

Abstract
Topoisomerases are involved in controlling and maintaining the topology of DNA and are present in all kingdoms of life. Unlike all other types of topoisomerases, similar type IB enzymes have only been identified in bacteria and eukarya. The only putative type IB topoisomerase in archaea is represented by Methanopyrus kandleri topoisomerase V. Despite several common functional characteristics, topoisomerase V shows no sequence similarity to other members of the same type. The structure of the 61 kDa N-terminal fragment of topoisomerase V reveals no structural similarity to other topoisomerases. Furthermore, the structure of the active site region is different, suggesting no conservation in the cleavage and religation mechanism. Additionally, the active site is buried, indicating the need of a conformational change for activity. The presence of a topoisomerase in archaea with a unique structure suggests the evolution of a separate mechanism to alter DNA.
Keywords: helix–hairpin–helix motif, helix–turn–helix domain, Methanopyrus kandleri, topoisomerase IB, topoisomerase V
Introduction
Topoisomerases are ubiquitous proteins found in bacteria, archaea, and eukarya. They change the topology of DNA by transiently breaking one (type I) or two (type II) DNA strands and passing another single or double strand through the break, respectively. Topoisomerases have been involved in several cellular processes and the importance of their cellular role is underscored by the fact that they are the target of several cancer chemotherapeutic agents and antibiotics (for a recent review, see Champoux, 2001). Type I enzymes have been further classified into two different families, types IA and IB, depending on whether they form a transient covalent bond with the 5′ or 3′ end of the broken DNA strand. Type IA enzymes have been identified in bacteria, archaea, and eukarya, and show marked sequence and structural similarities, with a characteristic toroidal fold that has been observed in two bacterial enzymes, Escherichia coli topoisomerases I and III (Lima et al, 1994; Changela et al, 2001), and in a hyperthermophilic archaeal enzyme, Archaeoglobus fulgidus reverse gyrase (Rodriguez and Stock, 2002).
Type IB enzymes have been identified in eukaryotes, poxviruses and some bacteria. Eukaryotic type IB molecules are large, typically over 90 kDa, while viral and bacterial type IB molecules are relatively small, typically around 36 kDa. Despite their differences in size, bacterial and viral type IB enzymes and eukaryotic type IB molecules share a common fold and catalytic mechanism (Cheng et al, 1998; Redinbo et al, 1998). Furthermore, these similarities extend to the tyrosine recombinase family, suggesting a common ancestor for both type IB topoisomerases and tyrosine recombinases (Cheng et al, 1998; Redinbo et al, 1998; Corbett and Berger, 2004). The only putative type IB enzyme identified in an archaeal organism is DNA topoisomerase V (Slesarev et al, 1993). Initially, it was found only in the hyperthermophile Methanopyrus kandleri isolated from a deep-water ‘black smoker' chimney in the Gulf of California (Huber et al, 1989). Later, its presence was confirmed in other Methanopyrus isolates from around the world (A Slesarev, unpublished results). Topoisomerase V possesses all the biochemical properties associated with type IB enzymes, including cleaveage of a single DNA strand, formation of a covalent intermediate with the 3′ end of the broken strand and the ability to relax both positive and negative supercoils in a magnesium-independent manner (Slesarev et al, 1993). Despite these common properties, topoisomerase V shows no sequence similarity to other type IB enzymes.
Topoisomerase V is a large enzyme, over 100 kDa, with many unusual characteristics. It has maximal topoisomerase activity at ∼108°C and is still active at 122°C (Kozyavkin et al, 1995). It is also active in high ionic strength conditions (>3 M potassium glutamate or ∼0.65 M NaCl) (Belova et al, 2002). In addition to its topoisomerase activity (Slesarev et al, 1993), topoisomerase V is also involved in DNA repair (Belova et al, 2001). These two unrelated catalytic activities are found in different regions of the same polypeptide; topoisomerase activity resides in the N-terminus of the protein, while the C-terminus of the protein possesses apurinic/apyrimidinic (AP) site-processing activity, normally associated with base excision DNA repair (Belova et al, 2002). Topoisomerase V contains 24 helix–hairpin–helix (HhH) DNA-binding motifs arranged in 12 tandem (HhH)2 domains (Shao and Grishin, 2000) at the C-terminus of the protein. Fragments containing fewer HhH motifs show optimal activity in a narrower set of temperature and salt conditions, and also exhibit a marked loss of processivity (Belova et al, 2002). Fragments as small as 44 kDa comprising the topoisomerase domain and only three HhH motifs retain full topoisomerase activity (Belova et al, 2002).
Here we report the structure of a 61 kDa fragment of topoisomerase V (designated topo-61) that spans the N-terminus 532 residues of topoisomerase V. The structure reveals a fold that is not related to the one observed in other topoisomerases, tyrosine recombinases or any other known protein. The active site contains a similar set of amino acids in topoisomerase V and in type IB molecules, but their spatial arrangement is different, suggesting a different mechanism of catalysis. The structure suggests that a large conformational change in the protein is needed for activity, an observation that is supported by biophysical experiments. All these observations taken together indicate that topoisomerase V is structurally and mechanistically different from type IB topoisomerases. Topoisomerase V hence represents a topoisomerase unique to archaea, with some similarities to other topoisomerases, but with important structural and mechanistic differences.
Results
Crystallization and structure determination
For our studies, we chose the topo-61 fragment that has been described previously (Belova et al, 2001). This construct exhibits full topoisomerase activity, but not AP lyase activity (Belova et al, 2002). Purified topo-61 protein was used for crystallizations to obtain crystal Form I (Table I). A small reduction in protein size in the crystallization drop was shown by mass spectrometry. Reproducible crystals could be obtained only upon inclusion of dilute amounts of trypsin in the crystallization drop to promote proteolysis. N-terminus sequencing showed that this cleavage occurs at the C-terminus of topo-61. C-terminus deletion constructs of topo-61 were made to increase the reproducibility of the crystals. As trypsin preferentially cleaves after arginines or lysines, four constructs were prepared with different C-termini (residues 513, 519, 523 or 530 and named topo-61dC1–4, respectively). Only the second construct, topo-61dC2, crystallized in the absence of trypsin.
Table 1.
