel-8 mutants might be a secondary consequence resulting from the increased synaptic abundance of GLR-1. In this model, overexpression of ubiquitin results in decreased GLR-1 abundance and thus decreased cluster number. Mutations in kel-8 can only partially block this effect, allowing enough of an increase in GLR-1 abundance to raise the detectable number of GLR-1 clusters but not enough to enlarge the size of GLR-1 clusters. We think that the latter model is more likely for several reasons. First, the shifts in both GLR-1 cluster size and number between nuIs89[MUb] animals, kel-8 mutants, and nuIs89[MUb]; kel-8 doubles are present at all points along their respective distributions, suggesting that ubiquitination and KEL-8 do not regulate subsets of clusters but rather regulate the global population of clusters (our unpublished data). Second, we see an increase in GLR-1 cluster size in transgenic lines that express GLR-1 to higher levels than the nuIs25 transgene used in our study, suggesting that as GLR-1 abundance increases, the excess receptors spread beyond the postsynaptic region and appear as large clusters (our unpublished data).
KEL-8 cannot be the sole factor through which overexpressed ubiquin mediates it affect on GLR-1 localization. Indeed, mutations in components of the APC result in a subtle (∼20%) increase in GLR-1 abundance, although GLR-1 is not the direct ubiquitination target of the APC (Juo and Kaplan, 2004). Thus, the subtle changes in GLR-1 abundance in APC mutants might be a secondary consequence of disrupted APC function. The LIN-23 ubiquitin ligase has recently been shown to regulate GLR-1 abundance by ubiquitinating BAR-1 β-catenin (Dreier et al., 2005). BAR-1::GFP levels are elevated in lin-23 mutants, and bar-1 mutations can partially suppress the GLR-1 accumulation observed in lin-23 mutants. Mutations that stabilize BAR-1 result in a subtle (∼20%) increase in GLR-1 abundance, suggesting that Wnt signaling plays a minor role in regulating GLR-1 abundance. In contrast to APC and Wnt signaling mutants, kel-8 mutants showed a dramatic enough increase (200–300% compared with wild type) to allow us to identify kel-8 in a forward genetic screen. Moreover, KEL-8 is expressed specifically in GLR-1-containing neurons, whereas APC and Wnt signaling components are broadly expressed. We currently do not know the substrate for KEL-8, and we have not been able to detect a binding interaction between KEL-8 and GLR-1 (our unpublished data), although the transient nature of the ubiquitination reaction makes this difficult to interpret. Together, our findings suggest that KEL-8 is a major, dedicated regulator of GLR-1 abundance.
What is the physiological relevance of KEL-8 acting to negatively regulate GLR-1 at the synapse? In nematode interneurons, GLR-1 signals to regulate the direction of movement by triggering a reversal in direction either in response to nose-touch stimuli or occasionally spontaneously (Hart et al., 1995; Maricq et al., 1995; Zheng et al., 1999; Mellem et al., 2002). Spontaneous reversals seem to allow the animals to change their overall direction of travel. Elevated levels of GLR-1 protein or GLR-1 activity result in an increase in spontaneous reversal frequency, suggesting that GLR-1 abundance is regulated to control locomotion behavior (Zheng et al., 1999; Mellem et al., 2002). Similar to these observations, we found that kel-8 mutants have a high frequency of spontaneous reversal, consistent with the observed increase in GLR-1 synaptic abundance in these mutants. Because kel-8 mutants have increased GLR-1 protein levels, our results suggest that a significant fraction of GLR-1 receptors is normally degraded. We speculate that the physiological role of KEL-8 is to regulate locomotory behavior by regulating GLR-1 synaptic levels.
KEL-8 Is Localized Adjacent to GLR-1
We found that KEL-8::GFP is localized in clusters along the ventral cord neurites. A KEL-8::GFP chimeric protein containing the BTB domain alone is also localized in clusters; however, the localization of this chimera requires an endogenous copy of wild-type kel-8. This observation is similar to that of Drosophila KELCH protein, which is localized to actin-rich ring canals (Robinson and Cooley, 1997; Kelso et al., 2002). The BTB domain of KELCH is also localized to ring canals via its interaction with endogenous KELCH through BTB homodimerization (Robinson and Cooley, 1997). Because the KEL-8 BTB domain also requires endogenous kel-8 for proper localization, we speculate that KEL-8 homodimerizes through its BTB domains.
