

Abstract
Interferon regulatory factor 3 (IRF3) plays a crucial role in mediating cellular responses to virus intrusion. The protein kinase TBK1 is a key regulator inducing phosphorylation of IRF3. The regulatory mechanisms during IRF3 activation remain poorly characterized. In the present study, we have identified by yeast two-hybrid approach a specific interaction between IRF3 and chaperone heat-shock protein of 90 kDa (Hsp90). The C-terminal truncation mutant of Hsp90 is a strong dominant-negative inhibitor of IRF3 activation. Knockdown of endogenous Hsp90 by RNA interference attenuates IRF3 activation and its target gene expressions. Alternatively, Hsp90-specific inhibitor geldanamycin (GA) dramatically reduces expression of IRF3-regulated interferon-stimulated genes and abolishes the cytoplasm-to-nucleus translocation and DNA binding activity of IRF3 in Sendai virus-infected cells. Significantly, virus-induced IRF3 phosphorylation is blocked by GA, whereas GA does not affect the protein level of IRF3. In addition, TBK1 is found to be a client protein of Hsp90 in vivo. Treatment of 293 cells with GA interferes with the interaction of TBK1 and Hsp90, resulting in TBK1 destabilization and its subsequent proteasome-mediated degradation. Besides maintaining stability of TBK1, Hsp90 also forms a novel complex with TBK1 and IRF3, which brings TBK1 and IRF3 dynamically into proximity and facilitates signal transduction from TBK1 to IRF3. Our study uncovers an essential role of Hsp90 in the virus-induced activation of IRF3.
INTRODUCTION
Toll-like receptors (TLRs) played a crucial role in innate immunity by recognizing structurally conserved bacterial and viral components termed pathogen-associated molecular patterns (PAMPs) (Medzhitov and Janeway, 1998). Eleven TLRs had been cloned in mammals, and each receptor had been reported to recognize a unique set of PAMPs (Akira and Takeda, 2004). Many studies have shown that TLR3 mediated the response to the viral-associated PAMPs (e.g., the double-stranded RNA [dsRNA]), whereas TLR4 recognized the bacterial-associated components, including lipopolysaccharide (LPS) and Gram-positive lipoteichoic acids (Takeuchi et al., 1999; Alexopoulou et al., 2001; Takeuchi and Akira, 2001). On stimulation by corresponding PAMPs, both TLR3 and TLR4 had been shown to activate interferon regulatory factor 3 (IRF3) through the MyD88-independent pathway (Akira and Takeda, 2004; Boehme and Compton, 2004; Palsson-McDermott and O'Neill, 2004; Bowie and Haga, 2005).
IRF3, originally identified in a variety of tissues based on homology with other known IRF family members, was expressed constitutively without activity in the cytoplasm; and no change in the relative levels of IRF3 mRNA was observed in virus-infected cells (Au et al., 1995). Recent investigations found that IRF3 was an important transcriptional regulator of antiviral immune response, mediating the expression of type I IFN and other Interferon stimulated genes (ISGs) (Schafer et al., 1998; Doyle et al., 2002; Elco et al., 2005). After LPS stimulation or virus infection (such as Sendai virus [Sv] or Newcastle disease virus], IRF3 was posttranslationally phosphorylated on specific serine residues in the C-terminal domain (Lin et al., 1998; Yoneyama et al., 1998; Kawai et al., 2001; Mori et al., 2004). Consequently, the phosphorylated IRF3 formed a homodimer and translocated from cytoplasm into nucleus, where it initiated its specific DNA binding and transcriptional stimulation with the help of such coactivators as CBP/p300 (Lin et al., 1998; Yoneyama et al., 1998), leading to the induction of anti-growth, anti-viral responses (Takeuchi and Akira, 2001; Doyle et al., 2002). Various genes had been identified as IRF3 immunoregulatory targets, such as ISG15, ISG56, regulated on activation normal T cell expressed and secreted (RANTES), and interferon (IFN)-β (Schafer et al., 1998; Lin et al., 1999; Grandvaux et al., 2002; Elco et al., 2005).
TANK binding kinase I (TBK1), also called nuclear factor-κB (NF-κB)-activating kinase (Tojima et al., 2000) or tumor necrosis factor receptor-associated factor (TRAF) 2-associated kinase (T2K) (Bonnard et al., 2000), was originally identified as an activator of NF-κB signaling pathway. It has recently been shown that TBK1 could phosphorylate IRF3 in vitro (Sharma et al., 2003; McWhirter et al., 2004) and played an essential role in IRF3 activation and interferon production induced by double-stranded RNA or virus in the innate immune response, as evidenced in TBK1-deficient mice or knockdown experiments (Fitzgerald et al., 2003; Perry et al., 2004). However, it remains controversial how TBK1 phosphorylates IRF3 in vivo.
Heat-shock proteins (HSPs) were a conserved protein family whose major roles seemed to promote the correct folding and assembly of target proteins as well as prevent their aggregation (Jakob et al., 1995; Agashe and Hartl, 2000; Bukau et al., 2000; Feldman and Frydman, 2000). Expressed in mammalian cells were several members that included Hsp110, -90, -70, -60, and -40 (Craig et al., 1993), among which Hsp90 was the most abundant cytosolic proteins, amounting to 1–2% of soluble protein (Welch and Feramisco, 1982; Lindquist and Craig, 1988; Welch, 1991). Unlike other HSPs, Hsp90 seemed to be more specific in terms of client selection. It had been reported that Hsp90 interacted mainly with proteins involved in transcriptional regulation and signal transduction, such as steroid hormone receptors (Giguere et al., 1986; Cadepond et al., 1991), transcriptional factor (Zou et al., 1998; Xu et al., 2004), and protein kinases (Buchner, 1999; Caplan, 1999; Mayer and Bukau, 1999; Pearl and Prodromou, 2000; Smith, 2000). The best characterized was the mechanism with which Hsp90 regulated maturation of glucocorticoid receptor (GR). In the absence of its ligand, GR was inactive and sequestered by Hsp90 in the cytoplasm. On binding to glucocorticoid, GR was released from the complex and translocated into the nucleus to promote the specific transcriptional programs (Giguere et al., 1986; Cadepond et al., 1991). A recent research demonstrated that Hsp90 acetylation regulated the chaperone-dependent activation of GR (Kovacs et al., 2005).
Several chemical products, including radicicol (RAD) and ansamycin antibiotics geldanamycin (GA) and 17-allylamino-17-demethoxygeldanamycin, had been routinely used to probe the functions of Hsp90. These drugs bound tightly to the Hsp90 ATP/ADP pocket (Prodromou et al., 1997; Stebbins et al., 1997; Schulte et al., 1998), thus preventing the process of client protein refolding and leading to proteasome-dependent degradation of these immature proteins, such as Akt, Raf, RIP, IRAK-1, and MLK3 (Schneider et al., 1996; Lewis et al., 2000; Basso et al., 2002; Zhang et al., 2004; De Nardo et al., 2005). Apparently, Hsp90 stabilized these proteins and kept them in a conformation amenable to activation under appropriate conditions.
In the present study, we started to find new candidate proteins that could regulate IRF3 activity in response to virus infection. Through yeast two-hybrid approach, we identified the specific interaction between IRF3 and chaperone Hsp90. Our data showed that C-terminal truncation mutant of Hsp90 was a strong dominant-negative inhibitor of IRF3 activation and that geldanamycin dramatically reduced the expression levels of IRF3-regulated ISGs and abolished the DNA binding activity of IRF3 in Sendai virus-infected cells. In addition, knockdown of endogenous Hsp90 by RNA interference attenuated IRF3 activation and its target gene expressions. Significantly, the virus-induced phosphorylation of IRF3 was blocked by GA, whereas GA did not affect the protein level of IRF3. Furthermore, our investigation found that TBK1 was a client protein of Hsp90 in vivo. Treatment of 293 cells with GA interfered with the interaction of TBK1 and Hsp90, resulting in TBK1 destabilization and its subsequent proteasome-mediated degradation. Further experiments revealed that, besides maintaining the stabilization of TBK1, Hsp90 formed a novel complex with TBK1 and IRF3, which brought TBK1 and IRF3 dynamically into proximity and facilitated signal transduction from TBK1 to IRF3. Together, this study uncovered an essential role of Hsp90 in the virus-induced activation of IRF3.
MATERIALS AND METHODS
Reagents
The antibodies against IRF3, hemagglutinin (HA), and His were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The monoclonal antibody (mAb) against TBK1 was purchased from Upstate Biotechnology (Lake Placid, NY). The antibody to human Hsp90 was produced by immunization of mouse with the human recombinant full-length Hsp90 protein and purified according to standard protocol. Anti-FLAG M2 mAb, anti-FLAG M2 affinity gel, protein A bead, anti-β-actin mAb, anti-Sp1 antibody, geldanamycin, and radicicol were purchased from Sigma-Aldrich (St. Louis, MO).