Crystallographic data
| Form I: SAD data | Form II | |
|---|---|---|
| Crystal and data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | a=70.7 Å, b=89.8 Å, c=189.1 Å | a=75.5 Å, b=86.4 Å, c=175.7 Å |
| Resolution (Å)a | 26.6–2.3 (2.38–2.3) | 28.8–2.9 (3.0–2.9) |
| Number of unique reflections | 53 175 | 25 958 |
| Completeness (%) | 98.1 (94.8) | 99.2 (98.5) |
| Data redundancy | 6.7 (6.0) | 3.7 (3.2) |
| Rsym (%)b | 6.4 (26.2) | 9.5 (32.8) |
| I/σ(I) | 9.6 (2.9) | 6.7 (2.3) |
| Phasing and refinement | ||
| Figure of merit (27.7–2.5 Å) (after SOLVE, after RESOLVE) | 0.31, 0.46 | |
| No. of protein atoms | 8507 | 8390 |
| No. of water molecules | 510 | — |
| R.m.s.d. bond lengths (Å) | 0.017 | 0.007 |
| R.m.s.d. bond angles | 1.6° | 1.1° |
| Wilson B-factor (Å2) | 46.5 | 62.6 |
| Average B-factor | ||
| Protein atoms (Å2) | 39.3 | 52.1 |
| Water molecules (Å2) | 42.1 | — |
| R-factor (%)c | 21.7 | 23.6 |
| Rfree (%)d | 29.2 | 30.8 |
| DPIe (Å) | 0.28 | 0.51 |
| Ramachandran plot | ||
| Most favored regions (%) | 92.1 | 90.6 |
| Disallowed regions (%) | 0.2 | 0.2 |
| aNumbers in parenthesis correspond to the highest resolution shell. | ||
| bRsym=∑∣I−〈I〉∣/∑I, where I is the observed intensity and 〈I〉 the average intensity obtained from multiple measurements. | ||
| cR-factor=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣, where ∣Fo∣ is the observed structure factor amplitude and ∣Fc∣ the calculated structure factor amplitude. | ||
| dRfree: R-factor based on 5% of the data excluded from refinement. | ||
| eDPI (diffraction-component precision index) (Cruickshank, 1999) calculated with the program SFCHECK (Collaborative Computational Project, 1994). | ||
The structure of topo-61 was determined to 2.3 Å resolution by single-wavelength anomalous dispersion (SAD) phasing from a single seleno-methionyl protein crystal and the coordinates refined to a final Rwork of 21.7% and Rfree of 29.2%. There are two molecules in the asymmetric unit with a different relative arrangement of the topoisomerase and multiHhH domains. Tight noncrystallographic symmetry restraints were applied to the topoisomerase domains, but not to the multiHhH domains (see Materials and methods). Crystals of topo-61dC2 were obtained reproducibly in the same form as trypsinized topo-61 and also in a new crystal form, Form II (Table I). Only the structure of Form II of topo61dC2 was solved and is designated as topo-61 crystal Form II hereafter (Table I). The structure of topo-61 crystal Form II was solved by Molecular Replacement to 2.9 Å resolution using the coordinates of residues 1–282 of Form I as the search model, and refined to a final Rwork of 23.6% and an Rfree of 30.8%. Form II also contains two molecules in the asymmetric unit and the topoisomerase domains were tightly restrained during all stages of the refinement.
The two molecules in the asymmetric unit of crystal Form I contact each other via the N-termini of two helices of one molecule and the β-sheet of the other, and bury less than 300 Å2 of solvent-accessible surface area between them. This figure is too small for a physiological dimer and indicates that the molecules interact with each other only as a result of crystal packing.
Overall architecture
Topo-61 can be described as a two-domain molecule. The N-terminus or topoisomerase domain forms the main body of the protein, while the C-terminus or multiHhH domain curls around it (Figure 1). The topoisomerase domain comprises residues 1–268 and matches very well the domain boundaries demarcated earlier by limited proteolysis (Belova et al, 2002). The C-terminus domain (residues 296–519) consists of eight HhH repeats arranged in tandem. The two domains are joined by a long bent α-helix (residues 269–295) designated as the linker helix.
Figure 1.
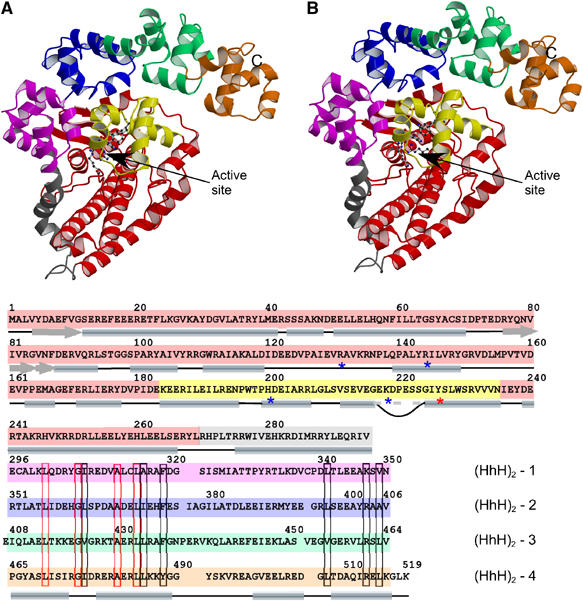
Overall structure of the 61 kDa amino terminal fragment of M. kandleri topoisomerase V (topo-61). (A) Stereo diagram of the structure showing the topoisomerase domain (red), the linker helix (gray) and the four (HhH)2 domains (pink, blue, green and orange). The HTH motif containing some of the active site residues is shown in yellow. The putative active site residues are shown as ball and stick. The C-terminus is marked with a C. The N-terminus resides at the beginning of the β-strand behind the HTH domain and is not marked for clarity. (B) Sequence of the topo-61 fragment showing the location of the different domains. The position of the secondary structure elements is shown by cylinders and arrows. The loop inserted in the recognition helix of the HTH motifs is shown by a curved line; the recognition helix is continuous despite this insertion. The putative active site residues are marked with an asterisk (*). The sequences of the (HhH)2 domains were aligned based on the structure. The position of the helices in the HhH motif is shown at the bottom of the alignment, but for clarity the helix linking the HhH motifs in an (HhH)2 domain is not shown. The alignment shows marked sequence similarities in the first HhH motif, but not in the second motif. Similar residues are boxed in black, identical residues are boxed in red.
The topoisomerase domain is essentially all α-helical, except for a short stretch at the N-terminus of the protein, where three β-strands are found arranged in an antiparallel fashion. A search with DALI (Holm and Sander, 1993) using the entire topoisomerase domain did not reveal any similarities to any other proteins. It appears that the structure of the topoisomerase domain does not resemble the structure of any known protein. Nevertheless, some local structural similarities to other proteins could be recognized. A three-helix bundle with a left-handed twist and a single crossover can be recognized at the N-terminus. A search with DALI (Holm and Sander, 1993) with this helical bundle showed significant similarities with ferritin-like four-helical bundle domains in the SCOP database (Murzin et al, 1995). However, only the first three helices of these four-helix bundles superposed with the three-helix bundle of topoisomerase V. Towards the C-terminus of the topoisomerase domain, a helix–turn–helix (HTH) domain (Aravind et al, 2005) is present, although it contains an insertion not normally observed in HTH domains.