KEL-8::GFP is localized to clusters that are adjacent to the postsynaptic clusters of GLR-1::RFP. One possible explanation for the proximal but not overlapping colocalization of KEL-8 and GLR-1 is that KEL-8 is at presynaptic terminals. We think this unlikely for several reasons. KEL-8::GFP is expressed in the same neurons as GLR-1::RFP. Moreover, expression of kel-8 cDNA by the glr-1 promoter rescues the kel-8 mutant phenotype, indicating that KEL-8 functions in the same cells as GLR-1. The GLR-1-expressing neurons make some interneuron-to-interneuron synaptic connections, accounting for ∼38% of clusters in the anterior portion of the ventral cord (White et al., 1986; Burbea et al., 2002). By contrast, >90% of GLR-1 clusters are adjacent to KEL-8 clusters, which is far more than can be explained if KEL-8 were at the presynaptic terminals of the interneuron-to-interneuron synapses.
Interestingly, recent studies of mammalian AMPARs indicate that these receptors move laterally and undergo endocytosis in membrane regions that are adjacent to the postsynaptic density (Racz et al., 2004). Thus, one possible explanation for KEL-8 localization adjacent to GLR-1 clusters is that KEL-8 is localized to tangential sites of endocytosis for GLR-1 receptors, similar to what has been observed for mammalian receptors.
KEL-8 Is a Substrate Receptor for CDL3
KEL-8 binds to a CUL-3 ubiquitin ligase subunit. BTB domain proteins have recently been shown to function as substrate receptors for CDL3 ubiquitin ligases (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003; Zhang et al., 2004). The BTB domain of this new class of substrate receptor (e.g., MEL-26) interacts with the Cullin repeats of Cul3, analogous to the manner by which Skp1 interacts with Cul1 in the CDL1/SCF complex. CUL-3 in turn recruits RING domain proteins and E2-conjugating enzymes into the complex, where the conjugating enzymes transfer ubiquitin to the substrate. Cullin-RING ligases are thought to assemble with numerous different substrate receptors, allowing them to recognize a large repertoire of substrates; however, the characterization of most Cullin-RING ligases has been limited to their role in cell division (Willems et al., 2004; Petroski and Deshaies, 2005). An exception is the BTB-Kelch protein Keap1, which interacts with Cullin 3 and regulates the levels of the transcription factor Nrf2 in response to oxidative stress (Zhang et al., 2004). There are 10 BTB-Kelch proteins in the C. elegans genome and 125 in the human genome. However, how many of these proteins assemble with Cullin-RING ligases, and the nature of their biological functions remains unclear. We speculate that KEL-8 represents a new group of BTB-Kelch superfamily members that function as Cullin substrate receptors and that many of these proteins will have critical postmitotic functions. In postmitotic neurons, rapid degradation of many synaptic proteins has been observed, and Cullin-RING ligases such as CDL3KEL-8 might provide a mechanism underlying some forms of synaptic plasticity.
Acknowledgments
We thank Alivia Dey for assistance with the yeast two-hybrid plasmids. We thank A. Fire, A. Fraser (Sanger Institute, London, United Kingdom), W. Harper, R. Herman (Caenorhabditis Genetics Center [CGC]), J. Kaplan, Y. Kohara, V. Maricq, J. Mendel, C. Mello, M. Nonet, L. Pintard, T. Stiernagle (CGC), R. Tsien, and L. Xu for reagents and strains. We thank Doreen Glodowski, Barth Grant, Bonnie Firestein, Junho Lee, and Ruth Steward for comments on the manuscript. C. R. is a Pew Scholar in the Biomedical Sciences. Funding was provided by the National Institutes of Health Grant R01 NS42023 and a Grant-In-Aid from the American Heart Association.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–08–0794) on January 4, 2006.