Plasmids and Proteins
Human IRF3 cDNA was obtained by PCR from human thymus plasmid cDNA library (Clontech, Mountain View, CA) and confirmed by sequencing. pcDNA3.1-N-Flag was described previously (Diao et al., 2005) and contained a FLAG-tag at the N-terminal sequence. pcDNA-flag-IRF3 was constructed by subcloning full-length IRF3 cDNA into pcDNA3.1-N-Flag at the EcoRI/XhoI sites. The cDNA encoding constitutively active IRF3 5D was a gift from Professor John Hiscott ((McGill University, Montreal, Quebec, Canada) and was subcloned into the pcDNA3.1-N-Flag vector. For truncation mutants of IRF3, the corresponding IRF3 mutant cDNAs were created by PCR and were cloned into pcDNA3.1-N-Flag in frame with the Flag-green fluorescence protein chimerical expression plasmids were generated by subcloning wild-type or mutated forms of IRF3 in frame into downstream region of enhanced green fluorescent protein (EGFP) in pEGFP-C1 vector (Clontech). The Gal4-BD-IRF3 was obtained by cloning IRF3 cDNA into pCMV-BD (Clontech) at the EcoRI/XhoI sites. Human Hsp90 cDNA was also obtained by PCR from the human liver cDNA library. The wild-type and truncated Hsp90 were cloned into the pcDNA3 containing an HA tag at the BamHI/XhoI sites. The Gal4-AD-Hsp90 was obtained by cloning the Hsp90 cDNA into pCMV-AD (Clontech) at the BamHI/XhoI sites. Human TBK1 cDNA was amplified from the human thymus plasmid cDNA library and cloned into pcDNA3. Mutagenesis for the replacement of the active-site lysine to alanine of TBK1 (TBK1 K38A) was performed by using a QuikChange XL site-directed mutagenesis method. The pSUPERHsp90i and control plasmid were kindly provided by Professor Kou-Juey Wu (National Yang-Ming University, Taipei, Taiwan) and experiment was carried out as described previously to knockdown endogenous Hsp90 successfully (Teng et al., 2004). All constructs were confirmed by sequencing.
Yeast Two-Hybrid Screening
Residues 1–328 of IRF3 were cloned into bait vector pGBKT7 containing the GAL4 DNA-binding domain at the N terminus. pGBKT7-IRF3 (1-328) was transformed into the yeast strain AH109, and this strain was transformed with a human thymus plasmid cDNA library in pACTII (Clontech) and selected on yeast synthetic media lacking histidine, leucine, tryptophan, and adenine. Colonies surviving after 4–6 d at 30°C were tested for β-galactosidase activity, and plasmid DNA was prepared from positive colonies and sequenced for identification of cDNA clones.
Cell Culture, Transfection, and Infection
Human embryonic kidney (HEK) 293T cells, 293 cells, and mouse RAW264.7 cells were cultured using DMEM plus 10% fetal calf serum (FCS), supplemented with antibiotics. Transfection of HEK293T cells was carried out according to the calcium phosphate precipitation method. 293 cells were infected with 80 hemagglutination units per milliliter of Sv/2–3 × 107 cells in serum-free medium. After adsorption for 1 h, virus inoculum was removed, and the cells were washed and fed with DMEM medium containing 10% FCS. On infection with Sv for indicated time, HEK293 cell lysates were also prepared for other experiments.
Nuclear Extracts
After treatment, cells were washed with phosphate-buffered saline (PBS) and lysed on ice for 15 min in hypotonic buffer (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 1 mM Na3VO4, 20 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride). Lysates were vortexed vigorously and centrifuged at 5000 × g for 5 min at 4°C. The pellet was washed once with cold PBS, and then the nuclei were extracted in a high salt buffer (450 mM NaCl,1.5 mM MgCl2, 0.2 mM EDTA, 1 mM Na3VO4, and 20 mM sodium fluoride) and shaken for 30 min at 4°C. Nuclear extracts were obtained by centrifugation at 15,000 × g for 5 min. Protein concentration was calibrated by the Bio-Rad protein assay (Bio-Rad, Hercules, CA).
IRF3 Reporter Gene Assays
The HEK293T cells (1 × 105 cells/well) were seeded into 12-well plates. Cells were transfected with the p561-Luc reporter gene plasmid (Jiang et al., 2004) by the calcium phosphate precipitation method for 24 h after seeding, along with each expression vector as indicated. The total DNA concentration was kept constant by supplementing with empty vector pcDNA3. pTK-Renilla was transfected at the same time for normalizing transfection efficiencies. Twenty-four hours after transfection, luciferase activity was determined with the dual-luciferase assay system (Promega, Madison, WI). The values represented the average of three independent experiments with variability shown by the error bars.
Electrophoretic Mobility Shift Assay (EMSA)
EMSAs were performed using a 32P end-labeled probe corresponding to the interferon-stimulated response element (ISRE) of the ISG15 promoter (5′-GATCCATGCCTCGGGAAAGGGAAACCGAAACTGAAGCC-3′). Equal amounts of protein were incubated with poly(dI-dC) and labeled oligonucleotides in ISRE binding buffer (40 mM KCl, 20 mM HEPES, pH 7.0, 1 mM MgCl2, 0.1 mM EGTA, 0.5 mM dithiothreitol, and 0.02% Nonidet P-40). Electrophoresis was performed on 6% nondenaturing Tris borate-EDTA-PAGE, and the gels were dried and subjected to autoradiography. For supershift experiments, nuclear extracts were incubated on ice with the specified antibody against IRF3 for 1 h at 4°C before the addition of the labeled oligonucleotide.
Immunoprecipitation and Immunoblotting
Cultures of HEK293T cells in 6-cm-diameter dishes were transfected with various combinations of plasmids. Forty-eight hours after this, cells were washed using PBS before lysed in 300 μl of buffer containing 50 mM Tris-HCl, pH 7.5, 0.5% Nonidet P-40, and 150 mM NaCl. After centrifugation for 5 min at 13,000 × g, the supernatant was removed. Various mutants of Flag-IRF3 or HA-Hsp90 were immunoprecipitated from 150 μl of cell lysate using 2 μl anti-FLAG M2 affinity gel or HA antibody bound to protein A beads. After 2-h incubation at 4°C, beads were washed three times using immunoprecipitation buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100 and then twice using Tris-buffered saline buffer. The immunoprecipitates were probed with various antibodies. For immunoblotting, the immunoprecipitates or whole-cell lysates were resolved on SDS-PAGE gels and transferred to polyvinylidene difluoride membranes (Bio-Rad). The membranes were immunoblotted with various antibodies, and the bound antibodies were visualized with alkaline phosphatase (AP)-conjugated antibodies against mouse IgG by using NBT/BCIP Western blotting system (Promega).
Immunofluorescence
The green fluorescent protein (GFP)-IRF3 expression plasmids were transiently transfected into 293 cells by the calcium phosphate coprecipitation method. Ten hours after transfection, transfected cells were infected with Sendai virus or with GA (500 nM) for another 16 h. Cells were fixed in 4% formaldehyde, permeabilized in 0.5% Triton-X 100, blocked, and nucleus was stained with 4,6-diamidino-2-phenylindole (DAPI). GFP fluorescence was analyzed with an Olympus fluorescence microscope by using a 40× objective.
RESULTS
Identification of Hsp90 as an IRF3 Interacting Protein
Although activation of IRF3 was essential in the establishment of anti-viral state, the regulatory mechanism underlying this activation was not adequately characterized. To identify potential proteins that might regulate IRF3 activity, a yeast two-hybrid screen was performed using as bait the IRF3 (1–328aa) fused in frame downstream of Gal4 DNA-binding domain and cotransfecting with a human thymus cDNA library fused to the GAL4 activation domain. We screened 6 × 105 transformants via auxotroph, of which 39 clones were 5-bromo-4-chloro-3-indolyl-β-d-galactoside positive and confirmed to be the N terminus of human Hsp90 (our unpublished data). Previously, Hsp90 was shown to modulate its client proteins important in transcriptional regulation and signal transduction (Lindquist and Craig, 1988; Pearl and Prodromou, 2000; Smith, 2000). This led us to hypothesize that Hsp90 might play a role in IRF3 activation. To confirm the interaction between IRF3 and Hsp90 in mammalian system, Flag-IRF3 and HA-Hsp90 were coexpressed in HEK293T cells. As was shown in Figure 1A, HA-Hsp90 was coimmunoprecipitated with Flag-IRF3. In contrast, neither was HA-cyan fluorescent protein coimmunoprecipitated with Flag-IRF3 nor was HA-Hsp90 with Flag-TRAF6. These results suggested that interaction between IRF3 and Hsp90 was highly specific. In addition, endogenous Hsp90 was also coimmunoprecipitated with Flag-IRF3 in HEK293T cells (Figure 1B). Alternatively, we explored mammalian two hybrid assay system to further confirm the interaction in vivo, using a Gal4-Luc reporter gene. On transfecting Gal4-BD-IRF3 with the reporter gene alone, only marginal luciferase activity was observed. In contrast, robust luciferase activity was detected when Gal4-BD-IRF3 and Gal4-AD-Hsp90 were cotransfected along with Gal4-Luc reporter gene. Notably, the induced luciferase activity was markedly increased in a Gal4-AD-Hsp90 dose-dependent manner, whereas Gal4-AD-Hsp90 alone did not display any effect on Gal4-Luc reporter gene (Figure 1C), which again substantiated the interaction between Hsp90 and IRF3. To rule out the possibility that interaction between Hsp90 and IRF3 was an artifact of overexpression, we indeed detected endogenous IRF3 in the immunoprecipitate when immunoprecipitating endogenous Hsp90 from HEK293T cells (Figure 1D). This was also true in RAW264.7 cells, supporting the physiological relevance of Hsp90 and IRF3 interaction (Figure 1D).