The C-terminus multiHhH domain consists of loosely packed (HhH)2 domains rendering the overall multiHhH domain more variable in comparison to the topoisomerase domain (Figure 2). Consequently, while the topoisomerase domains are virtually identical in the four molecules observed and were treated as such during the refinement (with the major differences found in the connecting loops), the r.m.s.d. among the four multiHhH domains is much higher and varies from 1.27 to 2.04 Å in the four structures (Figure 2). The multiHhH domain also contains a disulfide bond (Cys-314 to Cys-338), which is unusual for a protein produced in the reducing environment of bacteria, but not so unusual for an archaeal protein (Kadokura et al, 2003).
Figure 2.

The multiHhH domain is flexible, while the topoisomerase domain is rigid. Schematic diagram showing the superposition of the two monomers in the asymmetric unit of crystal Form I. The active site residues are shown in yellow. In (A) only the multiHhH domains were used for the superposition, while in (B) the topoisomerase domains were used for the superposition. The figure illustrates the variability in the multiHhH domains (Supplementary data). For clarity, in each monomer the topoisomerase domain and the multiHhH domain are shown in different shades of the same color.
Active site of topoisomerase V
Tyr-226 identifies the active site of topoisomerase V (Belova et al, 2001) and allows the identification of other residues that could be involved in cleavage and religation of DNA. Several candidates have been proposed earlier as possible catalytic residues of topo-61 (Belova et al, 2001). In the structure, most of these are not in the vicinity of the active site tyrosine, ruling out their role in cleavage and religation of DNA.
To identify candidate active site residues, the active site tyrosines of human topoisomerase I (Tyr-723) (Redinbo et al, 1998) and vaccinia virus topoisomerase I (Tyr-274) ((Cheng et al, 1998) were used to search for amino acids of similar chemical type and also for groups of residues in a similar spatial arrangement. The structure of the region around the active site in both human and vaccinia virus topoisomerase I is highly conserved and consists of a similar constellation of amino acids surrounding the active site tyrosines Arg-488, Arg-590, Lys-532 and His-632 in human topoisomerase I, and Arg-130, Lys-167, Arg-223 and His-265 in vaccinia topoisomeraseI. All residues of the catalytic pentad of human topoisomerase I have an equivalent in the vicinity of the topoisomerase V active site tyrosine, namely Arg-131, Arg-144, His-200 and Lys-218 (Figure 3A). However, the five residues in topoisomerase V are not spatially arranged in the same manner as in the type IB enzymes. Interestingly, three of the residues of the putative catalytic pentad (Tyr-226, His-200 and Lys-218) of topoisomerase V are present in the HTH domain, while the remaining two arginines are present in an intervening loop and in a helix. There is no equivalent HTH domain in vaccinia virus or human topoisomerase I. Overall, the lack of structural conservation of the putative active site residues in topoisomerase V and the active site residues in type IB topoisomerases argues against a conserved mechanism of cleavage and religation. Nevertheless, the similarity in the type of residues surrounding the catalytic tyrosine helps to identify residues that could be involved in catalysis.
Figure 3.
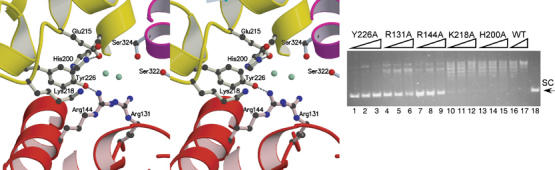
Topoisomerase V active site. (A) Stereo diagram of the active site region. The location is defined by the presence of the active site tyrosine, Tyr226. Other putative residues involved in catalysis are shown, including Arg-131, Arg-144, His-200 and Lys-218. A network of interactions involving water molecules (shown in green) is present. The first (HhH)2 domain blocks access to the active site and would have to move away in order to expose the active site. The coloring of the structure corresponds to the scheme in Figure 1. (B) Mutational analysis of the putative active site residues. The amino acids were mutated to alanine and tested for relaxation activity. Five mutants were tested: Y226A, R131A, R144A, K218A and H200A. In the assay, 0.2 μg of negatively supercoiled pBR322 DNA was incubated at 75°C for 15 min with 1, 2 or 5 U of protein, respectively. Lanes 1–3, Y226A; lanes 4–6, R131A; lanes 7–9, R144A; lanes 10–12, K218A; lanes 13–15, H200A. As a control, the relaxation by 0.7 and 5 U of wild-type protein (WT) is also shown (lanes 16 and 17). Lane 18 shows plasmid DNA with no protein. The position of the supercoiled DNA is labeled as SC.
To assess whether the five amino acids are indeed forming part of the active site, they were separately mutated to alanines and each mutant tested for topoisomerase activity. As shown in Figure 3B, Y226A, R131A and R144A show complete or marked reduction of topoisomerase activity (lanes 1–9), while K218A and H200A show a marginal reduction in topoisomerase activity. None of the mutants had a complete loss of activity, but this is not unusual and has been observed in other topoisomerases. In the case of vaccinia virus topoisomerase I, mutating the different residues in the catalytic pentad resulted in widely different levels of activity reduction (Cheng et al, 1997; Petersen and Shuman, 1997; Wittschieben and Shuman, 1997).
A search for additional residues that may be a part of the active site reveals the presence of a glutamate residue (Glu-215) at 3.0–4.0 Å from Tyr-226. An E215A mutation in topo-61 did not lead to any discernible loss of activity. Aside from residues forming part of the first (HhH)2 domain, there are no other amino acids in the vicinity of the active site tyrosine. As the structure suggests that the multiHhH domain needs to move in order to provide access to the active site, the residues in the first (HhH)2 domain were not considered as part of the active site.
DNA binding
Two different DNA-binding motifs can be identified in the topo-61 structure: an HTH motif in the topoisomerase domain and eight tandem HhH motifs (or four (HhH)2 domains) in the multiHhH domain. The four (HhH)2 domains are structurally similar, but there are variations within the domains. The relative disposition of the two motifs as well as the length and structure of the helix linking them varies from one (HhH)2 domain to another. Interestingly, there is noticeable sequence conservation in the first HhH motif of the (HhH)2 domains: all motifs are of the same length and contain several completely or highly conserved amino acids (Figure 1). The second HhH motifs vary in sequence length and do not show strong sequence conservation.