References
- Aarts, M. M., and Tymianski, M. (2004). Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr. Mol. Med. 4, 137–147. [DOI] [PubMed] [Google Scholar]
- Adams, J., Kelso, R., and Cooley, L. (2000). The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 10, 17–24. [DOI] [PubMed] [Google Scholar]
- Ahmad, K. F., Engel, C. K., and Prive, G. G. (1998). Crystal structure of the BTB domain from PLZF. Proc. Natl. Acad. Sci. USA 95, 12123–12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork, P., and Doolittle, R. F. (1994). Drosophila kelch motif is derived from a common enzyme fold. J. Mol. Biol. 236, 1277–1282. [DOI] [PubMed] [Google Scholar]
- Bredt, D. S., and Nicoll, R. A. (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40, 361–379. [DOI] [PubMed] [Google Scholar]
- Brenner, S. (1974). The genetics of C. elegans. Genetics 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie, P. J., Madsen, D. M., Zheng, Y., Mellem, J., and Maricq, A. V. (2001). Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J. Neurosci. 21, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbea, M., Dreier, L., Dittman, J. S., Grunwald, M. E., and Kaplan, J. M. (2002). Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron 35, 107–120. [DOI] [PubMed] [Google Scholar]
- Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 99, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies, R. J. (1999). SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell. Dev. Biol. 15, 435–467. [DOI] [PubMed] [Google Scholar]
- Dingledine, R., Borges, K., Bowie, D., and Traynelis, S. F. (1999). The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61. [PubMed] [Google Scholar]
- Dong, H., O'Brien, R. J., Fung, E. T., Lanahan, A. A., Worley, P. F., and Huganir, R. L. (1997). GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386, 279–284. [DOI] [PubMed] [Google Scholar]
- Dreier, L., Burbea, M., and Kaplan, J. M. (2005). LIN-23-mediated degradation of beta-catenin regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Neuron 46, 51–64. [DOI] [PubMed] [Google Scholar]
- Ehlers, M. D. (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525. [DOI] [PubMed] [Google Scholar]
- Ehlers, M. D. (2004). Deconstructing the axon: Wallerian degeneration and the ubiquitin-proteasome system. Trends Neurosci. 27, 3–6. [DOI] [PubMed] [Google Scholar]
- Esteban, J. A., Shi, S. H., Wilson, C., Nuriya, M., Huganir, R. L., and Malinow, R. (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143. [DOI] [PubMed] [Google Scholar]
- Firestein, B. L., Brenman, J. E., Aoki, C., Sanchez-Perez, A. M., El-Husseini, A. E., and Bredt, D. S. (1999). Cypin: a cytosolic regulator of PSD-95 postsynaptic targeting. Neuron 24, 659–672. [DOI] [PubMed] [Google Scholar]
- Furukawa, M., He, Y. J., Borchers, C., and Xiong, Y. (2003). Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat. Cell Biol. 5, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Geyer, R., Wee, S., Anderson, S., Yates, J., and Wolf, D. A. (2003). BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol. Cell 12, 783–790. [DOI] [PubMed] [Google Scholar]
- Glodowski, D. R., Wright, T., Martinowich, K., Chang, H. C., Beach, D., and Rongo, C. (2005). Distinct LIN-10 domains are required for its neuronal function, its epithelial function, and its synaptic localization. Mol. Biol. Cell 16, 1417–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, K. L., and Dixon, J. E. (1991). Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 192, 262–267. [DOI] [PubMed] [Google Scholar]
- Haglund, K., Di Fiore, P. P., and Dikic, I. (2003). Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 28, 598–603. [DOI] [PubMed] [Google Scholar]
- Hart, A. C., Sims, S., and Kaplan, J. M. (1995). Synaptic code for sensory modalities revealed by C. elegansGLR-1 glutamate receptor. Nature 378, 82–84. [DOI] [PubMed] [Google Scholar]