Figure 1.

Identification of Hsp90 as a new IRF3 interacting protein. (A) 293T cells were cotransfected with different combinations of HA-tagged and Flag-tagged protein constructs and equal amounts of cell lysates were immunoprecipitated with anti-FLAG beads. The immunoprecipitates were immunoblotted with the indicated antibodies. (B) 293T cells were transfected with mock or Flag-IRF3. The cell lysates were incubated with anti-FLAG M2 agarose. The immunoprecipitates were immunoblotted with the indicated antibodies. (C) pCMV-BD-IRF3 and pCMV-AD-Hsp90 were transfected into 293T cells along with 3xGAL4 reporter gene as indicated. Twenty-four hours after transfection, luciferase activity was determined with the dual-luciferase assay system. (D) RAW264.7 and 293 cell lysates were incubated with protein A/G-agarose conjugated with control mouse IgG or mouse anti-Hsp90 antibodies. The immunoprecipitated proteins were immunoblotted with the indicated antibodies.
Identification of the Domains Responsible for IRF3–Hsp90 Interaction
IRF3 is composed of N-terminal DNA-binding domain and C-terminal transactivation domain. To map the Hsp90-binding domain in IRF3, we generated a series of Flag-tagged truncation mutants of IRF3 (as indicated in Figure 2A) and tested their binding capability to HA-Hsp90 in 293T cells using coimmunoprecipitation assay. We first examined whether the transactivation domain was responsible for interacting with Hsp90. The set of Flag-tagged IRF3 C-terminal truncation mutants were transfected into 293T cells along with HA-tagged Hsp90; whole cell extracts were immunoprecipitated with anti-FLAG beads, and the beads were probed with anti-HA immunoblotting. As shown in Figure 2B, all the C-terminal truncation mutants, including IRF3 (1-140aa), IRF3 (1-147aa), IRF3 (1-201aa), IRF3 (1-357aa), and IRF3 (1-382aa), retained binding capability and interacted with Hsp90 as well as the wild type, suggesting that the Hsp90-binding domain was located within the DNA-binding domain of IRF3. To confirm this possibility, we made additional N-terminal truncation mutants of IRF3, including IRF3 (141-427aa), IRF3 (148-427aa), and IRF3 (201-427aa) and performed the same binding assay as with the C-terminal truncation mutants. Analysis revealed that all the N-terminal truncation mutants failed to display any binding affinity to Hsp90, once the domain spanning amino acid residues 1–140 of IRF3 was deleted (Figure 2C). Together, these data indicated that IRF3 N-terminal domain (1-140aa) was both necessary and sufficient to interact specifically with Hsp90.
Figure 2.
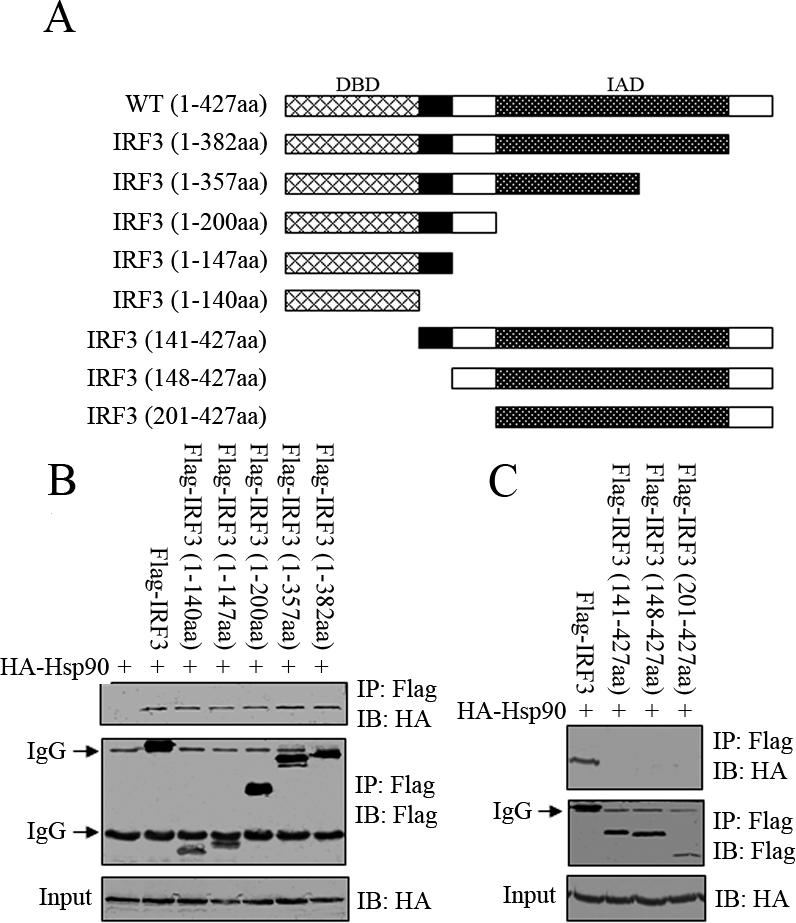
Identification of the IRF3 domain responsible for binding to Hsp90. (A) Schematic diagram of IRF3 truncation mutants used in this study. HA-Hsp90 (WT) was transiently transfected into 293T cells together with the indicated Flag-IRF3 wild type or truncation mutants (B and C). The Flag-tagged proteins were immunoprecipitated and immunoblotted with the indicated antibodies.
We went on to map IRF3-binding domain within Hsp90. As in the case of IRF3 analysis, we generated a series of Hsp90 truncation mutants (Figure 3A) and tested their binding affinity to IRF3. Deletion of Hsp90 C-terminal domain retained its ability to bind to IRF3. In contrast, N-terminal truncation mutants of Hsp90, such as Hsp90 (402-732aa) and Hsp90 (233-732aa), failed to display binding capability to IRF3. Previously, both N-terminal and middle domain of Hsp90 had been implicated as important in interaction between Hsp90 and its client protein (Scheibel et al., 1998). To further delineate Hsp90 individual domains, we generated Hsp90 (233-620aa) that harbored the middle domain. Coimmunoprecipitation assay found that it did not show any detectable binding to IRF3 at all, which suggested that IRF3-binding motif was within the N-terminal domain of Hsp90 (Figure 3B). This was confirmed through using Hsp90 truncation mutants Hsp90 (1-232aa) and Hsp90 (1-401aa) that contained the N-terminal domain and both interacted with IRF3, as was shown in Figure 3C. Together, these data indicated that Hsp90 N-terminal domain (1-232aa) was both necessary and sufficient to interact specifically with IRF3.
Figure 3.
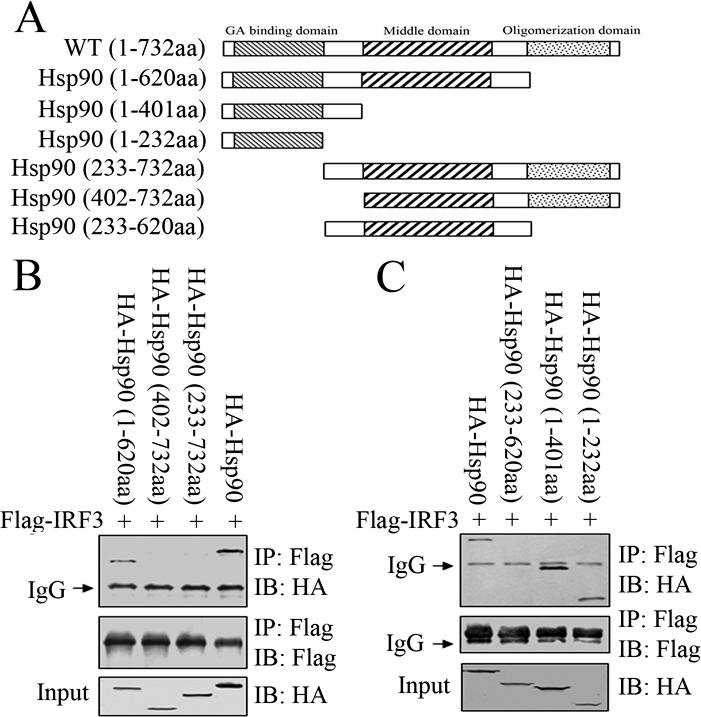
Identification of the Hsp90 domain responsible for binding to IRF3. (A) Schematic diagram of Hsp90 truncation mutants used in this study. Flag-IRF3 (WT) was transiently transfected into 293T cells together with the indicated HA-Hsp90 wild type or truncation mutants (B and C). The Flag-tagged proteins were immunoprecipitated and immunoblotted with the indicated antibodies.