From previous structures, it is known that HhH motifs interact with DNA mainly through residues in the hairpin linking the two helices forming the motif (Shao and Grishin, 2000). In the topoisomerase V structure, the hairpins in the two HhH motifs in each (HhH)2 domain are separated by 15–23 Å, compared with the ∼20 Å separation observed in complexes of (HhH)2 with DNA. Modeling of DNA binding to the (HhH)2 domains using a known structure of a (HhH)2/DNA complex (Ariyoshi et al, 2000) shows that each (HhH)2 domain alone is arranged in a manner that is compatible with DNA binding. However, not all (HhH)2 domains in topoisomerase V could bind to the same DNA molecule simultaneously without a large conformational change in the protein, the DNA or both (Supplementary data). If the DNA is modeled as a canonical, straight B-DNA molecule, it appears that the first two (HhH)2 domains could bind DNA without a large conformational change, while the last two (HhH)2 domains could also bind without a large change, but all four domains could not bind to the same DNA molecule simultaneously.
A second DNA-binding motif in the topo-61 structure is an HTH-like domain found towards the C-terminus of the topoisomerase domain. An interesting variation to the HTH domain in topoisomerase V is the insertion of a small loop at the N-terminus of the recognition helix. This loop contains several polar residues that could take part in additional interactions with the phosphate backbone of DNA and also contains one of the residues involved in catalysis, Lys-218. In all, three of the five residues that form the active site are found in the HTH domain.
In the structure of topo-61, it is impossible to model-build DNA bound to the HTH domain without removing the (HhH)2 domain blocking the active site. Removal of the (HhH)2 domains and the linker region from the topoisomerase domain exposes a large groove that represents a potential DNA-binding surface and also exposes the HTH domain. A surface charge potential indicates that the proposed DNA-binding cavity of topo-61 is positively charged (Figure 4A). Once the (HhH)2 domains and the linker helix are removed, DNA interactions with the HTH of topoisomerase V can be modeled. DNA can be model-built to bind to the HTH, with the major groove of DNA interacting with the third helix of the HTH domain (Figure 4B). In the model, the active site residues are all close to the DNA backbone and in a position suitable for catalysis. In particular, the active site tyrosine points directly to the phosphodiester backbone. Some regions of the protein interfere with binding, but only small changes would be needed to accommodate the DNA. In this model, around two full DNA turns would interact with the topoisomerase domain. Additionally, one of the helices in the protein far away from the active site sits in the major groove of DNA.
Figure 4.

Model for a topoisomerase domain–DNA complex. (A) Electrostatic surface representation of the topoisomerase domain. The diagram shows that the topoisomerase has a large positively charged groove in one face of the protein centered around the active site. The insets correspond to 90° views of the molecule and show that there are no additional large positively charged regions in the protein. The electrostatic potential was calculated with the program APBS (Baker et al, 2001), with a dielectric constant of 80 for the solvent and 20 for the protein. The surface is colored with a blue to red gradient from +5 to −5 KbT/e. (B) The diagram shows a model of the topoisomerase domain in complex with B-DNA. The coloring scheme is the same as in Figure 1. To create the model, the linker helix and the multiHhH domain were removed. DNA was docked using the HTH motif of human Pax6-paired domain–DNA complex (Xu et al, 1999), but replacing the DNA with a canonical, straight B-DNA model. No attempts were made to prevent steric clashes. In the model, the active site tyrosine is ideally placed to interact with the phosphodiester backbone and the other putative active site residues are also in the vicinity of DNA. The model suggests a potential way for topo-61 to interact with DNA, making extensive interactions.
Conformational changes at high temperature are needed for activity
M. kandleri is a hyperthermophile methanogen that grows under extreme temperature conditions, around 110°C. Not surprisingly, topoisomerase V is active at high temperature, with optimal activity around 108°C, but inactive at temperatures below ∼60°C (Slesarev et al, 1993). This is somewhat surprising as other enzymes from thermophiles are active at lower temperatures, albeit somewhat less efficiently. One possible explanation for the temperature dependence of topoisomerase V is that the enzyme changes its conformation at higher temperatures. Moreover, it was observed before that an additional proteolytic cleavage site in the second (HhH)2 domain appears above 80°C and results in an N-terminus 380 residue fragment, topo-44 (Belova et al, 2002). This proteolytic site is absent at lower temperatures, suggesting that there is a conformational change in topoisomerase V at high temperature that facilitates protease accessibility to this site.
In order to explore the possibility of a temperature-dependent change in conformation of the topo-61 fragment, tryptophan fluorescence of the protein was monitored as a function of temperature. Topo-61 contains four tryptophans, three in the topoisomerase domain and one in the linker helix. A small blue shift in the fluorescence emission maximum of topo-61 was observed upon heating (Figure 5A). This shift may correspond to a highly specific and local conformational change at a temperature around 65°C, which may be sufficient to enable DNA binding in the active site, since DNA relaxation assays with topo-61 at varying temperatures follow a similar trend and show increasing activity at higher temperatures (Figure 5C). The reversibility of this shift, in addition to its subtle nature, is also suggestive of its biological significance.
Figure 5.
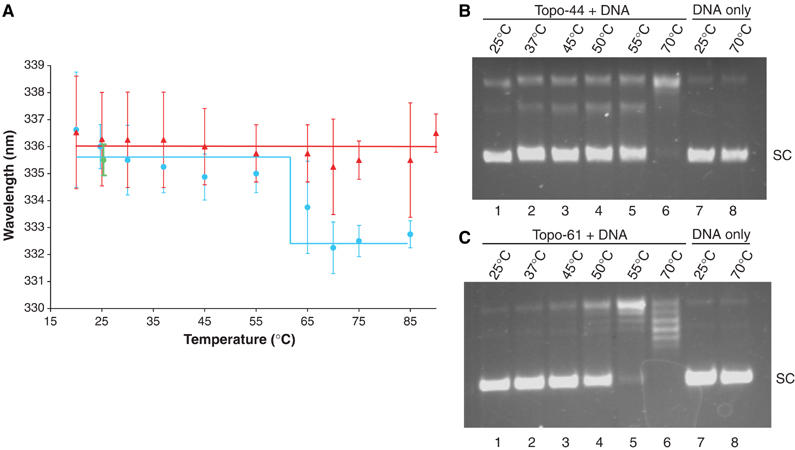
The 61 kDa N-terminal fragment of topoisomerase V undergoes a temperature-dependent conformational change. (A) Plot of the wavelengths of maximum emission versus temperature. A small blue shift is observed in the fluorescence emission maximum of topo-61, but not topo-44 as the temperature is increased. The blue circles and red triangles show the position of emission maximum at each temperature for topo-61 and for topo-44, respectively. A line is used as a guide to identify the two conformational states of topo-61 or the single conformational state of topo-44, respectively. The wavelength of maximum emission returns to its original value upon cooling the protein back to 25°C (green circle). The error bars correspond to the standard deviations of the measurements. (B, C) Temperature dependence of the relaxation activity of topo-44 and topo-61. Relaxation assays were performed at the different temperatures indicated. Topo-61 is not active below 50°C (lanes 1–3 in (C)), above which its activity increases with temperature (lanes 4–6 in (C)). Similarly, topo-44 shows no activity below 55°C (lanes 1–4 in (B)), above which its activity increases with temperature (lanes 5–6 in (B)). Lanes 7 and 8 in both (B) and (C) show DNA with no protein at 25 and 70°C. The conformational change in topo-61 correlates with the temperature dependence of its relaxation activity.