- Hegde, A. N. (2004). Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog. Neurobiol. 73, 311–357. [DOI] [PubMed] [Google Scholar]
- Hegde, A. N., Inokuchi, K., Pei, W., Casadio, A., Ghirardi, M., Chain, D. G., Martin, K. C., Kandel, E. R., and Schwartz, J. H. (1997). Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89, 115–126. [DOI] [PubMed] [Google Scholar]
- Hicke, L., and Dunn, R. (2003). Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell. Dev. Biol. 19, 141–172. [DOI] [PubMed] [Google Scholar]
- Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108. [DOI] [PubMed] [Google Scholar]
- Joazeiro, C. A., and Weissman, A. M. (2000). RING finger proteins: mediators of ubiquitin ligase activity. Cell 102, 549–552. [DOI] [PubMed] [Google Scholar]
- Juo, P., and Kaplan, J. M. (2004). The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr. Biol. 14, 2057–2062. [DOI] [PubMed] [Google Scholar]
- Kaplan, J. M., and Horvitz, H. R. (1993). A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 90, 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso, R. J., Hudson, A. M., and Cooley, L. (2002). Drosophila Kelch regulates actin organization via Src64-dependent tyrosine phosphorylation. J. Cell Biol. 156, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, A., Kang, M. I., Okawa, H., Ohtsuji, M., Zenke, Y., Chiba, T., Igarashi, K., and Yamamoto, M. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz, T., Pintard, L., Willis, J. H., Hamill, D. R., Gonczy, P., Peter, M., and Bowerman, B. (2002). Cytoskeletal regulation by the Nedd8 ubiquitin-like protein modification pathway. Science 295, 1294–1298. [DOI] [PubMed] [Google Scholar]
- Malenka, R. C. (2003). Synaptic plasticity and AMPA receptor trafficking. Ann. N Y Acad. Sci. 1003, 1–11. [DOI] [PubMed] [Google Scholar]
- Malinow, R. (2003). AMPA receptor trafficking and long-term potentiation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricq, A. V., Peckol, E., Driscoll, M., and Bargmann, C. I. (1995). Mechanosensory signalling in C. elegans mediated by the GLR-1 glutamate receptor. Nature 378, 78–81. [DOI] [PubMed] [Google Scholar]
- Mattson, M. P. (2003). Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol. Med. 3, 65–94. [DOI] [PubMed] [Google Scholar]
- Meldrum, B. S. (2000). Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J. Nutr. 130, 1007S–1015S. [DOI] [PubMed] [Google Scholar]
- Mellem, J. E., Brockie, P. J., Zheng, Y., Madsen, D. M., and Maricq, A. V. (2002). Decoding of polymodal sensory stimuli by postsynaptic glutamate receptors in C. elegans. Neuron 36, 933–944. [DOI] [PubMed] [Google Scholar]
- Moghaddam, B. (2003). Bringing order to the glutamate chaos in schizophrenia. Neuron 40, 881–884. [DOI] [PubMed] [Google Scholar]
- Nonet, M. L. (1999). Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J. Neurosci. Methods 89, 33–40. [DOI] [PubMed] [Google Scholar]
- Osten, P., Khatri, L., Perez, J. L., Kohr, G., Giese, G., Daly, C., Schulz, T. W., Wensky, A., Lee, L. M., and Ziff, E. B. (2000). Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron 27, 313–325. [DOI] [PubMed] [Google Scholar]
- Papa, F. R., and Hochstrasser, M. (1993). The yeast DOA4 gene encodes a deubiquitinating enzyme related to a product of the human tre-2 oncogene. Nature 366, 313–319. [DOI] [PubMed] [Google Scholar]
- Petroski, M. D., and Deshaies, R. J. (2005). Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 6, 9–20. [DOI] [PubMed] [Google Scholar]
- Pintard, L., et al. (2003). The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature 425, 311–316. [DOI] [PubMed] [Google Scholar]
- Racz, B., Blanpied, T. A., Ehlers, M. D., and Weinberg, R. J. (2004). Lateral organization of endocytic machinery in dendritic spines. Nat. Neurosci. 7, 917–918. [DOI] [PubMed] [Google Scholar]