Specific Inhibitors of Hsp90 Attenuated IRF3 Activation Induced by Sendai Virus
Because Hsp90 was constitutively and abundantly expressed inside mammalian cells, the specific inhibitors of it were powerful research tool to probe its function. It was known that GA was able to specifically bind to the ATP binding pocket of Hsp90 (Stebbins et al., 1997) and inhibited the Hsp90-mediated conformational maturation, resulting in the degradation of Hsp90 client proteins. To see whether GA had the ability to interfere with the interaction of Hsp90 and IRF3, we pretreated HEK293 cells with either dimethyl sulfoxide (DMSO) or GA and then immunoprecipitated from corresponding cell lysates endogenous Hsp90 with its specific antibody. Subsequent immunoblotting analysis of the immunoprecipitates showed that GA treatment markedly disrupted the interaction between IRF3 and Hsp90 but that DMSO exhibited no detectable effect (Figure 4A).
Figure 4.

GA attenuates IRF3 activation induced by Sv. (A) 293 cells were treated with mock or 2 μM GA for 16 h. Cell lysate was immunoprecipitated by anti-Hsp90 protein A/G bead, and the latter was probed with indicated antibodies. (B) 293 cells were transfected with p561-luc reporter and pTK-Renilla reporter. Sixteen hours after transfection, the transfected cells were treated with DMSO or GA in different concentrations 1 h before and during overnight treatment with Sv. Luciferase activities were measured as described in Materials and Methods. (C) 293 cells were treated with Sv and GA as indicated. Total RNAs were extracted and equal amount of RNA was amplified by RT-PCR, using specific primers for IFNβ, ISG15, and RANTES, respectively. (D) 293 cells were treated with Sv and GA in the indicated concentrations for overnight. Equal amount of cell lysate was probed with anti-ISG15 antibody. (E) 293 cells were infected with Sv for indicated time length (left), or 293 cells were infected with Sv for 20 h and treated with different concentrations of GA during infection (right). Nucleus extracts were subjected to EMSA assay using the ISRE of ISG15 as probe. (F) 293 cells were transfected with GFP-IRF3 plasmid. At 10 h posttransfection, cells were treated with Sv or GA for 16 h as indicated. The subcellular location of IRF3 was analyzed with an Olympus fluorescence microscope. The nucleus was visualized by DAPI staining.
Therefore, we used GA to investigate whether Hsp90 was important in IRF3 activation induced by Sv. IRF3 was previously demonstrated to regulate human p56 gene in response to Sv by targeting its ISRE (Guo et al., 2000; Elco et al., 2005), so transient transfection assays were set up to monitor endogenous IRF3 activity, in which was used a p561-Luc reporter gene that harbored firefly luciferase under the control of the ISRE promoter and was responsive to IRF3 activity (Jiang et al., 2004). As was shown in Figure 4B, infection of the p561-luc transfected cells with Sv for 24h led to very strong induction of p561 promoter. However, treating p561-luc-transfected cells with GA before Sv infection resulted in the marked reduction of p561 promoter activity compared with the control. This negative regulation was GA dose dependent, too. DMSO, by contrast, had no such inhibitory effect.
Another way to monitor IRF3 activation was to check IRF3-dependent gene expression upon Sv infection by reverse transcription (RT)-PCR, such as mRNA transcripts of IFNB, RANTES, and ISG15 (Schafer et al., 1998; Lin et al., 1999; Elco et al., 2005). As was shown in Figure 4C, Sv infection drastically augmented the mRNA expression levels of IFNB, RANTES, and ISG15 in 293 cells. Notably, these inductions were almost completely suppressed by GA in a dose-dependent manner (the differences of the sensitivity to GA treatment were probably because of the intrinsic configurations of the different promoters of the target genes). This was also true for RAD treatment, which was another inhibitor of Hsp90 that was structurally unrelated to GA but had the same inhibitory effect (our unpublished data). We also tested the protein level of ISG15, which was reported as one of the most abundant genes induced by Sendai virus (Elco et al., 2005). Consistent with the above-mentioned results, the amount of ISG15 and ISG15 conjugates in response to Sv were dramatically reduced by GA (Figure 4D).
IRF3 was formerly proposed to dimerize and bind to its cognate DNA elements upon activation (Yoneyama et al., 1998). We therefore tested whether GA could interfere with IRF3 DNA binding activity using EMSA. As shown in Figure 4E, a strong protein-DNA band occurred from cell lysates collected from ∼16 h after Sv infection, which extended to 20 h after Sv infection. This band was confirmed to be because of IRF3-DNA complex by incubation with anti-IRF3 antibody. Consistently, this DNA binding activity was significantly reduced when cells were treated with GA before harvesting, which again substantiated the importance of Hsp90 in IRF3 activation.
Previous studies had shown that IRF3 translocated from cytoplasm into nucleus upon Sv infection (Lin et al., 1998). To explore whether Hsp90 was important for this process, IRF3-GFP plasmid was transfected into 293 cells. Consistent with previous reports (Lin et al., 1998; Yoneyama et al., 1998), in situ immunofluorescence experiments showed that GFP-IRF3 localized exclusively in the cytoplasm in resting cells, and Sv infection resulted in translocation of IRF3 into the nucleus. As expected, virus-induced cytoplasm-to-nucleus translocation of GFP-IRF3 was abrogated when 293 cells were treated with GA for 1 h before and during virus infection (Figure 4F). Together, these results revealed that GA suppressed IRF3 activation induced by Sv and strongly suggested that Hsp90 played an essential role in the process.
Repression of Endogenous Hsp90 by RNA Interference (RNAi) Impaired Sv-induced IRF3 Activation
So far, the above-mentioned results had established that IRF3 activation in response to virus infection was sensitive to geldanamycin. We went on to address whether Hsp90 was indeed essential for IRF3 activation and responsible for this effect. Previously, it was shown that the technique of RNA interference could be used to effectively knock down the protein level of endogenous Hsp90 (Teng et al., 2004). To confirm this result, the plasmids pSuperHsp90i and pSuper (as a control) were transfected into 293 cells, respectively. Forty-eight hours posttransfection, the cell lysates were collected to analyze the protein level of endogenous Hsp90. As was expected, Hsp90 expression was significantly knocked down in pSuperHsp90i-transfected cells. In contrast, there was no observable effect to endogenous Hsp90 in pSuper-transfected cells (Figure 5A). To further explore whether IRF3 activation was impaired by this knockdown, we transfected 293 cells with p561-luc reporter in the presence or absence of pSuperHsp90i plasmids and then infected the cells with Sendai virus. As shown in Figure 5B, luciferase assay revealed that the strong induction of p561 promoter was markedly attenuated in a dose-dependent way in Hsp90i-transfected cells. pSuper, by contrast, had no such inhibitory effect. Consistently, GA dramatically inhibited p561-luc induction in response to Sv infection, whereas DMSO had no such effect. In addition, the effect of Hsp90 knock down was investigated via RT-PCR in terms of IRF3-regulated gene expression induced by Sv. Similar to GA interference, the mRNA expression levels of IFNB, RANTES, and ISG15 were significantly reduced in Hsp90 knockdown cells upon Sv infection (Figure 5C). We also tested whether IRF3 DNA binding activity induced by Sv was affected in Hsp90 knockdown cells using EMSA. 293 cells were transfected with pSuper and pSuperHsp90i, respectively, for 48 h, followed by Sv infection. After 20 h of infection, the nuclear extracts were subject to EMSA assay using IRF3 target probe. Consistently, the IRF3 DNA binding activity was significantly reduced in pSuperHsp90i-transfected cells compared with pSuper-transfected cells (Figure 5D). Together, these data strongly indicated that Hsp90 played an essential role in the process of IRF3 activation induced by Sv.
Figure 5.

Knockdown of Hsp90 by small interfering RNA attenuates Sv-induced IRF3 activation. (A) 293 cells were transfected with mock, pSuper-Hsp90i construct, or control pSuper vector. Forty-eight hours posttransfection, equal amounts of cell lysates were immunoblotted with indicated antibodies. (B) 293 cells were transfected with p561-luc reporter and pTK-Renilla reporter, along with pSuper-Hsp90i construct or control pSuper vector. Forty-eight hours after transfection, the transfected cells were treated with Sv overnight. The infected cells were treated with DMSO or GA as a control. Luciferase activities were measured as described in Materials and Methods. (C) 293 cells transfected with pSuper-Hsp90i construct or control pSuper vector were infected with Sv as indicated. Total RNAs were extracted, and an equal amount of RNA was amplified by RT-PCR, using specific primers for IFNβ, ISG15, and RANTES, respectively. (D) 293 cells were transfected with pSuperHsp90i construct or control pSuper vector. Forty-eight hours posttransfection, these cells were infected with Sv for 20 h. Other cells infected with Sv were treated with 2 μM GA as a control. Nucleus extracts were subjected to EMSA assay using the ISRE of ISG15 as probe.