DNA relaxation assays with topo-44 also follow a temperature-dependent activity profile (Figure 5B). However, unlike topo-61, topo-44 does not show the blue shift in fluorescence emission maximum at higher temperatures (Figure 5A). This difference in the two proteins can be attributed to the presence of fewer HhH motifs in topo-44, as both topo-44 and topo-61 contain the same subset of tryptophans. The observation is also in agreement with the proteolysis experiments and suggests that the conformational change may involve the HhH domains following topo-44. In the absence of DNA, topo-44 behaves as a compact core subdomain of topoisomerase V, even at higher temperatures.
To confirm that the blue shift observed upon heating is not due to unfolding of the protein, topo-61 was unfolded at room temperature with guanidium chloride, which results in a red shift of the fluorescence emission maximum to 350 nm. This was further confirmed by 8-anilino-1-naphthalene sulfonate (ANS) binding and potassium iodide (KI) quenching experiments. There is no enhancement of ANS fluorescence upon heating the protein. In addition, as much as 85% quenching of fluorescence signal by KI occurs upon unfolding with guanidium chloride in comparison to only ∼20% quenching upon heating, further supporting the occurrence of a conformational change at temperatures >65°C, which does not involve its unfolding.
Discussion
Topoisomerase V contains several potential DNA-binding sites
The structure of the 61 kDa fragment of topoisomerase V reveals several interesting features of this enzyme. The protein is arranged into two distinct domains that interact extensively with each other, but that probably need to detach from each other for activity. Topo-61 is the only protein containing more than three HhH motifs or more than one (HhH)2 domain in the same polypeptide, whose structure is known. Recently, in the structure of Aeropyrum pernix XPF endonuclease it was observed that (HhH)2 domains can bind DNA in tandem, although in this case each (HhH)2 domain comes from a different polypeptide (Newman et al, 2005). Interestingly, in XPF endonuclease, the (HhH)2 domain is linked to the nuclease domain by a flexible linker and a domain rearrangement is needed to allow DNA binding (Newman et al, 2005).
The (HhH)2 domains have been shown to be responsible for nonsequence specific DNA binding in a number of proteins and the DNA-binding mode has been observed in several instances (Ariyoshi et al, 2000; Hollis et al, 2000; Newman et al, 2005). In topo-61, the DNA-binding loops in the (HhH)2 domains face away from the active site. Additionally, it is impossible to model build a complex of a straight DNA molecule with all four (HhH)2 domains at once. This may mean that DNA has to be highly deformed, that the DNA-binding motifs have to change conformation, or a combination of both. Another possibility is that the different (HhH)2 domains bind the same DNA molecule by wrapping around the DNA, in a manner reminiscent of tandem repeats of zinc-fingers bound to DNA (Pavletich and Pabo, 1991). Furthermore, the protein may bind DNA without all (HhH)2 domains binding DNA simultaneously, as was observed for GLI, another protein with tandem repeats of zinc-fingers (Pavletich and Pabo, 1993). The latter possibility is supported by the observation that the second and third (HhH)2 domains share a helix that would have to move or melt to allow both domains to bind DNA simultaneously, even to highly deformed DNA.
Type IA and type II topoisomerases contain HTH domains that harbor the active site tyrosine and termed 5Y-CAP domains (Berger et al, 1998). However, the tyrosine in topoisomerase V is found at a different location in the HTH domain, in the middle of the recognition helix, as opposed to a loop following the recognition helix. The 5Y-CAP domains and the HTH domain in topoisomerase V are structurally similar, but the functionally important residues are not spatially conserved. HTH domains are ubiquitous, and hence the presence of an HTH domain in type IA and type II enzymes and in topoisomerase V may be just fortuitous.
Type IB topoisomerases and topoisomerase V have different active sites
Like type IB enzymes, topoisomerase V forms a 3′ phosphotyrosine bond and relaxes DNA in steps of one. However, the structure of topo-61 shows that topoisomerase V has an overall fold different from other type IB enzymes. It shares a similar set of amino acids surrounding the active site tyrosine, but they are not in equivalent spatial positions. In other enzymes where a tyrosine is also involved in DNA cleavage, similar groups of amino acids have been implicated, but the catalytic mechanism is different. For example, in telomerase resolvase ResT, a catalytic pentad of identical composition has been identified, but the mechanism of catalysis is different from the one used by tyrosine recombinases and type IB topoisomerases (Deneke et al, 2004). In topoisomerase V, aside from the active site tyrosine, mutagenesis studies implicate two arginines in catalysis, Arg-131 and Arg-144, which show a marked reduction in catalytic activity (>102 loss). Additionally, mutation of a lysine and a histidine also shows a modest reduction in activity (∼5–10-fold loss). In contrast, mutation of Glu-315 resulted in no loss of activity, serving as a control that not all mutations result in activity reduction. In comparison, in vaccinia virus topoisomerase I, the two arginines, one lysine and the other histidine, show a reduction of activity of 105, 105, 104 and 102, respectively (Cheng et al, 1997; Petersen and Shuman, 1997; Wittschieben and Shuman, 1997). The measurements for topoisomerase V are crude and further work is needed to ascertain more carefully the effect of these mutations, but it is already clear that the effects observed for the lysine and histidine are small and probably indicate that these two amino acids are involved in but are not crucial for catalysis. Overall, the structural and mutagenesis studies indicate that type IB topoisomerases and topoisomerase V do not share a common mechanism of cleavage and religation, even though both form 3′ phosphotyrosine bonds.