- Robinson, D. N., and Cooley, L. (1997). Drosophila Kelch is an oligomeric ring canal actin organizer. J. Cell Biol. 138, 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongo, C., and Kaplan, J. K. (1999). CaMKII regulates the density of central glutamatergic synapses in vivo. Nature 402, 195–199. [DOI] [PubMed] [Google Scholar]
- Rongo, C., Whitfield, C. W., Rodal, A., Kim, S. K., and Kaplan, J. M. (1998). LIN-10 is a shared component of the polarized protein localization pathways in neurons and epithelia. Cell 94, 751–759. [DOI] [PubMed] [Google Scholar]
- Sans, N., Racca, C., Petralia, R. S., Wang, Y. X., McCallum, J., and Wenthold, R. J. (2001). Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J. Neurosci. 21, 7506–7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, M., and Hyoung Lee, S. (2003). AMPA receptor trafficking and synaptic plasticity: major unanswered questions. Neurosci. Res. 46, 127–134. [DOI] [PubMed] [Google Scholar]
- Shim, J., Umemura, T., Nothstein, E., and Rongo, C. (2004). The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol. Biol. Cell 15, 4818–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, I., Kamboj, S., Xia, J., Dong, H., Liao, D., and Huganir, R. L. (1998). Interaction of the N-ethylmaleimide-sensitive factor with AMPA receptors. Neuron 21, 393–400. [DOI] [PubMed] [Google Scholar]
- Srivastava, S., et al. (1998). Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron 21, 581–591. [DOI] [PubMed] [Google Scholar]
- Stogios, P. J., and Prive, G. G. (2004). The BACK domain in BTB-kelch proteins. Trends Biochem. Sci. 29, 634–637. [DOI] [PubMed] [Google Scholar]
- Swaminathan, S., Amerik, A. Y., and Hochstrasser, M. (1999). The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol. Biol. Cell 10, 2583–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke, T. M. (2002). Glutamatergic mechanisms in different disease states: overview and therapeutical implications–an introduction. Amino Acids 23, 147–152. [DOI] [PubMed] [Google Scholar]
- Umemura, T., Rapp, P., and Rongo, C. (2005). The role of regulatory interactions in UNC-43 CaMKII localization and trafficking. J. Cell Sci. 118, 3327–3338. [DOI] [PubMed] [Google Scholar]
- White, J. G., Southgate, E., Thomson, J. N., and Brenner, S. (1986). The structure of the nervous system of C. elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1–340. [DOI] [PubMed] [Google Scholar]
- Willems, A. R., Schwab, M., and Tyers, M. (2004). A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 1695, 133–170. [DOI] [PubMed] [Google Scholar]
- Wood, W. B. (1988). The nematode C. elegans, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Xia, J., Zhang, X., Staudinger, J., and Huganir, R. L. (1999). Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron 22, 179–187. [DOI] [PubMed] [Google Scholar]
- Xu, L., Wei, Y., Reboul, J., Vaglio, P., Shin, T. H., Vidal, M., Elledge, S. J., and Harper, J. W. (2003). BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 425, 316–321. [DOI] [PubMed] [Google Scholar]
- Zhang, D. D., Lo, S. C., Cross, J. V., Templeton, D. J., and Hannink, M. (2004). Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24, 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D. D., Lo, S. C., Sun, Z., Habib, G. M., Lieberman, M. W., and Hannink, M. (2005). Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteosome-independent pathway. J. Biol. Chem. 280, 30091–30099. [DOI] [PubMed] [Google Scholar]
- Zheng, N., et al. (2002). Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709. [DOI] [PubMed] [Google Scholar]
- Zheng, Y., Brockie, P. J., Mellem, J. E., Madsen, D. M., and Maricq, A. V. (1999). Neuronal control of locomotion in C. elegans is modified by a dominant mutation in the GLR-1 ionotropic glutamate receptor. Neuron 24, 347–361. [DOI] [PubMed] [Google Scholar]