Hsp90 Interacted with TBK1 and Regulated the IRF3 Activity Induced by TBK1
Normally, Hsp90 functioned as chaperone to stabilize its client proteins. Geldanamycin competed with ATP for its binding pocket to inhibit this activity and thus destabilized the client proteins. To address whether stability of IRF3 depended on the chaperone activity of Hsp90, 293 cells were incubated with GA to test its effect on protein steady state of endogenous IRF3. As was shown in Figure 6A, GA did not influence the steady state of IRF3 nor did it induce any degradation of IRF3, which suggested that although Hsp90 interacted with IRF3, it did not function to stabilize IRF3. So we turned to other possible mechanisms by which Hsp90 might explore regulation of IRF3 activation. One of the earliest steps of IRF3 activation was its being phosphorylated at specific C-terminal Ser/Thr residues (Lin et al., 1998). When analyzing IRF3 phosphorylation status in response to Sv infection, we found that the virus-induced phosphorylation of IRF3 was significantly inhibited by GA compared with control treatment, which suggested that Hsp90 might regulate the process of IRF3 phosphorylation (Figure 6B). Previous studies had suggested that TBK1 could phosphorylate IRF3 directly (McWhirter et al., 2004) in vitro and played an essential role in the activation of IRF3 induced by Sv and dsRNA (Fitzgerald et al., 2003; McWhirter et al., 2004). In addition, it had been reported that Hsp90 could interact with protein kinases in other signaling pathways (Lewis et al., 2000; Basso et al., 2002; Zhang et al., 2004). This led us to speculate that Hsp90 might associate with TBK1, which was confirmed by the observation that immunoprecipitates of endogenous Hsp90 from 293 cellular lysates contained endogenous TBK1 (Figure 6C). In addition, we also tested the binding ability of Hsp90 to wild-type TBK1 and kinase-defective TBK1 (K38A), respectively. It turned out that no difference was observed with these two forms of TBK1, although they had different kinase activity (Figure 6D). These data indicated that TBK1 activity did not influence its interaction with Hsp90. Previously, we have shown that GA could interfere with IRF3 DNA binding activity upon virus stimulation. To examine whether GA could inhibit the IRF3 DNA binding activity induced directly by TBK1, Flag-IRF3 and Myc-TBK1 were cotransfected into HEK293T cells and the cells were subsequently treated with DMSO or GA in different concentrations. Nuclear extracts from these cells were assessed by EMSA using ISRE of ISG15 as a probe. As was expected, the IRF3 DNA binding activity was significantly reduced with GA treatment in comparison with DMSO treatment (Figure 6E). Given that GA attenuated TBK1 activity and Hsp90 interacted with the latter, we explored whether truncation mutants of Hsp90 might display any dominant-negative effect on p561-Luc reporter gene expression stimulated by TBK1. As was shown in Figure 6F, this luciferase activity was greatly inhibited by overexpressing Hsp90 (1-232aa), Hsp90 (1-401aa), and Hsp90 (1-620aa) that all harbored the domain responsible for binding to IRF3. In contrast, overexpression of Hsp90 (233-732aa), Hsp90 (402-732aa), and Hsp90 (233-620aa), which lost the ability to bind to IRF3, did not display any effect on p561-Luc reporter gene expression (Figure 6F). These experiments indicated that TBK1 interacted with Hsp90 and that this interaction played a regulatory role in IRF3 activation.
Figure 6.

Hsp90 interacts with TBK1 and regulates the IRF3 activity induced by TBK1. 293 cells were treated with the indicated concentrations of GA for 20 h (A) and 293 cells were infected with Sv in the presence of DMSO or 2 μM GA for various time as indicated (B). Equal amount of cell lysates were probed with anti-IRF3 antibodies. (C) 293 cell lysates were incubated with protein A/G agarose conjugated with control mouse IgG or mouse anti-Hsp90 antibodies. The immunoprecipitated proteins were immunoblotted with the indicated antibodies. (D) Flag-TBK1 (WT) or Flag-TBK1 (K38A) was transfected into 293T cells with HA-Hsp90 (WT). Cell lysates were incubated with anti-FLAG M2 agarose, and the immunoprecipitates were immunoblotted with the indicated antibodies. (E) 293T cells were transfected with Myc-TBK1 and Flag-IRF3 as indicated. Six hours after transfection, the transfected cells were treated with GA in different concentrations for 16 h. Nuclear extracts were assayed for DNA binding activities by EMSA using the ISRE of ISG15 as probe. (F) TBK1 and a series of Hsp90 truncation mutants were cotransfected into HEK293T cells along with the p561-luc reporter and pTK-Renilla reporter. Twenty-four hours after transfection, luciferase activities were measured as described in text.
Stability of TBK1 Was Dependent on Hsp90 Activity
Because TBK1 interacted with Hsp90, we wondered whether the chaperon activity of Hsp90 was essential for the stability of TBK1. So, Flag-TBK1 was transfected into 293T cells, and the cells were treated with GA or DMSO for the indicated time. Time-course experiments revealed that the protein level of Flag-TBK1 started to reduce within 1 h after GA treatment (300 nM) and diminished significantly by 3 h (Figure 7A). In addition, GA treatment resulted in a dose-dependent decrease in the protein level of Flag-TBK1. This phenomenon was also observed with RAD treatment, another specific inhibitor of Hsp90 (our unpublished data). By contrast, treatment of the cells with DMSO barely changed the protein level of Flag-TBK1 (Figure 7B). To see whether this functional relationship also existed between endogenous TBK1 and Hsp90, HEK293 cells were treated with different concentrations of GA for 16 h, and the protein levels of endogenous TBK1 were evaluated by Western blotting. The treatment of 293 cells with either GA or RAD induced a dose-dependent decrease in the protein level of endogenous TBK1 compared with DMSO treatment (Figure 7C). In the above-mentioned sections, it had been shown that Hsp90 was essential for Sv-induced IRF3 activation; thus, we wondered whether this was partly because of its chaperone effect on TBK1. Thus, we measured the protein level of TBK1 after Sv and GA treatment. As a control for IRF3 activation in this experiment, the protein level of ISG15 was also examined. As expected, ISG15 was robustly induced, and GA treatment abolished the induction by the Sv infection. Consistent with this observation, TBK1 was degraded in response to GA treatment (Figure 7D). Interestingly, we observed reproducibly that the concentration of GA effective for TBK1 degradation was lower when it was activated either by overexpression or virus infection than that when TBK1 was in rest. For most of the Hsp90 client proteins, this degradation was mediated by 26S proteasome (Sepp-Lorenzino et al., 1995; Lewis et al., 2000). To investigate whether the proteasome was also involved in GA-induced TBK1 degradation, we tested the ability of proteasome inhibitor MG132 to block TBK1 degradation. MG132 was added to the culture medium 1 h before treating cells with GA. Cells were collected and whole cell lysates was used for Western blotting to determine TBK1 quantity. As shown in Figure 7E, the presence of MG132 antagonized GA-induced TBK1 degradation. Together, these results indicated that Hsp90 was a chaperone of TBK1 and that it regulated IRF3 activation partly by stabilizing TBK1.
Figure 7.

Stability of TBK1 is dependent on Hsp90 activity. (A) 293T cells transiently expressing Flag-tagged TBK1 were treated with 300 nM GA for the indicated times. The cells were lysed and probed with anti-FLAG antibody. (B) 293T cells transiently expressing Flag-tagged TBK1 were treated 6 h posttransfection with the indicated concentrations of GA or equivalent volume of DMSO for 16 h. The cells were lysed and probed with ant-FLAG antibody. (C) 293T cells were treated with the indicated concentrations of GA, RAD, or DMSO for 16 h, lysed, and then immunoblotted with the indicated antibodies. (D) 293 cells were treated with GA in various concentrations for 2 h before and overnight (20-h) incubation with Sv. ISG15 and TBK1 were measured by Western blot in total lysate. (E) 293T cells were treated with the proteasome inhibitor MG132 (25 μM) 1 h before and during overnight treatment with 500 nM GA. Cells were collected and subjected to Western blotting analysis for TBK1 content.
The TBK1–Hsp90–IRF3 Complex Was Important for IRF3 Activation
As was demonstrated above, Hsp90 interacted with IRF3 and TBK1, which suggested that these three proteins resided in a protein complex in vivo. To investigate this possibility, we tested whether endogenous IRF3 and TBK1 were both present in the immunoprecipitate when immunoprecipitating endogenous Hsp90 from HEK293 cells. In addition, we also tested whether endogenous IRF3 could coimmunoprecipitate TBK1 or vice versa (Figure 8, A and D). Our results revealed that this was indeed the case and Hsp90 formed a novel complex with IRF3 and TBK1 in vivo. Because IRF3 was first phosphorylated by TBK1 and then translocated from cytoplasm into nucleus (Lin et al., 1998; Fitzgerald et al., 2003; McWhirter et al., 2004), we wondered whether the phosphorylation could lead IRF3 to disassociate from the complex. To examine this possibility, HA-Hsp90 was transiently transfected into HEK293T cells along with Flag-wt-IRF3 or Flag-IRF3 (5D), respectively. IRF3 (5D) was previously shown to mimic phosphorylated IRF3 and be constitutively active inside nucleus. The above-mentioned cell extracts were immunoprecipitated with anti-FLAG beads to analyze the affinity of IRF3 (WT) or IRF3 (5D) to Hsp90. As was shown in Figure 8B, HA-Hsp90 interacted much more weakly with Flag-IRF3 (5D) than with Flag-wt-IRF3.
Figure 8.