Conformational changes are needed for topoisomerase V activity
It is clear from the structure that the protein has to change conformation in order to allow access of DNA to the active site. In addition, the changes observed in the intrinsic fluorescence emission maximum at higher temperatures suggest that the protein undergoes a change near the same temperature, where it starts to become active. These changes could correspond to movements triggered by the rearrangement of the HhH motifs to allow DNA binding in the active site. Alternately, the changes could be localized to another region close to the active site to enlarge the cavity for DNA binding, although this possibility appears to be less likely. The presence of a buried active site is reminiscent of type IA enzymes that also contain buried active sites (Changela et al, 2001), but different from type IB enzymes that have more accessible active sites (Cheng et al, 1998; Redinbo et al, 1998). Large conformational rearrangements have been observed in topoisomerase III in complex with DNA (Changela et al, 2001) and in structures of topoisomerase I (Feinberg et al, 1999) and topoisomerase II (Fass et al, 1999), indicating that topoisomerases are enzymes that require to change conformation for activity. It is thus not surprising that topoisomerase V may also have to undergo a conformational change for activity.
Topoisomerase V represents a new structural family of topoisomerases
Type I topoisomerases are divided into type IA and IB and this classification has been accurate so far, with all the enzymes of the same type sharing structural and mechanistic similarities (Figure 6). Surprisingly, the overall structure of the topoisomerase domain of topoisomerase V is different from that of all other previously observed topoisomerase structures. Due to their biochemical similarities (Slesarev et al, 1993), it was expected that topoisomerase V would share some structural similarities with type IB enzymes despite their low sequence similarity. It is hence surprising to find that it does not show any structural similarity to type IB topoisomerases.
Figure 6.

Topoisomerase V represents a new family of type IB topoisomerases. The figure summarizes some of the properties of the three families of type I topoisomerases. Each column shows the overall structure and the active site region of a representative of the respective family (type IA, E. coli topoisomerase III (Changela et al, 2001); type IB, human topoisomerase I (Redinbo et al, 1998); topo V, 61 kDa N-terminal fragment of topoisomerase V). Type IA enzymes form a very distinct family with no sequence, structural or mechanistic similarities to the other families. Type IB enzymes and topoisomerase V share many overall features, but there is no sequence, structure or apparent mechanistic similarities between these two families.
The structure of topoisomerase V provides few clues on the possible mechanism of DNA relaxation by this enzyme. It appears that topoisomerase V shares with type IA and type II enzymes the need to change conformation for activity, as large protein rearrangements will be necessary to bind DNA in the active site. Type IA and type II enzymes change the DNA topology by passing strands, and hence require the presence of holes in the protein to accommodate the passing strands. There are no similar regions in topoisomerase V in the conformation observed, suggesting that it is not likely that this enzyme uses a strand passage mechanism. All type IB enzymes have been proposed to relax DNA through a ‘controlled rotation' mechanism (Champoux, 2001), where the protein embraces the DNA and allows one strand to rotate around the other. Topoisomerase V shows a large groove that could accommodate DNA and make extensive interactions, but it is not clear whether the protein can surround DNA. Nevertheless, as the structure of topoisomerase V does not have any holes, it is more likely that it uses a variation of the ‘controlled rotation' mechanism employed by type IB enzymes.
Topoisomerase V has no sequence, structural and probably mechanistic similarities to any other topoisomerase or protein. It does not appear to be evolutionary related to type IA or IB enzymes, tyrosine recombinases or any other protein. Instead, it appears that some archaeal organisms, such as M. kandleri, have a completely different topoisomerase whose metabolic role is still unknown. The need for this new family of topoisomerases is not clear, as the genome of M. kandleri not only encodes for topoisomerase V but also for a reverse gyrase and a type IA enzyme (Slesarev et al, 2002). Topoisomerase V must have evolved to fulfill a specific role in some extremophiles and, in doing so, a new way of changing DNA topology emerged. Whether topoisomerase V has an ancestor in bacteria or is conserved in specific eukaryotes remains to be determined, but so far it appears that topoisomerase V represents an enzyme unique to archaea, with a unique structure and a unique mechanism.
Materials and methods
Purification and crystallization
An N-terminus fragment spanning residues 1–532 of M. kandleri topoisomerase V (topo-61) had been cloned earlier into PET14b (Novagen) (Belova et al, 2001). It was transformed into E. coli BL21(DE3) and the transformed cells were grown at 37°C in LB medium containing 100 μg/ml ampicillin to an optical density A600 of ∼0.6. Protein production was induced by adding 1 mM IPTG and then cells were allowed to grow for 4 h before harvesting by centrifugation. The cell pellet was resuspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 0.5 mM EDTA and 1 mM DTT), lysed by sonication, and the lysed cells clarified by centrifugation at 30 000 g. The supernatant was heated at 75°C for 30 min to precipitate other cellular proteins. The precipitated proteins were removed by centrifugation at 30 000 g, while topo-61 was retained in the supernatant. A further purification step over a HiTrap heparin column, as described earlier (Slesarev et al, 1994), resulted in pure protein. Seleno-methionine-substituted topo-61 was prepared using the suppression of methionine biosynthesis procedure (Doublie, 1997). Purification of the seleno-methionine-substituted topo-61 followed the wild-type protein purification procedure, except that all buffers contained 5 mM DTT. In both cases, the purified protein was finally dialyzed against 50 mM Tris, pH 8.0, 300 mM NaCl, 0.2 mM EDTA and 1 mM DTT (5 mM DTT for the Se-Met protein), and concentrated to 5 mg/ml for crystallization.
Topo-61 was crystallized by the sitting drop vapor diffusion method by mixing 3 μl of topo-61 with 3 μl of reservoir solution (12% PEG 3350, 100 mM Na-cacodylate, pH 6.5 and 50 mM MgCl2) at 22°C. To grow reproducible crystals, a 1:1000 dilution of 1 U of trypsin was included in the crystallization drop. Crystals of approximate size 0.3 × 0.2 × 0.1 mm3 were obtained in 5–7 days. For data collection, crystals were flash frozen in liquid nitrogen after transfers into increasing PEG 3350 solutions to a final PEG concentration of 35% in incremental steps of 5% with 30-s incubations between each transfer.
The topo-61dC1–4 constructs spanning residues 1–513, 519, 523 or 530, respectively, and a topo-44 construct spanning residues 1–380 were prepared by PCR amplification from the topo-61 clone. The proteins were expressed and purified as the full-length topo-61. Crystals of topo-61dC2 were obtained by the sitting drop vapor diffusion method in identical conditions to topo-61, but without trypsin (crystal form I); all other protein constructs crystallized only in the presence of trypsin. A second crystal form was found with 100 mM sodium acetate buffer, pH 4.6, and 2 M sodium formate as precipitant. Form I crystals were flash frozen identically to topo61, while the new crystal form was flash frozen in liquid nitrogen with 20% glycerol in the crystallization buffer as a cryoprotectant.