Hsp90, TBK1, and IRF3 form a dynamic protein complex. (A) 293 cell lysates were incubated with protein A/G-agarose conjugated with control mouse IgG, mouse anti-Hsp90, or mouse anti-TBK1 antibodies as indicated. The immunoprecipitated proteins were immunoblotted with the indicated antibodies. (B) HA-Hsp90 (WT) was transfected into 293T cells along with Flag-IRF3 (WT) or Flag-IRF3 (5D). The cell lysates were incubated with anti-FLAG M2 agarose. The immunoprecipitated proteins were immunoblotted with the indicated antibodies. (C) HA-Hsp90 (WT) was transfected into 293T cells along with indicated combinations of plasmids. The cell lysates were immunoprecipitated with anti-FLAG M2 agarose. The immunoprecipitates were probed with indicated antibodies. (D) Flag-IRF3 (WT) or Flag-IRF3 (5D) was transfected into 293T cells. The cell lysates were incubated with anti-FLAG M2 agarose. The immunoprecipitated proteins were immunoblotted with the indicated antibodies. (E) 293 cells were infected with mock or Sv for 20 h. Hsp90 was immunoprecipitated and immunoblotted with the indicated antibodies.
Because overexpression of TBK1 could lead to phosphorylation of IRF3, Flag-IRF3 and HA-Hsp90 were cotransfected into HEK293T cells with or without Myc-TBK1, to test whether interaction between IRF3 and Hsp90 was also affected by phosphorylation process in vivo. Consistent with the above-mentioned observation, IRF3 was phosphorylated by TBK1, and this phosphorylation apparently blocked the interaction between IRF3 and Hsp90 (Figure 8C). We also examined the interaction between Flag-IRF3 (WT) or Flag-IRF3 (5D) with endogenous Hsp90 and TBK1. Consistent with above-mentioned results, no obvious interaction of Flag-IRF3 (5D) with Hsp90 and TBK1 was detected compared with Flag-IRF3 (WT) (Figure 8D). To address whether this functional relationship also existed in virus-infected cells, endogenous immunoprecipitation assay was performed in 293 cells with or without Sv infection. As was expected, it was found that the interaction between Hsp90 and IRF3 was apparently weakened upon Sv infection (Figure 8E). The interaction between Hsp90 and TBK1, by contrast, had no such negative effect (Figure 8E), which strongly suggested that interaction between Hsp90 and IRF3 was dynamically regulated, whereas Hsp90 constitutively interacted with TBK1. This led us to speculate that Hsp90 probably played a role in bringing TBK1 and IRF3 into proximity and facilitating TBK1 to phosphorylate IRF3.
Previously, it was known that a protein called CHIP could, via its TPR domain, specifically bind to Hsp90 C-terminal domain and effectively competed away proteins interacting with Hsp90 (Sumanasekera et al., 2003). However, a point mutation of CHIP (K30A) in the TPR domain deprived its ability to bind to Hsp90 (Xu et al., 2002). So, we investigated the effect of CHIP on interfering with IRF3-Hsp90-TBK1 complex formation and also IRF3 activation. HA-CHIP or HA-CHIP(K30A) was transfected into HEK293T cells, respectively. Endogenous Hsp90, IRF3, or TBK1 was immunoprecipitated from the cell lysates by corresponding antibodies bound to protein A/G beads. The immunoprecipitates were probed with anti-HA antibody to check the presence or absence of the CHIP and CHIP(K30A). As was expected, HA-CHIP but not HA-CHIP (K30A) was coimmunoprecipitated with Hsp90 (Figure 9A). In contrast, neither TBK1 nor IRF3 had any affinity to HA-CHIP or HA-CHIP(K30A) (Figure 9, B and C). Interestingly, the endogenous IRF3–Hsp90–TBK1 complex was disrupted significantly when CHIP was expressed inside the cell. This was apparently because of the competitive binding of CHIP to Hsp90, because CHIP(K30A) was unable to affect the integrity of this complex (Figure 9D). Consistent with this interference, cotransfecting CHIP with TBK1 resulted in an apparent inhibition of the TBK1-inducible IRF3 transcriptional activity. More importantly, CHIP(K30A) had not such an inhibitory effect on IRF3 activation (Figure 9E). Based on these experiments, we concluded that Hsp90 formed a functional complex with IRF3 and TBK1 in vivo; and the complex was important for IRF3 activation.
Figure 9.

The TBK1–Hsp90–IRF3 complex was important for IRF3 activation. (A) 293T cells were transfected with mock, HA-CHIP, or HA-CHIP(K30A). The cell lysates were incubated with protein A/G-agarose conjugated with indicated antibodies (B–D). The immunoprecipitated proteins were immunoblotted with the indicated antibodies. (E) TBK1 was transfected into HEK293T cells with CHIP or CHIP(K30A) as indicated, along with the p561-luc reporter gene and pTK-Renilla internal control. After 24 h, luciferase activity was measured as described in text.
DISCUSSION
IRF3 was expressed constitutively and latently in a variety of tissues, and activated after virus infection. Its activation involved consecutively multiple steps: phosphorylation at multiple serine and threonine residues in the carboxy terminus, cytoplasm-to-nucleus translocation, DNA binding, and transcriptional promoting of target gene expression (Lin et al., 1998; Yoneyama et al., 1998). It was known that Sendai virus-induced IRF3 activation-mediated transcription and synthesis of multiple ISG genes important in antiviral defense, cell growth regulation, and immune activation (Lin et al., 1998; Yoneyama et al., 1998; Schafer et al., 1998). This activation was independent of both interferon production and virus replication (Collins et al., 2004). A recent report demonstrated that TLR3, although essential for gene induction by dsRNA, was dispensable for gene induction by Sv (Elco et al., 2005). Therefore, it will be both interesting and important to investigate how IRF3 is regulated in response to Sv infection. It has recently been shown that TBK1 could phosphorylate IRF3 in vitro (McWhirter et al., 2004) and played an essential role in IRF3 activation induced by virus in the innate immune response (Fitzgerald et al., 2003). However, it remains controversial how TBK1 phosphorylates IRF3 in vivo. In this report, we demonstrated that Hsp90 formed a dynamic complex with IRF3 and TBK1, which was essential for IRF3 activation in response to Sendai virus. These results revealed an important physiological function of the Hsp90 chaperone in transducing IRF3 activation signals.
First, we identified Hsp90 as a new IRF3 regulatory protein by yeast two-hybrid approach and confirmed its interaction in vitro and in vivo. Hsp90 is an abundant and highly conserved protein involved in a diverse array of cellular processes. In contrast to other heat-shock proteins, most of the identified Hsp90 client proteins were regulatory proteins such as transcription factors and protein kinases (Zou et al., 1998; Lewis et al., 2000; Basso et al., 2002; Xu et al., 2004; De Nardo et al., 2005). Frequently, Hsp90 acted as a chaperone for unstable client proteins and kept them poised for activation. Further analysis in this study indicated that the N-terminal domain (1-140) of IRF3 was responsible for its binding to Hsp90. This domain was also found to be important for DNA binding in previous research (Schafer et al., 1998; Mamane et al., 1999). In addition, we found that the N-terminal domain (1-233) of Hsp90 was necessary and sufficient to mediate its binding to IRF3. This domain was also responsible for ATP binding and was the target sites of Hsp90-specific inhibitors, such as GA and RAD (Stebbins et al., 1997; Schulte et al., 1998).
On Sv infection, IRF3 was phosphorylated and translocated from cytoplasm into nucleus, inducing transcription of downstream genes (Yoneyama et al., 1998; Elco et al., 2005). The physiological relevance of IRF3 and Hsp90 interaction was established by the following findings. First, geldanamycin and radicicol could inhibit Hsp90 binding to IRF3, which in turn attenuated both mRNA and protein expression of IRF3 responsive reporter gene and IRF3 target gene. Second, interference of Hsp90 binding to IRF3 resulted in loss of IRF3 translocation from cytoplasm into nucleus, reduction of the ability of IRF3 to bind to its cognate ISRE DNA sequence. Third, knock down of Hsp90 expression by specific RNAi also impaired the mRNA expression of IRF3 target gene and suppressed the IRF3 DNA binding activity upon Sv infection. Fourth, Sv-induced IRF3 phosphorylation was inhibited when Hsp90 and IRF3 interaction was disrupted. Fifth, C-terminal truncation mutants of Hsp90 that could bind to IRF3 were strong dominant-negative inhibitor of IRF3 activation, whereas the N-terminal truncation mutants of Hsp90 that failed to bind to IRF3 could not do so. Significantly, our data revealed that interaction between IRF3 and Hsp90 was not served to maintain the stability of IRF3, as was shown by the observation that disruption of this interaction did not affect the steady state of IRF3, nor was IRF3 degraded. Interestingly, similar phenomenon was reported in the case of the transcription factor HSF1 (Zou et al., 1998). These results indicated that Hsp90 was far more than a chaperone for IRF3, and it might regulate the upstream kinase of IRF3.
As noted above, Hsp90 interacted with some protein kinases in other signaling pathways and contributed to their protein integrities (Donze et al., 2001; De Nardo et al., 2005). Previous research suggested that an unknown kinase activity detected in dsRNA- or virus-induced cells was sensitive to geldanamycin treatment (Iwamura et al., 2001). Recently, TBK1 was implicated to be the kinase for IRF3 activation in response to dsRNA and Sv stimulation (Sharma et al., 2003; McWhirter et al., 2004). So we investigated the possibility that TBK1 was a client protein of Hsp90. This was found to be the case in this study. First, Hsp90 formed stable complex with TBK1 in vitro and in vivo, and this interaction was not dependent on the activation status of TBK1. We had tried to dissect functional domains of TBK1 for this interaction and found that no discrete part of TBK1 could be definitely assigned this role (Unpublished data). Most likely, a three-dimensional juxtaposed domain is involved. Second, TBK1 required Hsp90 to activate IRF3. GA treatment suppressed the DNA binding activity of IRF3 induced by overexpression of TBK1. Third, it was suggested recently that once bound to its specific antagonist GA, Hsp90 lost the ability to maintain its client proteins in a functional conformation, resulting instead in their rapid destabilization and degradation, frequently by the 26S proteasome. For example, a recent study reported that disruption of RIP and Hsp90 interaction resulted in RIP destabilization and its subsequent proteasome-mediated degradation (Lewis et al., 2000). Significantly, our present study showed that Hsp90 regulated the integrity of TBK1. Disruption of the interaction between them profoundly affected the stability and steady state of TBK1 inside cells. In contrast, Hsp90 did not regulate the stability of IRF3. In addition, this degradation process was also mediated by 26S proteasome, as the proteasome specific inhibitor MG-132 blocked the GA-induced TBK1 degradation. Interestingly, we observed reproducibly that the concentration of GA effective for TBK1 degradation was lower when it was activated either by overexpression or virus infection than that when TBK1 was in rest. As a rule, protein kinases normally experienced conformational change when they were activated. This intriguing phenomenon might reflect this dynamic process and implied that there was an active regulatory process during TBK1 activation, which needs to be explored in the future. Fourth, after inhibition of Hsp90, the loss of TBK1 expression in 293 cells severely compromised the ability of Sv to trigger the activation of IRF3 and expression of ISG15. Therefore, Hsp90 is indeed a specific chaperone for TBK1, and this activity is important for TBK1 stability and IRF3 activation in response to Sendai virus infection.
Besides serving as a chaperone for TBK1, the present investigation also implicated Hsp90 to be an important adaptor protein for signal transduction. As was shown above, Hsp90 formed a novel complex with IRF3 and TBK1. Significantly, this complex was formed only in resting cell and subjected to change upon virus infection. It was found in this study that upon Sv infection, the interaction between Hsp90 and IRF3 was diminished, whereas that between Hsp90 and TBK1 remained intact. Interestingly, IRF3 (5D), a mutant mimicking phosphorylated IRF3 and displaying constitutive activity, was unable to bind to Hsp90. Furthermore, it was observed that overexpression of TBK1 in 293T cells was enough to disrupt the interaction between IRF3 and Hsp90 in vivo because TBK1 directly induced IRF3 phosphorylation. In addition, the specific Hsp90 binding protein CHIP was able to compete away Hsp90 from IRF3–Hsp90–TBK1 protein complex and significantly inhibited IRF3 activation. In contrast, CHIP (K30A), a point mutation of CHIP that was deprived of its ability to bind to Hsp90, had not such an inhibitory effect. These results strongly suggest that Hsp90 played a role in bringing IRF3 and TBK1 into proximity and facilitating signaling transduction from TBK1 to IRF3.
In the context of virus–host interaction, virus infection could lead to activation of several signal transduction pathways and transcription factors such as NF-κB, AP1, STAT, and IRF. Coordinated regulation of these transcription factors resulted in the induction of antiviral state (Sen, 2001). Hsp90 was implicated to be important in the regulation of these signaling pathways. For example, Hsp90 was found to regulate the activation of NF-κB signaling pathway (Chen et al., 2002). It was possibly responsible for constitutive activation of IKK and NF-κB in Hodgkin's lymphoma cells (Broemer, et al., 2004). Hsp90 also interacted with Stat3 and regulated the process of fever (Shah, et al., 2002). It is apparent that the regulatory function of Hsp90 is not restricted to only one pathway or one protein. So, it will be a great challenge to understand how Hsp90 works in response to virus infection in a global perspective.
This study uncovered an essential role of Hsp90 in the virus-induced activation of IRF3. Hsp90 formed a complex with IRF3 and TBK1 in vivo. This complex was dynamic and subjected to change during IRF3 activation. In addition, Hsp90 was important for stabilizing TBK1 and promoting IRF3 phosphorylation by TBK1 in response to virus infection. It will be especially intriguing to address how virus regulates this complex in future studies.
Acknowledgments
We thank professors John Hiscott (McGill University, Montreal, Quebec, Canada), Xiao-xia Li (Cleveland Clinic Foundation, Cleveland, OH), Kou-Juey Wu (National Yang-Ming University, Taiwan), Zhi-min Yin (Nan Jing Normal University, China), and You-hua Xie (Shanghai Institute of Biochemistry, Shanghai, China) for providing plasmids for this study. We thank Professor Lin Li and Bo-liang Li (Shanghai Institute of Biochemistry) for instructive discussions and advice as well as professor Zhu-chuan Zhang (Shanghai Institute of Biochemistry) for technical assistance. C. W. was a scholar of “The Rising Star Program” from Shanghai Municipal Government and of “The Hundred Talents Program” from Chinese Academy of Sciences. This work was supported in part by “The Distinguished Young Scholars Program” from National Natural Science Foundation of China (30225013) and 973 Project (2002CB513003).
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–09–0853) on January 4, 2006.
References
- Agashe, V. R., and Hartl, F. U. (2000). Roles of molecular chaperones in cytoplasmic protein folding. Semin. Cell Dev. Biol. 11, 15–25. [DOI] [PubMed] [Google Scholar]
- Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. [DOI] [PubMed] [Google Scholar]
- Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- Au, W. C., Moore, P. A., Lowther, W., Juang, Y. T., and Pitha, P. M. (1995). Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 92, 11657–11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso, A. D., Solit, D. B., Chiosis, G., Giri, B., Tsichlis, P., and Rosen, N. (2002). Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277, 39858–39866. [DOI] [PubMed] [Google Scholar]
- Boehme, K. W., and Compton, T. (2004). Innate sensing of viruses by toll-like receptors. J. Virol. 78, 7867–7873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnard, M., et al. (2000). Deficiency of T2K. leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19, 4976–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie, A. G., and Haga, I. R. (2005). The role of Toll-like receptors in the host response to viruses. Mol. Immunol. 42, 859–867. [DOI] [PubMed] [Google Scholar]
- Broemer, M., Krappmann, D., and Scheidereit, C. (2004). Requirement of Hsp90 activity for IkappaB kinase (IKK) biosynthesis and for constitutive and inducible IKK and NF-kappaB activation. Oncogene 23, 5378–5386. [DOI] [PubMed] [Google Scholar]
- Buchner, J. (1999). Hsp90 & Co. - a holding for folding. Trends Biochem. Sci. 24, 136–141. [DOI] [PubMed] [Google Scholar]
- Bukau, B., Deuerling, E., Pfund, C., and Craig, E. A. (2000). Getting newly synthesized proteins into shape. Cell 101, 119–122. [DOI] [PubMed] [Google Scholar]
- Cadepond, F., Schweizer-Groyer, G., Segard-Maurel, I., Jibard, N., Hollenberg, S. M., Giguere, V., Evans, R. M., and Baulieu, E. E. (1991). Heat shock protein 90 as a critical factor in maintaining glucocorticosteroid receptor in a nonfunctional state. J. Biol. Chem. 266, 5834–5841. [PubMed] [Google Scholar]
- Caplan, A. J. (1999). Hsp90's secrets unfold: new insights from structural and functional studies. Trends Cell Biol. 9, 262–268. [DOI] [PubMed] [Google Scholar]
- Chen, G., Cao, P., and Goeddel, D. V. (2002). TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 9, 401–410. [DOI] [PubMed] [Google Scholar]
- Collins, S. E., Noyce, R. S., and Mossman, K. L. (2004). Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 78, 1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig, E. A., Gambill, B. D., and Nelson, R. J. (1993). Heat shock proteins: molecular chaperones of protein biogenesis. Microbiol. Rev. 57, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo, D., Masendycz, P., Ho, S., Cross, M., Fleetwood, A. J., Reynolds, E. C., Hamilton, J. A., and Scholz, G. M. (2005). A central role for the Hsp90.Cdc37 molecular chaperone module in interleukin-1 receptor-associated-kinase-dependent signaling by toll-like receptors. J. Biol. Chem. 280, 9813–9822. [DOI] [PubMed] [Google Scholar]