Data collection and structure determination
Diffraction data were collected at the Dupont Northwestern Dow and the Life Science Collaborative Access Team stations (DND-CAT and LS-CAT) at the Advanced Photon Source in Argonne National Laboratory. Data collection statistics are shown in Table I. Both crystal forms of topo61 are orthorhombic and each contains two molecules in the asymmetric unit.
The structure of topo-61 was solved by SAD using data collected at the peak energy of the X-ray absorption spectrum of selenium. Data were processed and integrated using XDS (Kabsch, 1993) and scaled with SCALA (Collaborative Computational Project, 1994). Two of the 14 possible Se sites in the asymmetric unit were located using SOLVE (Terwilliger, 2003). Ten additional sites were located after refinement of the partial heavy atom model with SHARP (delaFortelle and Bricogne, 1997). Final phasing calculations were performed with SHARP. Solvent flattening yielded good-quality maps that allowed automatic building of 46% of amino acids with RESOLVE (Terwilliger, 2003). The remaining amino acids in the structure were built manually using O (Jones et al, 1991). Iterative cycles of model building and refinement using REFMAC5 (Murshudov et al, 1997) resulted in a final R-factor of 0.217 and Rfree of 0.292 (see Table I). Throughout the refinement, the topoisomerase domains were tightly restrained to be noncrystallographically symmetry related, except for a few loops. It became apparent during the last stages of model building that the (HhH)2 domains are loosely packed against each other and that the multiHhH domains are not noncrystallographically symmetry related. At the end of the refinement the restraints were removed, but this resulted only in a modest improvement in the refinement statistics and hence the restrained model was accepted. Hence, the topoisomerase domains are virtually identical. The final model consists of two monomers, spanning residues 3–519 each, 510 water molecules and one Mg ion.
The structure of topo-61dC2 in crystal form II was solved by Molecular Replacement using the topoisomerase domain of topo-61 (residues 1–282) as the search model, with B-factors set to 20 Å2. Rotation and translation searches using data to 3 Å resulted in a single solution in CNS (Brunger et al, 1998). Rigid-body refinement in Refmac5 improved the maps and helped locate the density for the HhH repeats. The remaining model was built using O with interspersed refinement cycles using REFMAC5, while the resolution was extended to 2.9 Å resolution. Tight noncrystallographically symmetric restraints for the topoisomerase domain were used during all stages of the refinement. The final R-factor and Rfree are 0.236 and 0.308, respectively (see Table I). The final model consists of two monomers spanning residues 3–518.
Mutagenesis of active site residues
The putative catalytic residues forming the active site, namely, Arg-131, Arg-144, His-200 and Lys-218, along with Glu-215, were mutated to alanines by using the Quickchange site-directed mutagenesis kit (Stratagene) on the topo-61dC2 construct. Additionally, Tyr-226 was also mutated to alanine as a control. The presence of the desired mutation was confirmed by DNA sequencing. The mutant proteins were purified as described above.
Relaxation assays
Topoisomerase activity assays were performed by incubating the protein (topo61-dC2, topo-44, or the active site mutants) with 0.2 μg of negatively supercoiled pBR322 DNA in 30 mM Tris–HCl, pH 7.0, 30 mM NaCl and 5 mM MgCl2. The reactions were carried out at the desired temperature for 15 min and terminated by cooling and addition of SDS to 1%. The products were analyzed by electrophoresis on a 1% agarose gel and visualized by ethidium bromide staining.
Fluorescence measurements
Fluorescence measurements were performed with a Perkin-Elmer LS50B luminescence spectrometer in a 1 ml quartz cuvette in the temperature range of 20–90°C. For tryptophan fluorescence measurements, an excitation wavelength of 295 nm was used and the emission spectrum recorded between 300 and 400 nm with both excitation and emission slit widths set at 8 nm. The concentration of the protein (topo61-dC2 or topo-44) for intrinsic fluorescence measurements was kept at 40 μg/ml in 30 mM Tris, pH 8.0, 300 mM NaCl, and the protein incubated for 5 min at each temperature point before taking the scan.
KI quenching experiments were performed by incubating 1 μM of topo61-dC2 with 1.5 M KI for at least 1 h before taking scans and all steps carried out in the dark. An additional incubation of 15 min was allowed at each temperature point before taking the scan.
Topo61-dC2 was unfolded by incubating with 5 M guanidium chloride for 3 h before measuring the spectra. To monitor quenching of tryptophan fluorescence upon unfolding, 1.5 M KI was included in the unfolding reaction.
ANS was used as an extrinsic fluorescence probe to analyze exposure of hydrophobic regions in topo61-dC2 due to possible unfolding of the protein upon heating. ANS is a hydrophobic dye that shows poor fluorescence in polar solutions, but upon binding to hydrophobic surfaces results in a multiple-fold increase in its fluorescence emission. ANS was used at a concentration of 40 μM, the excitation wavelength was 380 nm and the emission wavelength scanned from 400 to 600 nm. The excitation and emission slit widths were 8 nm each, as above, and the concentration of topo61-dC2 was 1 μM.
In all experiments, appropriate buffer baselines were subtracted for all measured spectra and the spectra normalized for comparison. Each spectrum was an average of three scans. The fluorescence intensities were collected from at least two different protein samples to confirm the results.
Protein data bank accession codes
The final structure coordinates of topo-61 and topo-61dC2 have been deposited in the Protein Data Bank, with accession codes 2CSB and 2CSD, respectively.
Supplementary Material
Supplementary Table I
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary References
Acknowledgments
We thank M Backovic, A Krasilnikov, S Maar and G Minasov for comments and advice, RI MacDonald for her comments, help, and advice with the fluorescence experiments, and G Belova for her advice with expression plasmids. We acknowledge the use of equipment at the Keck Biophysics Facility at Northwestern University and DND and LS CAT at the APS. Support from the RH Lurie Comprehensive Cancer Center of Northwestern University to the Structural Biology Facility is acknowledged. Research was supported by NIH grants GM51350 (to AM) and 2R44CA101566 (to AS). Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center at the Advanced Photon Source (APS). DND-CAT is supported by Dupont, DOW and the NSF. Use of the APS is supported by the Department of Energy (DOE).