- Diao, L., Zhang, B., Fan, J., Gao, X., Sun, S., Yang, K., Xin, D., Jin, N., Geng, Y., and Wang, C. (2005). Herpes virus proteins ICP0 and BICP0 can activate NF-kappaB by catalyzing IkappaBalpha ubiquitination. Cell Signal 17, 217–229. [DOI] [PubMed] [Google Scholar]
- Donze, O., Abbas-Terki, T., and Picard, D. (2001). The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J. 20, 3771–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, S., et al. (2002). IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17, 251–263. [DOI] [PubMed] [Google Scholar]
- Elco, C. P., Guenther, J. M., Williams, B. R., and Sen, G. C. (2005). Analysis of genes induced by Sendai virus infection of mutant cell lines reveals essential roles of interferon regulatory factor 3, NF-kappaB, and interferon but not toll-like receptor 3. J. Virol. 79, 3920–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman, D. E., and Frydman, J. (2000). Protein folding in vivo: the importance of molecular chaperones. Curr. Opin. Struct. Biol. 10, 26–33. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., Coyle, A. J., Liao, S. M., and Maniatis, T. (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Giguere, V., Hollenberg, S. M., Rosenfeld, M. G., and Evans, R. M. (1986). Functional domains of the human glucocorticoid receptor. Cell 46, 645–652. [DOI] [PubMed] [Google Scholar]
- Grandvaux, N., Servant, M. J., tenOever, B., Sen, G. C., Balachandran, S., Barber, G. N., Lin, R., and Hiscott, J. (2002). Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76, 5532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J., Peters, K. L., and Sen, G. C. (2000). Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 267, 209–219. [DOI] [PubMed] [Google Scholar]
- Iwamura, T., Yoneyama, M., Yamaguchi, K., Suhara, W., Mori, W., Shiota, K., Okabe, Y., Namiki, H., and Fujita, T. (2001). Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells 6, 375–388. [DOI] [PubMed] [Google Scholar]
- Jakob, U., Lilie, H., Meyer, I., and Buchner, J. (1995). Transient interaction of Hsp90 with early unfolding intermediates of citrate synthase. Implications for heat shock in vivo. J. Biol. Chem. 270, 7288–7294. [DOI] [PubMed] [Google Scholar]
- Jiang, Z., Mak, T. W., Sen, G., and Li, X. (2004). Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc. Natl. Acad. Sci. USA 101, 3533–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai, T., Takeuchi, O., Fujita, T., Inoue, J., Muhlradt, P. F., Sato, S., Hoshino, K., and Akira, S. (2001). Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 167, 5887–5894. [DOI] [PubMed] [Google Scholar]
- Kovacs, J. J., Murphy, P. J., Gaillard, S., Zhao, X., Wu, J. T., Nicchitta, C. V., Yoshida, M., Toft, D. O., Pratt, W. B., and Yao, T. P. (2005). HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607. [DOI] [PubMed] [Google Scholar]
- Lewis, J., Devin, A., Miller, A., Lin, Y., Rodriguez, Y., Neckers, L., and Liu, Z. G. (2000). Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 275, 10519–10526. [DOI] [PubMed] [Google Scholar]
- Lin, R., Heylbroeck, C., Genin, P., Pitha, P. M., and Hiscott, J. (1999). Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell Biol. 19, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, R., Heylbroeck, C., Pitha, P. M., and Hiscott, J. (1998). Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist, S., and Craig, E. A. (1988). The heat-shock proteins. Annu. Rev. Genet. 22, 631–677. [DOI] [PubMed] [Google Scholar]
- Mamane, Y., Heylbroeck, C., Genin, P., Algarte, M., Servant, M. J., LePage, C., DeLuca, C., Kwon, H., Lin, R., and Hiscott, J. (1999). Interferon regulatory factors: the next generation. Gene 237, 1–14. [DOI] [PubMed] [Google Scholar]
- Mayer, M. P., and Bukau, B. (1999). Molecular chaperones: the busy life of Hsp90. Curr. Biol. 9, R322–R325. [DOI] [PubMed] [Google Scholar]
- McWhirter, S. M., Fitzgerald, K. A., Rosains, J., Rowe, D. C., Golenbock, D. T., and Maniatis, T. (2004). IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 101, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov, R., and Janeway, C. A., Jr. (1998). Innate immune recognition and control of adaptive immune responses. Semin. Immunol. 10, 351–353. [DOI] [PubMed] [Google Scholar]
- Mori, M., Yoneyama, M., Ito, T., Takahashi, K., Inagaki, F., and Fujita, T. (2004). Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 279, 9698–9702. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott, E. M., and O'Neill, L. A. (2004). Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl, L. H., and Prodromou, C. (2000). Structure and in vivo function of Hsp90. Curr. Opin. Struct. Biol. 10, 46–51. [DOI] [PubMed] [Google Scholar]
- Perry, A. K., Chow, E. K., Goodnough, J. B., Yeh, W. C., and Cheng, G. (2004). Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199, 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou, C., Roe, S. M., O'Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75. [DOI] [PubMed] [Google Scholar]
- Schafer, S. L., Lin, R., Moore, P. A., Hiscott, J., and Pitha, P. M. (1998). Regulation of type I interferon gene expression by interferon regulatory factor-3. J. Biol. Chem. 273, 2714–2720. [DOI] [PubMed] [Google Scholar]
- Scheibel, T., Weikl, T., and Buchner, J. (1998). Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc. Natl. Acad. Sci. USA 95, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C., Sepp-Lorenzino, L., Nimmesgern, E., Ouerfelli, O., Danishefsky, S., Rosen, N., and Hartl, F. U. (1996). Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc. Natl. Acad. Sci. USA 93, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte, T. W., Akinaga, S., Soga, S., Sullivan, W., Stensgard, B., Toft, D., and Neckers, L. M. (1998). Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 3, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, G. C. (2001). Viruses and interferons. Annu. Rev. Microbiol. 55, 255–281. [DOI] [PubMed] [Google Scholar]
- Sepp-Lorenzino, L., Ma, Z., Lebwohl, D. E., Vinitsky, A., and Rosen, N. (1995). Herbimycin A induces the 20 S proteasome- and ubiquitin-dependent degradation of receptor tyrosine kinases. J. Biol. Chem. 270, 16580–16587. [DOI] [PubMed] [Google Scholar]
- Shah, M., Patel, K., Fried, V. A., and Sehgal, P. B. (2002). Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J. Biol. Chem. 277, 45662–45669. [DOI] [PubMed] [Google Scholar]
- Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R., and Hiscott, J. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Smith, D. F. (2000). Chaperones in progesterone receptor complexes. Semin. Cell Dev. Biol. 11, 45–52. [DOI] [PubMed] [Google Scholar]
- Stebbins, C. E., Russo, A. A., Schneider, C., Rosen, N., Hartl, F. U., and Pavletich, N. P. (1997). Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89, 239–250. [DOI] [PubMed] [Google Scholar]
- Sumanasekera, W. K., Tien, E. S., Davis, J. W., 2nd, Turpey, R., Perdew, G. H., and Vanden Heuvel, J. P. (2003). Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARbeta activity. Biochemistry 42, 10726–10735. [DOI] [PubMed] [Google Scholar]
- Takeuchi, O., and Akira, S. (2001). Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 1, 625–635. [DOI] [PubMed] [Google Scholar]
- Takeuchi, O., Hoshino, K., Kawai, T., Sanjo, H., Takada, H., Ogawa, T., Takeda, K., and Akira, S. (1999). Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451. [DOI] [PubMed] [Google Scholar]
- Teng, S. C., Chen, Y. Y., Su, Y. N., Chou, P. C., Chiang, Y. C., Tseng, S. F., and Wu, K. J. (2004). Direct activation of HSP90A transcription by c-Myc contributes to c-Myc-induced transformation. J. Biol. Chem. 279, 14649–14655. [DOI] [PubMed] [Google Scholar]
- Tojima, Y., et al. (2000). NAK is an IkappaB kinase-activating kinase. Nature 404, 778–782. [DOI] [PubMed] [Google Scholar]
- Welch, W. J. (1991). The role of heat-shock proteins as molecular chaperones. Curr. Opin. Cell Biol. 3, 1033–1038. [DOI] [PubMed] [Google Scholar]
- Welch, W. J., and Feramisco, J. R. (1982). Purification of the major mammalian heat shock proteins. J. Biol. Chem. 257, 14949–14959. [PubMed] [Google Scholar]
- Xu, W., Marcu, M., Yuan, X., Mimnaugh, E., Patterson, C., and Neckers, L. (2002). Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA 99, 12847–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W., Yu, F., Yan, M., Lu, L., Zou, W., Sun, L., Zheng, Z., and Liu, X. (2004). Geldanamycin, a heat shock protein 90-binding agent, disrupts Stat5 activation in IL-2-stimulated cells. J. Cell. Physiol. 198, 188–196. [DOI] [PubMed] [Google Scholar]
- Yoneyama, M., Suhara, W., Fukuhara, Y., Fukuda, M., Nishida, E., and Fujita, T. (1998). Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Wu, W., Du, Y., Santos, S. J., Conrad, S. E., Watson, J. T., Grammatikakis, N., and Gallo, K. A. (2004). Hsp90/p50cdc37 is required for mixedlineage kinase (MLK) 3 signaling. J. Biol. Chem. 279, 19457–19463. [DOI] [PubMed] [Google Scholar]
- Zou, J., Guo, Y., Guettouche, T., Smith, D. F., and Voellmy, R. (1998). Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480. [DOI] [PubMed] [Google Scholar]