References
- Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM (2005) The many faces of the helix–turn–helix domain: transcription regulation and beyond. FEMS Microbiol Rev 29: 231–262 [DOI] [PubMed] [Google Scholar]
- Ariyoshi M, Nishino T, Iwasaki H, Shinagawa H, Morikawa K (2000) Crystal structure of the holliday junction DNA in complex with a single RuvA tetramer. Proc Natl Acad Sci USA 97: 8257–8262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belova GI, Prasad R, Kozyavkin SA, Lake JA, Wilson SH, Slesarev AI (2001) A type IB topoisomerase with DNA repair activities. Proc Natl Acad Sci USA 98: 6015–6020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belova GI, Prasad R, Nazimov IV, Wilson SH, Slesarev AI (2002) The domain organization and properties of individual domains of DNA topoisomerase V, a type 1B topoisomerase with DNA repair activities. J Biol Chem 277: 4959–4965 [DOI] [PubMed] [Google Scholar]
- Berger JM, Fass D, Wang JC, Harrison SC (1998) Structural similarities between topoisomerases that cleave one or both DNA strands. Proc Natl Acad Sci USA 95: 7876–7881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Champoux JJ (2001) DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70: 369–413 [DOI] [PubMed] [Google Scholar]
- Changela A, DiGate RJ, Mondragón A (2001) Crystal structure of a complex of a type IA DNA topoisomerase with a single-stranded DNA molecule. Nature 411: 1077–1081 [DOI] [PubMed] [Google Scholar]
- Cheng C, Kussie P, Pavletich N, Shuman S (1998) Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell 92: 841–850 [DOI] [PubMed] [Google Scholar]
- Cheng C, Wang LK, Sekiguchi J, Shuman S (1997) Mutational analysis of 39 residues of vaccinia DNA topoisomerase identifies Lys-220, Arg-223, and Asn-228 as important for covalent catalysis. J Biol Chem 272: 8263–8269 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Corbett KD, Berger JM (2004) Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct 33: 95–118 [DOI] [PubMed] [Google Scholar]
- Cruickshank DW (1999) Remarks about protein structure precision. Acta Crystallogr D 55: 583–601 [DOI] [PubMed] [Google Scholar]
- delaFortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol 276: 472–494 [DOI] [PubMed] [Google Scholar]
- Deneke J, Burgin AB, Wilson SL, Chaconas G (2004) Catalytic residues of the telomere resolvase ResT: a pattern similar to, but distinct from, tyrosine recombinases and type IB topoisomerases. J Biol Chem 279: 53699–53706 [DOI] [PubMed] [Google Scholar]
- Doublie S (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol 276: 523–530 [PubMed] [Google Scholar]
- Fass D, Bogden CE, Berger JM (1999) Quaternary changes in topoisomerase II may direct orthogonal movement of two DNA strands. Nat Struct Biol 6: 322–326 [DOI] [PubMed] [Google Scholar]
- Feinberg H, Lima CD, Mondragón A (1999) Conformational changes in E. coli DNA topoisomerase I. Nat Struct Biol 6: 918–922 [DOI] [PubMed] [Google Scholar]
- Hollis T, Ichikawa Y, Ellenberger T (2000) DNA bending and a flip-out mechanism for base excision by the helix–hairpin–helix DNA glycosylase, Escherichia coli AlkA. EMBO J 19: 758–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Huber R, Kurr M, Jannasch HW, Stetter KO (1989) A novel group of abyssal methanogenic archaebacteria (Methanopyrus) growing at 110°C. Nature 342: 833–834 [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kabsch W (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 26: 795–800 [Google Scholar]
- Kadokura H, Katzen F, Beckwith J (2003) Protein disulfide bond formation in prokaryotes. Annu Rev Biochem 72: 111–135 [DOI] [PubMed] [Google Scholar]
- Kozyavkin SA, Pushkin AV, Eiserling FA, Stetter KO, Lake JA, Slesarev AI (1995) DNA enzymology above 100 degrees C. Topoisomerase V unlinks circular DNA at 80–122 degrees C. J Biol Chem 270: 13593–13595 [DOI] [PubMed] [Google Scholar]
- Lima CD, Wang JC, Mondragón A (1994) Three-dimensional structure of the 67 K N-terminal fragment of E. coli DNA topoisomerase I. Nature 367: 138–146 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247: 536–540 [DOI] [PubMed] [Google Scholar]
- Newman M, Murray-Rust J, Lally J, Rudolf J, Fadden A, Knowles PP, White MF, McDonald NQ (2005) Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J 24: 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO (1991) Zinc finger-DNA recognition: crystal structure of a Zif268–DNA complex at 2.1 A. Science 252: 809–817 [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO (1993) Crystal structure of a five-finger GLI–DNA complex: new perspectives on zinc fingers. Science 261: 1701–1707 [DOI] [PubMed] [Google Scholar]
- Petersen BO, Shuman S (1997) Histidine 265 is important for covalent catalysis by vaccinia topoisomerase and is conserved in all eukaryotic type I enzymes. J Biol Chem 272: 3891–3896 [DOI] [PubMed] [Google Scholar]
- Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 279: 1504–1513 [DOI] [PubMed] [Google Scholar]
- Rodriguez AC, Stock D (2002) Crystal structure of reverse gyrase: insights into the positive supercoiling of DNA. EMBO J 21: 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Grishin NV (2000) Common fold in helix–hairpin–helix proteins. Nucleic Acids Res 28: 2643–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesarev AI, Lake JA, Stetter KO, Gellert M, Kozyavkin SA (1994) Purification and characterization of DNA topoisomerase V. An enzyme from the hyperthermophilic prokaryote Methanopyrus kandleri that resembles eukaryotic topoisomerase I. J Biol Chem 269: 3295–3303 [PubMed] [Google Scholar]
- Slesarev AI, Mezhevaya KV, Makarova KS, Polushin NN, Shcherbinina OV, Shakhova VV, Belova GI, Aravind L, Natale DA, Rogozin IB, Tatusov RL, Wolf YI, Stetter KO, Malykh AG, Koonin EV, Kozyavkin SA (2002) The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc Natl Acad Sci USA 99: 4644–4649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesarev AI, Stetter KO, Lake JA, Gellert M, Krah R, Kozyavkin SA (1993) DNA topoisomerase V is a relative of eukaryotic topoisomerase I from a hyperthermophilic prokaryote. Nature 364: 735–737 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2003) SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol 374: 22–37 [DOI] [PubMed] [Google Scholar]
- Wittschieben J, Shuman S (1997) Mechanism of DNA transesterification by vaccinia topoisomerase: catalytic contributions of essential residues Arg-130, Gly-132, Tyr-136 and Lys-167. Nucleic Acids Res 25: 3001–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO (1999) Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev 13: 1263–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table I
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary References