

Abstract
Deletion of Lsh perturbs DNA methylation patterns in mice yet it is unknown whether Lsh plays a direct role in the methylation process. Two types of methylation pathways have been distinguished: maintenance methylation by Dnmt1 occurring at the replication fork, and de novo methylation established by the methyltransferases Dnmt3a and Dnmt3b. Using an episomal vector in Lsh−/− embryonic fibroblasts, we demonstrate that the acquisition of DNA methylation depends on the presence of Lsh. In contrast, maintenance of previously methylated episomes does not require Lsh, implying a functional role for Lsh in the establishment of novel methylation patterns. Lsh affects Dnmt3a as well as Dnmt3b directed methylation suggesting that Lsh can cooperate with both enzymatic activities. Furthermore, we demonstrate that embryonic stem cells with reduced Lsh protein levels show a decreased ability to silence retroviral vector or to methylate endogenous genes. Finally, we demonstrate that Lsh associates with Dnmt3a or Dnmt3b but not with Dnmt1 in embryonic cells. These results suggest that the epigenetic regulator, Lsh, is directly involved in the control of de novo methylation of DNA.
Keywords: chromatin, DNA methylation, Dnmt3, epigenetic, Lsh
Introduction
DNA methylation regulates a number of biological processes, including genomic imprinting, X chromosome inactivation, silencing of tumor suppressor genes, and repression of retroviral elements (Bird, 2002; Li, 2002). Loss of methylation in mice results in severe developmental defects and early embryonic lethality (Li et al, 1992; Okano et al, 1999; Dennis et al, 2001). A number of human inherited diseases linked to faulty methylation pathways and exhibiting abnormal development include Rett, ICF, and ATRX syndromes (Amir et al, 1999; Okano et al, 1999; Gibbons et al, 2000). Moreover, aberrant methylation patterns are thought to be involved in tumorigenesis (Jones and Baylin, 2002; Chen et al, 2004; Yu et al, 2005) causing genomic instability, abnormal imprinting, and deregulated expression of oncogenes or tumor suppressor genes.
Two types of methylation pathways are functionally distinct: maintenance versus de novo methylation. During early mammalian embryogenesis, DNA methylation patterns are largely erased and re-established shortly after implantation in a wave of de novo methylation (Reik et al, 2001; Bird, 2002; Li, 2002). These newly established patterns are then thought to be faithfully copied after each round of replication onto the newly synthesized DNA strand. In contrast, genomic imprints, which are largely dependent on DNA methylation, are mostly established in germ cells and preserved throughout embryogenesis. Thus, maintenance activity is found in all somatic cells while the highest de novo methylation activity is found in embryonic cell lines, germ cells, or in postimplantation embryos.
Several DNA cytosine methyltransferases have been identified in mammalian cells (Chen and Li, 2004; Goll and Bestor, 2005). Dnmt1 is primarily responsible for maintenance methylation since Dnmt1 shows high affinity for hemimethylated substrates and is present at replication forks via its association with PCNA (Leonhardt et al, 1992; Chuang et al, 1997; Okano et al, 1998; Pradhan et al, 1999). De novo methylation activity is primarily dependent on Dnmt3a and Dnmt3b, two partially redundant Dnmt family members (Okano et al, 1999). Dnmt3a plays an additional crucial role in de novo methylation of imprinted sites in germ cells together with Dnmt3L, a Dnmt family member lacking catalytic activity (Hata et al, 2000; Bourc'his and Bestor, 2004; Kaneda et al, 2004). Although the enzymes responsible for methylation patterns have been identified, the precise molecular mechanisms including cofactors that lead to recruitment and efficient targeting of the enzymatic machinery to their appropriate sites are unknown.
We have previously reported that Lsh controls genomic methylation patterns in mice (Dennis et al, 2001, Muegge, 2005). Lsh belongs to the SNF2 family of proteins (Jarvis et al, 1996; Geiman et al, 1998), whose members participate in chromatin remodeling (Fyodorov and Kadonaga, 2001; Langst and Becker, 2004). Targeted disruption of Lsh in mice leads to developmental defects and early lethality (Geiman and Muegge, 2000; Dennis et al, 2001; Geiman et al, 2001; Fan et al, 2003; Sun et al, 2004). Lsh controls normal heterochromatin structure and function in mice, and upon deletion a number of epigenetic modifications are perturbed. For example, Lsh-deficient cells show genome wide CpG hypomethylation, altered histone H3 methylation, and increased acetylation levels for histone H3 and histone H4 (Dennis et al, 2001; Yan et al, 2003a, 2003b; Huang et al, 2004, Sun et al, 2004). Since epigenetic modifications are closely linked, it remains unclear which one is the initial epigenetic modification targeted by Lsh. In this report we attempt to determine the functional role of Lsh in either maintenance or de novo methylation, and to characterize the role of Lsh in the establishment of epigenetic modifications. We provide evidence that Lsh is required for de novo methylation of DNA and that it is directly involved in the methylation process.
Results
Lsh is required for methylation of episomal DNA in MEF cells
Lsh is a global regulator of DNA methylation in mice. To investigate further the functional role of Lsh in methylation, we used an episomal vector system (Figure 1A) that allows for the discrimination between de novo and maintenance of genomic methylation. The episomal vector can be transfected into mammalian cells to serve as a target for de novo methylation (Hsieh, 1999). The presence of novel CpG methylated sites can be determined using methylation-sensitive PCR once the episomal construct is recovered and completely digested with a methylation-sensitive restriction enzyme such as HpaII. Figure 1A illustrates the design of PCR primers to amplify regions of the episomal construct, pCEP4. Primer pair P5/6 surrounds multiple HpaII sites and can detect successful methylation. In contrast, the product of primer pair P1/P2 contains no HpaII sites, thus serving as an internal control for the amount of recovered episomal DNA. Primer pair P3/P4 amplifies a fragment that contains multiple DpnI sites and therefore is used as control to indicate successful replication in mammalian cells (DpnI can only digest bacterial Dam methylated DNA, and thus fails to cleave DNA that has replicated in mammalian cells).
Figure 1.
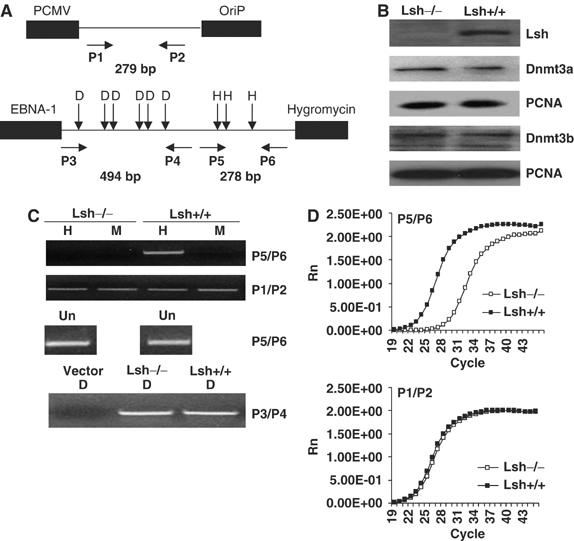
Lsh is required for methylation of an episomal vector in MEF cells. (A) Map of the episomal vector pCEP4 illustrating the location of HpaII/MspI and DpnI sites as well as the position of the primers used for methylation-sensitive PCR analysis. Primer pairs P5/P6 are designed for detection of methylation, P3/P4 for detection of successful replication, and P1/P2 as an internal control. The length of the expected PCR fragment is indicated in base pairs (bp). (B) Western blot analysis using nuclear extracts derived from Lsh−/− and Lsh+/+ embryonal fibroblasts (MEF) and specific antibodies against Lsh, Dnmt3a, Dnmt3b and PCNA (serving as control). (C) Methylation-sensitive PCR (upper panel). Episomal DNA derived from stably transfected Lsh−/− and Lsh +/+ MEF cells was digested with DpnI and then either with the methylation-sensitive enzyme HpaII (H) or the methylation independent enzyme MspI (M) followed by PCR analysis with indicated primers. Replication of the episomal vector was confirmed using DpnI (lower panel). DpnI cuts only DNA that has been methylated in bacteria by the dam methylase. The replicated episomal DNA in mouse cells should be DpnI resistant. For adjustment of input undigested DNA (Un) was used before digestion. (D) Episomal DNA was derived as in C. and subjected to real time-PCR analysis using methylation-sensitive primer pair P5/P6 (upper panel) or the internal control P1/P2 (lower panel).
The episomal construct pCEP4 was stably transfected into MEFs derived from Lsh−/− or Lsh+/+ embryos. Since the episomal construct serves as a target for de novo methylation by Dnmt3a and Dnmt3b, we first examined protein expression levels by Western blot analysis using nuclear extracts of stably transfected cell lines. As expected, Lsh protein levels were not detectable in Lsh−/− MEFs (Figure 1B). In contrast, DNA methyltransferases Dnmt3a and 3b were both expressed equally well in the presence or absence of Lsh, suggesting a comparable de novo methylation activity in both cell lines.
To examine the methylation status of the recovered episome, methylation-sensitive PCR analysis was performed as shown in Figure 1C. After quantification of recovered episomal DNA to adjust for equal input using primers P5/P6 (undigested samples), the DNA was digested with the methylation-sensitive restriction enzyme HpaII. The successful amplification, using P5/P6 primers, of Lsh+/+ derived DNA after treatment with HpaII indicated the presence of methylated CpG sites. Digestion with MspI (which cleaves DNA independent of methylation) served as control. In contrast, the P5/P6 PCR fragment was not detectable using DNA derived from Lsh−/− MEFs, suggesting an impaired gain of methylation in the absence of Lsh. To ensure that equal amounts of DNA were indeed present in wild type and Lsh−/− samples after digestion (since digestion and further handling can lead to unavoidable loss of DNA), the control primers P1/P2 were used which do not surround HpaII sites (Figure 1C). Using these internal control primers, the amplification was indistinguishable between Lsh wild type and Lsh−/− DNA. In order to quantify the appearance of methylation in the episome sequence, real-time PCR was performed. As shown in Figure 1D, the use of the methylation-sensitive primer set P5/P6 revealed a significant difference of methylation comparing wild type and Lsh−/− samples, whereas the internal control primers confirmed equal amounts of DNA after digestion. In an attempt to quantify methylation levels, a standard curve for PCR amplifications using known concentrations of the episomal DNA was performed and used to calculate the copy numbers using primers P5/P6 before and after digestion (Supplementary Figure 1). Wild-type samples were completely methylated at the examined sites of pCEP4. In contrast, Lsh−/− samples were only methylated about 1% by this calculation. These observations demonstrated that Lsh is essential for the acquisition of methylation on episomal DNA in MEFs.
Lsh does not play a role in maintenance of methylation in MEF cells
Gain of methylation on the episomal vector pCEP4 requires de novo methylation activity as well as the ability to maintain newly acquired methylation patterns. To differentiate between these two processes, we tested the ability of Lsh−/− cells to maintain methylation on previously in vitro methylated DNA. The episomal vector pCEP4 was treated with SssI methyltansferase and the degree of methylation was determined by digestion with the methylation-sensitive restriction enzyme, HpaII. Resistance to HpaII digestion indicated the successful methylation of the vector (Figure 2A).
Figure 2.
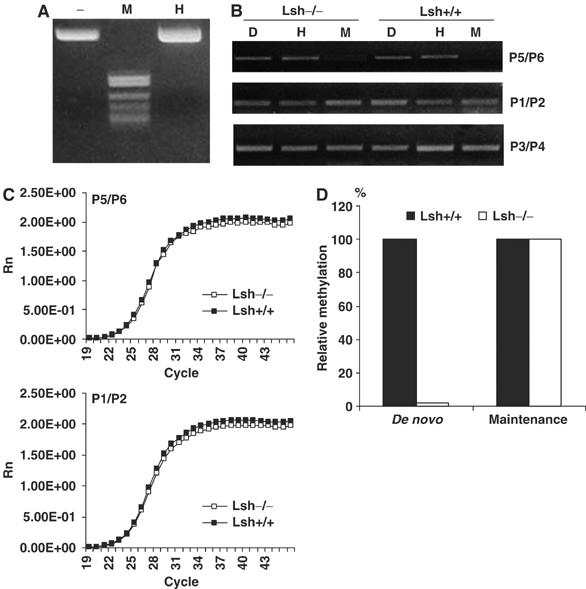
Lsh does not play a role in the maintenance of methylation in MEF cells. (A) Ethidium bromide stain of in vitro methylated episomal vector pCEP4 that has been digested with HpaII (H) and MspI (M). Resistance to HpaII digestion indicates that the episome has been fully methylated. (B) Methylation-sensitive PCR. Methylated episomal DNA was stably transfected into Lsh−/− and Lsh+/+ MEF cells and the recovered DNA was first digested with DpnI. Then the DNA was either digested with the methylation-sensitive enzyme HpaII (H) or the methylation independent enzyme MspI (M) followed by PCR analysis with the indicated primers as shown in Figure 1A. (C) Real-time PCR analysis. Episomal DNA was derived as described in (B). and subjected to real time-PCR analysis using the methylation-sensitive primer pair P5/P6 (upper panel) or the internal control primer pair P1/P2 (lower panel). (D) The relative copy numbers for real time-PCR products of HpaII digested episomal DNA were calculated based on the standard curve equation. Results of Figure 1 are represented as ‘de novo' and results from Figure 2 as ‘maintenance' methylation.
The methylated episomal DNA was stably transfected into Lsh−/− and Lsh+/+ MEFs and methylation-sensitive PCR performed after recovery of the episome. Episomal DNA derived from wild-type MEFs showed the expected PCR fragment using primer pair P5/P6 after HpaII digestion, indicating the presence of methylated sites on the episome (Figure 2B). Using episomal DNA derived from Lsh−/− MEFs, a comparable PCR amplification was observed suggesting a similar degree of methylation in the absence of Lsh (Figure 2B). These results were further confirmed using real-time PCR analysis. As shown in Figure 2C, there were no detectable differences in the amplification using primer pair P5/6 or the internal controls P1/P2 when comparing wild type and Lsh deficient cells indicating a similar ability to preserve methylation patterns. As summarized in Figure 2D, the calculated copy numbers which were amplified from premethylated plasmids were indistinguishable between Lsh+/+ and Lsh−/− cells. Thus, methylation patterns are faithfully maintained in the absence of Lsh, while the acquisition of a novel methylation mark depends on Lsh. We therefore conclude that Lsh plays a role in de novo methylation rather than in maintenance methylation.
Lsh is required for Dnmt3a or Dnmt3b mediated silencing of a retroviral transgene
De novo methylation is thought to be conferred by either Dnmt3a or Dnmt3b activity in mice (Hsieh, 1999; Okano et al, 1999). In order to understand whether Lsh is involved in the methylation process mediated by either DNA methyltransferase, we stably overexpressed both proteins in Lsh−/− and Lsh+/+ MEFs. By Western blot analysis, an increase in Dnmt3a or Dnmt3b proteins over endogenous methyltransferase levels was observed using nuclear extracts from Lsh−/− MEFs and Lsh +/+ MEF controls (Figure 3A). To investigate the silencing function caused by methylation, the retroviral reporter vector pMSCV-hGFP was used (kind gift of Dr Jonathan Keller, NCI, Frederick). Upon methylation, expression of the green fluorescence protein (GFP) is suppressed and can be monitored by either fluorescence microscopy or FACS analysis. After infection of Lsh−/− and Lsh+/+ MEFs with the retroviral vector pMSCV-hGFP, GFP expression was monitored by microscopy (Figure 3B). Lsh wild-type cells that were either stably transfected with Dnmt3a, Dnmt3b, or both showed a decrease in GFP expression within 8 days after infection. In contrast, Lsh deficient cells stably expressing Dnmt3a, Dnmt3b, or both were unable to significantly reduce GFP expression. Quantitative measurement of fluorescence expression by FACS analysis revealed a reduction of GFP intensity of about 25–30% when comparing wild-type cells with Lsh−/− MEFs, suggesting an impairment of reporter silencing in the absence of Lsh (Figure 3C). In contrast, untransfected MEFs (Un) that did not overexpress the Dnmt3 proteins were not able to silence the integrated reporter gene and thus, GFP expression levels were indistinguishable between wild type and Lsh deficient cells (Figure 3C). Lsh, therefore, plays an important role in Dnmt3a or Dnmt3b mediated silencing of retrovirus directed protein expression.
Figure 3.
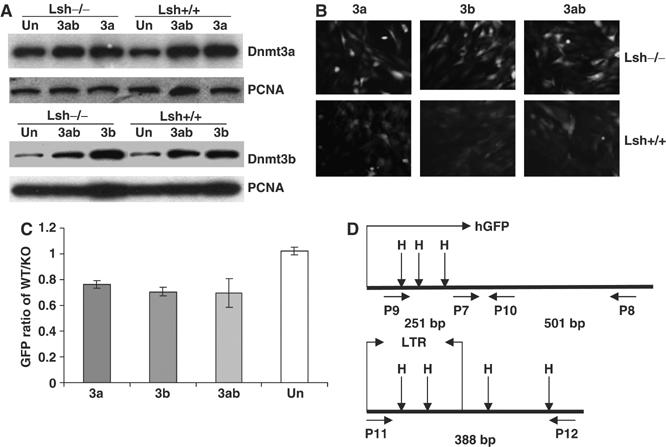
Lsh is required for silencing of Dnmt3a or Dnmt3b mediated silencing of a retroviral transgene. (A) Western blot analysis using nuclear extracts derived from Lsh−/− and Lsh+/+ mouse embryonal fibroblasts stably expressing Dnmt3a, Dnmt3b, Dnmt3a/Dnmt3b, or untransfected (Un) MEFs. For detection, specific antibodies were used against Dnmt3a, Dnmt3b, or PCNA as control. (B) Fluorescence analysis. Lsh+/+ and Lsh−/− MEFs that were stably expressing Dnmt3a, Dnmt3b, or Dnmt3a/Dnmt3b were infected with pMSCV-hGFP and examined after 8 days for GFP expression using a fluorescence microscope. (C) FACS analysis. The GFP intensity of Lsh+/+ and Lsh−/− MEF cells expressing Dnmt3a, Dnmt3b, Dnmt3a/Dnmt3b, or untransfected (Un) was measured by FACS analysis 8 days after retroviral infection. The difference in fluorescence intensity was expressed as GFP ratio of Lsh+/+ over Lsh−/−. (D) Map of the retroviral vector pMSCV-hGFP indicating the location of HpaII/MspI sites and the position of the primers used for methylation-sensitive PCR analysis. Primer pair P9/P10 detects methylation within the GFP region. Primer pair P7/P8 serves as internal control. Primer pair P11/P12 detects methylation in the 5′-LTR and the adjacent region. The length of the expected PCR fragments is indicated in base pairs (bp).
Lsh functionally cooperates with de novo methylation mediated by either Dnmt3a or Dnmt3b
Since Lsh plays an important role in the downregulation of GFP expression, we tested whether CpG hypermethylation was directly involved in gene silencing of the retroviral transgene using methylation-sensitive PCR. For detection of methylation, primers were designed around HpaII/MspI sites in the GFP gene (P9/P10) and the 5′-LTR region (P11/P12) of the vector (Figure 3D). The control primers P7/P8 did not surround HpaII sites thus serving as internal control.
After the undigested genomic DNA was quantified to adjust for equal input using primers P11/12 (Figure 4A), restriction enzyme digestion with the methylation-sensitive enzyme HpaII was performed. Lsh wild-type samples were successfully amplified and generated PCR fragments in the 5′LTR (primer P11/P12) and the GFP coding region (P9/P10), suggesting methylation within the amplified regions (Figure 4A). In contrast, Lsh−/− samples did not generate the expected PCR fragments indicating reduced methylation (Figure 4A). This decrease of methylation occurred in Dnmt3a, Dnmt3b, or Dnmt3a/b overexpressing Lsh−/− MEFs, suggesting that Lsh is required for methylation induced by either Dnmt3 protein. As control, to ensure equal amounts of DNA after digestion, the internal methylation independent primers were used (P7/P8). Distinct methylation levels when comparing wild type and Lsh−/− samples were further confirmed using real-time PCR (Figure 4B–D). Whereas the use of primer pair P7/P8 verified equal loading of DNA after digestion, use of primer pair P11/12 indicated significant differences in methylation at the 5′LTR region in wild type versus Lsh deficient samples. Without overexpression of the Dnmt3 proteins, Lsh+/+ and Lsh−/− samples were indistinguishable since methylation was undetectable (data not shown). In an attempt to quantify methylation levels, a standard curve for PCR amplifications using known concentrations of the template was performed and used to calculate the copy numbers using primers P11/12 before and after digestion (Supplementary Figure 2). Although the frequency of having all four sites methylated in wild-type cells was low, the deletion of Lsh revealed consistently a 10-fold reduction in Dnmt3a, Dnmt3b or Dnmt3a,b transfected cells. Possibly, the low methylation efficiency in the retroviral transgene in comparison to the full methylation of the episomal vector was due to distinct retroviral target sequences, the integration of the transgene or a difference in the experimental time frame. Taken together, the difference in CpG methylation levels between wild type and Lsh deficient cells correlated with the difference in silencing of GFP expression. These results suggest, therefore, that Lsh cooperates with either Dnmt3a or Dnm3b for de novo methylation.
Figure 4.
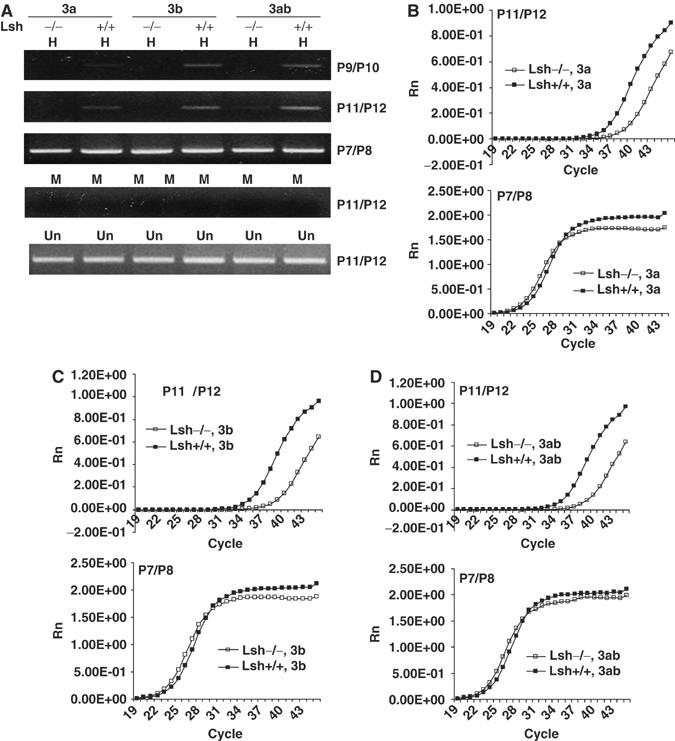
Lsh functionally cooperates with de novo methylation mediated by either Dnmt3a or Dnmt3b. (A) Methylation-sensitive PCR. At 8 days after the retroviral infection, genomic DNA from Lsh−/− and Lsh+/+ MEF cells stably expressing Dnmt3a, Dnmt3b, or Dnmt3a/Dnmt3b was extracted, digested with HpaII (H) or MspI (M) and subjected to PCR with the indicated primer pairs. For adjustment of DNA undigested DNA (Un) was used before digestion. (B–D). Real-time PCR analysis of HpaII digested DNA using primer pair P11/P12. Control primers P7/P8 are used in the lower panel of the graphs.
Silencing of Lsh in embryonal stem cells results in loss of de novo methylation
Embryonal stem (ES) cells are used for de novo methylation assays since they are rich in endogenous Dnmt3a/b proteins in contrast to somatic cells and readily methylate newly integrated retroviral DNA (Okano et al, 1999; Chen et al, 2002). To further investigate the molecular mechanism of Lsh in methylation, we established ES cell lines with low levels of the Lsh protein using RNA interference.
Stably expressing Lsh hairpin siRNA in ES cell lines (siLsh#1 and siLsh#2) significantly decreased Lsh protein levels by more than 90% as shown by Western blot analysis, whereas Dnmt3a and Dnmt3b levels remained unchanged (Figure 5A). Using Southern blot analysis, genomic DNA was examined for DNA hypomethylation at minor satellite repeat sequences (Figure 5B). Whereas control ES cells were fully methylated at these repeats, low levels of Lsh protein were associated with a small degree of hypomethylation after several passages in vitro. The delay in hypomethylation mimicked a similar time course generated by conditional deletion of Dnmt3b in ES cells, which suggests ongoing de novo methylation in ES cells at minor satellite sequences (Chen et al, 2003). In contrast to embryonal stem cells, 3T3 fibroblasts represent somatic cells that have generally lower levels of Dnmt3a or Dnmt3b proteins and de novo methylation activity (Chen et al, 2002). Silencing of Lsh in 3T3 cells using the same siLsh vector had no detectable effect on CpG methylation at minor satellite sequences (Supplementary Figure 3A,B) consistent with the idea of low or absent de novo methylation activity at pericentric sequences in somatic cells (Chen et al, 2002). To determine the effect of Lsh reduction on silencing of retroviral genes, siLsh ES cells were infected with the retroviral vector pMSCV-hGFP that controls expression of the green fluorescence protein (GFP). Within 48–72 h after infection, GFP mRNA levels were notably reduced in wild-type ES cells in contrast to siLsh ES cells, which maintained GFP mRNA levels as shown by RT–PCR analysis (Figure 5C). FACS analysis revealed a decrease in GFP protein of about 80% when comparing control ES cells to Lsh deficient ES cells, suggesting that successful silencing of a retroviral vector requires the presence of Lsh (Figure 5D). Using methylation-sensitive PCR analysis, the genomic DNA derived from control ES cells 72 h after infection demonstrated HpaII resistance indicating the acquisition of methylation (Figure 5E). In contrast, siLsh ES cells showed no detectable CpG methylation at the GFP gene (P9/P10) or the 5′-LTR (P11/P12) regions as demonstrated by sensitivity to HpaII digestion. Real-time PCR analysis confirmed the differences in methylation (P11/P12) seen in the presence (control ES) or absence of Lsh (siLsh) (Figure 5F). In contrast, the control PCR reactions (P7/P8) remained indistinguishable comparing control or siLsh derived DNA. In summary, the use of retroviral target sequences in Lsh deficient embryonic stem (ES) cells confirms a role for Lsh in de novo methylation.
Figure 5.
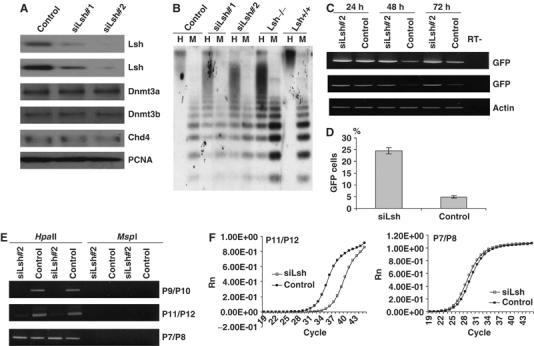
Silencing of Lsh in embryonic stem cells results in loss of de novo methylation. (A) Western analysis: nuclear extracts from two ES cell lines (#1 and #2 both received the same construct) stably expressing a silencing vector for Lsh (siLsh) were examined by Western analysis using specific antibodies against Lsh, Dnmt3a, Dnmt3b, Chd4 or PCNA as control. A scrambled sequence serves as the siRNA vector control. (B) Southern blot analysis: genomic DNA derived from two siLsh ES cell lines, an ES cell control, Lsh−/− MEFs, and Lsh+/+ MEF cells was digested with HpaII (H) or MspI (M), blotted, and probed for minor satellite sequence using the probe MR150. (C) RT–PCR analysis: siLsh ES cells were infected with the retroviral vector pMSCV-hGFP and after the indicated time points (24, 48, 72 h) RNA was extracted, reverse transcribed and analyzed by PCR for expression of GFP. β-Actin serves as a control. (D) FACS analysis: 72 h after retroviral infection of siLsh and control ES cells GFP expression was measured by FACS analysis. (E) Methylation-sensitive PCR: genomic DNA derived from the siLsh and control ES cells 3 days after retroviral infection was digested with HpaII or MspI and subjected to PCR analysis with the indicated primer pairs. (F) Real time-PCR analysis: as in (E), HpaII digested genomic DNA was subjected to real time-PCR using P11/P12. The right panel shows the internal control reaction with primers P7/P8.
Lsh is involved in de novo methylation of endogenous genes
Since Lsh participates in de novo methylation of episomal constructs and retroviral transgenes, we also wanted to test whether Lsh deletion in ES cells effects de novo methylation of endogenous genes such as the Oct-4 gene (Gidekel and Bergman, 2002). Using methylation-sensitive PCR at two distinct sites in the promoter region of the Oct-4 gene (Figure 6A), we had previously noticed that Lsh−/− MEFs have decreased methylation levels at site 2 in comparison with Lsh+/+ (Figure 6B) but show a similar degree of methylation at site 1 (data not shown). This suggested that Lsh played a role in methylation at selected sites of the Oct-4 gene. It has been reported that differentiation of ES cells in vitro is accompanied by CpG methylation at the endogenous Oct-4 gene (Lee et al, 2004). Using siLsh ES cells and control ES cells, we compared methylation levels at site 1 and site 2 before (0 day) and after differentiation (6 days) using treatment with retinoic acid. After quantification using undigested genomic DNA to ensure equal input (Figure 6C) digestion with methylation-sensitive restriction enzymes was performed. Site 1 and site 2 are both largely unmethylated in undifferentiated control ES cells or siLsh ES cells (0 day) (Figure 6C and D). On differentiation of control ES cells, site 1 and site 2 gain resistance to restriction enzyme digestion with HpyCh4IV (Hy) or HpaII (H) indicating de novo methylation (6 Days). In contrast, siLsh ES cells showed a decrease in methylation at site 2 consistent with the observed methylation defect in Lsh−/− MEFs. (Note that amplification of site 2 serves as internal control after digestion to guarantee equal amounts of DNA during the amplification reaction using methylation-sensitive primers for site 1 and vice versa.) These results suggested that Lsh deletion in ES cells leads to an impairment in the establishing methylation pattern at endogenous sequences, supporting its role in de novo methylation.
Figure 6.
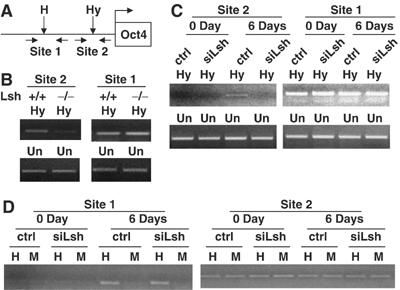
Lsh is involved in de novo methylation at the Oct-4 gene. (A) Map of murine Oct4 gene and its promoter indicating the location of HpaII/MspI site (H in Site 1) and HpyCh4 IV (Hy in Site 2) and the position of the primers used for methylation-sensitive PCR analysis. Due to different recognition sequences of HpaII and HpyCh4 IV, site 1 and site 2 can be used as internal control for each other in methylation-sensitive PCR. (B) Methylation-sensitive PCR. Genomic DNA derived from the Lsh wild type and Lsh−/− MEF cells was digested with HpyCh4 IV and subjected to PCR analysis with the indicated primer pairs. Site 1 served as internal control for site 2. Undigested DNA (Un) was used for adjustment of DNA input before digestion. (C) Methylation-sensitive PCR. Genomic DNA derived from the siLsh and control ES cells at 0 day and 6 days after differentiation was digested with HpaCh4 IV and subjected to PCR analysis with the indicated primer pairs. Site 1 served as internal control for site 2. Undigested DNA (Un) was used for adjustment of DNA input before digestion. (D) Methylation-sensitive PCR. Genomic DNA derived from the siLsh and control ES cells at 0 day and 6 days after differentiation was digested with HpaII and MspI and subjected to PCR analysis with the indicated primer pairs. Site 2 served as internal control for site 1.
Lsh interacts with Dnmt3a and Dnmt3b in ES cell
Since Lsh participates in the process of de novo methylation, we tested the idea of whether Lsh directly associates with the known de novo methyltransferases Dnmt3a or Dnmt3b. Using nuclear extracts from ES cells or from the embryonal cell line P19, we precipitated Lsh with affinity purified antisera directed against a C-terminal peptide of Lsh (Lsh#1) or directed against Lsh recombinant protein (Lsh#2). As shown by Western blot analysis, Dnmt3a as well as two isoforms of Dnmt3b could be co-immnunoprecipitated in the pluripotent embryonal carcinoma cell line P19 (Figure 7A) or ES cells (not shown) using either anti-Lsh antibody, while control rabbit serum served as a control. In contrast, the maintenance methyltransferase Dnmt1 was not precipitated together with Lsh under the same conditions (Figure 7A). In order to test for the reciprocal precipitation, specific antisera suitable for immunoprecipiation of either Dnmt3a or Dnmt3b were applied using nuclear extracts of the embryonal cell line P19. As shown in Figure 7B, either anti-Dnmt3a or anti-Dnmt3b antisera were able to precipitate endogenous Lsh. In contrast, a specific antiserum against Dnmt1 failed to precipitate Lsh, although it was suitable to precipitate endogenous Dnmt1. To further support the specific interaction of Lsh with Dnmt3a or Dnmt3b, the embryonal cell line P19 was stably transfected with plasmids expressing either Dnmt3a or Dnmt3b as myc-tagged fusion proteins. As expected, specific precipitation of Lsh with affinity purified antisera directed against a C-terminal peptide of Lsh (Lsh#1) or directed against Lsh recombinant protein (Lsh#2), but not a control serum was able to detect the Dnmt3a or Dnmt3b fusion proteins by Western analysis using specific anti-myc antibodies (Figure 7C). In a reciprocal approach, using specific anti-myc antibodies for immunoprecipitation, we were able to detect Lsh in the precipitate (Figure 7D). In summary, these results suggest that Lsh specifically interacts with Dnmt3a or Dnm3b, but not Dnmt1 in embryonal stem cells. This supports further the idea that Lsh is involved in de novo methylation in contrast to maintenance of methylation and furthermore suggests a direct role for Lsh in the process.
Figure 7.
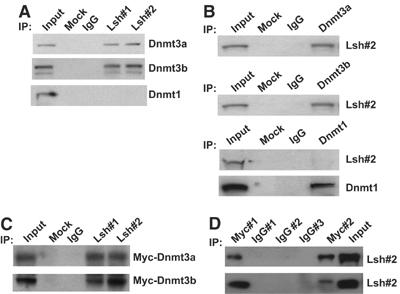
Lsh interacts with Dnmt3a and Dnmt3b. (A) Western analysis for detection of Dnmt3a, Dnmt3b or Dnmt1 after immunoprecipitation (IP) with Lsh-specific antibodies (Ab): Lsh#1 (C-terminal peptide, affinity purified) and Lsh#2 (recombinant protein, affinity purified) using P19 nuclear extracts. The blots are probed with a monoclonal anti-Dnmt3a, Dnmt3b and Dnmt1 antibodies. The negative controls are normal rabbit IgG or omission of antibodies (mock). Nuclear extracts before IP serve as positive controls. (B) Western analysis for detection of Lsh and Dnmt1 after IP with specific antibodies against Dnmt3a, Dnmt3b or Dnmt1 using P19 nuclear extracts. The following antibodies were used for Western analysis: anti-Lsh#2 (recombinant protein, affinity purified) Dnmt1. The negative controls are normal mouse IgG or omission of antibodies (mock). (C) Western analysis for detection of Myc-Dnmt3a and Myc-Dnmt3b fusion proteins after IP with anti-Lsh#1 (C-terminal peptide) and Lsh#2 (recombinant protein). The nuclear extracts were prepared from P19 cells stably expressing Myc-Dnmt3a (upper panel) or Myc-Dnmt3b (lower panel). The negative controls are normal rabbit IgG or omission of antibodies (mock). (D) Western analysis for detection of Lsh after IP with anti-Myc antibodies using nuclear extracts derived from P19 cells stably expressing Myc-Dnmt3a or Myc-Dnmt3b. Two different anti-myc antibodies were used for IP. The negative controls are normal mouse IgG (IgG#1), mouse IgG1 (IgG#2) or normal rabbit IgG (IgG#3).
Discussion
Here we present evidence that Lsh is involved in the process of de novo methylation at retroviral genes, episomal targets and endogenous sequences and that Lsh interacts with Dnmt3a and Dnmt3b.
We have previously demonstrated that Lsh deletion in mice leads to multiple epigenetic modifications including genomic hypomethylation, alterations in the pattern of histone methylation, and changes in acetylation levels for histone H3 and histone H4 (Muegge, 2005). Since cross talk exists between different types of histone modifications and DNA methylation, it was unclear which epigenetic modifications were altered first by Lsh. The association with Dnmt3a and b suggests that methylation changes are the primary molecular effect of Lsh.
There is no evidence to date that Lsh directly binds to Dnmt3a or Dnmt3b. Instead, Lsh and Dnmt3 are likely part of a yet unidentified large complex including other protein or possibly RNA components involved in de novo methylation. Lsh, being a member of the SNF2 chromatin remodeling family, may facilitate access to some chromatin templates and thus promote targeting of Dnmt3a or 3b to specific genomic targets. Alternatively, Lsh may facilitate the stable association of Dnmt3s with their specific targets and promote the methylation efficiency of nucleosomal templates or condensed chromatin.
The finding that Lsh is not required for maintenance of methylation at episomal plasmids and does not interact with Dnmt1 is consistent with previous observations. For example, the biologic phenotype of Dnmt1−/−mice differ greatly from Lsh−/− mice: Dnmt1−/− mice die around day 9–11 of gestation showing severe developmental defects (Li et al, 1992), whereas Lsh−/− mice die as newborns and are grossly morphologically normal (with the exception of renal and lymphoid development) (Muegge, 2005). Dnmt1 deletion perturbs imprints (Li et al, 1993), while Lsh−/− mice generally maintain imprints (with the exception of Cdkn1c) (Fan et al, 2005). Also, Lsh is associated with pericentric heterochromatin and unlike Dnmt1 is not continuously associated with the replication fork (Yan et al, 2003a). Thus, our data suggest that no functional link exists between Lsh and Dnmt1.
Also, the phenotypes of Dnmt3ab−/− mice differ somewhat from that of Lsh deleted mice (Okano et al, 1999; Dennis et al, 2001), suggesting that Lsh and Dnmt3s are not obligated to associate in one complex and can also have distinct biological functions. For example, loss of Dnmt3a and b, in contrast to Lsh deletion, affects the methylation pattern of imprinted genes (Chen et al, 2003). Since most methylation imprints are established in germ cells, it implies a specific role for Dnmt3s in the maintenance of methylation pattern at some imprinted loci. Lsh deletion, in contrast, does not in general affect imprinted genes with the exception of the paternal allele of the imprinted Cdkn1c (p57KIP2) gene. This site acquires de novo methylation during early embryogenesis and deletion of Lsh results in loss of methylation and silencing at the paternal allele and bi-allelec expression of the Cdkn1c gene (Fan et al, 2005). This finding is again consistent with a role for Lsh in de novo methyation versus maintenance of methylation.
Aberrant methylation patterns have been implicated in cancer development (Jones and Laird, 1999; Baylin et al, 2000). Loss of imprinting leading to overexpression of Igf2 increases the rate of tumor formation in mice (Sakatani et al, 2005). Genomic hypomethylation, causing chromosomal segregation defects, results in aggressive lymphoma development and an enhanced rate of sarcoma development in Nf1 and p53 mutant mice (Eden et al, 2003; Gaudet et al, 2003). Thus, a defect in nomal maintenance of methylation may be a crucial feature in tumorigensis. On the other hand, de novo methylation of tumor suppressor genes may also be involved in the pathogenesis of some tumors (Chen et al, 2004; Yu et al, 2005). In these scenarios, inhibition of de novo methylation may decrease tumorigenesis. Thus, it is crucial to determine the components involved in either maintenance or de novo methylation pathways, which could serve as potential molecular targets of intervention in cancer. The finding that Lsh participates in de novo methylation may be useful for the generation of selective molecular tools for the prevention of or interference with tumorigenesis.
Materials and methods
Plasmids
pCEP4 (Invitrogen) is an Epstein–Barr virus (EBV)-based plasmid vector carrying the EGV replication origin (oriP) and nuclear antigen (encoded by the EBNA-1 gene) to allow for extrochromosomal replication and stable expression in mammalian cells (Einav et al, 2003). The replication of pCEP4 was confirmed by digestion with DpnI since DpnI restriction sites require methylation by the bacterial dam methylase. Thus, episomal DNA becomes resistant to DpnI after replication in mammalian cells. The expression vectors pcDNA-Dnmt3a and pcDNA-Dnmt3b were generated by subcloning the corresponding cDNDAs into the EcoRI site of pcDNA6 (Chen et al, 2003). pcDN-Myc-Dnmt3a and pcDNA-Myc-Dnmt3b were gift from Dr T Chen, Novartis Institute for Biomedical research, Inc. MA. pMSCV-hGFP (kind gift of Dr Jonathan Keller, NCI, Frederick) has an hGFP reporter gene driven by the murine stem cell virus long terminal repeat (LTR). For RNA interference, the following target site of Lsh was used: 5′-ATTCGGTAGGCGAGACTTC-3′ and subcloned into pSEC-neo (Ambion). A scrambled DNA sequence served as negative control and was subcloned into pSEC-neo vector as well.
Tissue culture and transfection
Lsh−/− and Lsh+/+ mouse embryonic fibroblasts (MEF) were generated as described previously (Fan et al, 2003) and grown in DMEM (Invitrogen/GIBCO) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and antibiotics (Invitrogen/GIBCO).
The mouse ES cells (CCE) were cultured on gelatin-coated dishes without feeder cells in Knockout DMEM (Invitrogen/GIBCO) supplemented with 15% Knockout Serum Replacement, 0.1 mM nonessential amino acid, 0.1 mM β-mercaptoethanol, 50 U/ml of penicillin, 50 μg/ml of streptomycin (Invitrogen/GIBCO), and 1000 U/ml ESGRO (mouse leukemia inhibitor factor, Chemicon). For long-term culture, ES cells were trypsinized and passaged every other day. P19 mouse EC cells were grown in alpha-MEM with 10% fetal calf serum, with L-glutamine, and pen/strep. All transfections were performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instruction. Cells were subsequently selected in hygromycin (Invitrogen) for pCEP4, neomycin (Invitrogen) for pSEC-Lsh-neo, blasticidin for pcDNA-Myc-Dnmt3a or 3b, puromycin (Invitrogen) for co-transfection of in vitro methylated-pCEP4 and Dnmt3a or Dnmt3b with pBabepuro (ratio 1:10) conferring puromycin resistance. In vitro methylation was performed using CpG methylase Sss I (NEB) and successful methylation confirmed by HpaII and MspI digestion.
Differentiation of ES
CCE mouse ES cells were grown on 0.1% gelatin-coated plates in knockout DMEM media (Invitrogen) with 15% knockout serum replacement (Invitrogen), L-glutamine, pen/strep, nonessential amino acids, β-mercaptoethanol, and 1000 μ/ml Esgro (LIF, Chemicon). For differentiation, cells were placed in Petri dishes for aggregation in the absence of LIF for 4 days. ES cells were then grown for 2 more days in 1 μM all-trans retinoic acid (Sigma). Cells were harvested for nuclear extract at day 0 and day 6 of differentiation as previously described (Geiman and Muegge, 2000).
Immunoprecipitations
Nuclear extract was generated as previously described (Geiman and Muegge, 2000). Immunoprecipitations for Western blotting were performed by standard methods, in which the nuclear extract buffer was adjusted to a final concentration of 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40. In each case, 200 μg of nuclear extract was precleared for 30 min with protein G agarose (Invitrogen), incubated with 20 μl antibody for 2 h or overnight at 4°C with rotation. Washing was performed five times in 500 μl buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40) at 4°C, 5 min each on a rotator. Samples were separated on a 6% Tris–glycine SDS–PAGE gel and blotted onto Immobilon P membrane (Millipore). Antibodies used for immunoprecipitation Western blots were species matched normal IgG and IgG1 (Santa Cruz Biotechnology), anti-Lsh C-terminal peptide affinity purified antibody (Lsh#1), anti-Lsh recombinant protein affinity purified antibody (Lsh#2), Dnmt3a and Dnmt3b antibodies (Imgenex), Myc antibody (Roche). Western blot detection was performed according to the manufacturers' instructions (Amersham).
Western blot and antibodies
Nuclear protein extracts were prepared using Nuclear Pure kit (Sigma) and high salt buffer as described previously (Wang et al, 2004) (Figures 1, 3 and 5). The nuclear extracts were separated on 8% Tris–Glycine precast gels (Invitrogen) by electrophoresis and transferred onto PVDF membrane (Invitrogen). Western blotting was performed according to standard procedures using ECL detection reagents, according to the manufacturer's instructions (Amersham). The following primary antibodies were used: murine Dnmt3a (Imgenex); murine Dnmt3b (Imgenex); murine PCNA (Upstate); murine Chd4 (Gift from Dr W Wang, NIH/NIA/IRP); affinity purified rabbit antiserum against a C-terminal portion of Lsh (Lsh#1) (Geiman and Muegge, 2000); affinity purified rabbit antiserum against murine recombinant Lsh (Lsh#2). The following secondary antibodies were used: monoclonal mouse anti-rabbit HRP-conjugated IgG (Sigma), goat anti-mouse HRP-conjugated IgG (Upstate).
Recovery of episomal and genomic DNA, and methylation-sensitive PCR
Episomal DNA was recovered using the Hirt method (Hirt, 1967) and genomic DNA was extracted using the DNeasy kit (Qiagen). DNA was completely digested with HpaII, MspI, Hpych4IV, or DpnI. In order to analyze the methylation status of the episomal DNA vector pCEP4, PCR primer pairs P1/2, P3/4, and P5/6 were used:
P1 (forward, 5′ACGAACTAAACCTGACTACGACAT-3′);
P2 (reverse, 5′GTAAGAGCTTCAGCCAAGAGTTACA-3′);
P3 (forward, 5′AGCCCTCCCGTATCGTAGTT-3′);
P4 (reverse, 5′GCAGAGCGAGGTATGTAGGC-3′);
P5 (forward, 5′-CTGCCAGTGGCGATAAGTCGTGTCTT-3′);
P6 (reverse, 5′CGACAGGACTATAAAGATACCAGGCG-3′).
For analysis of the methylation status of the retroviral transgene pMSCV-hGFP, primer pairs P7/P8, P9/P10 and P11/P12 were used: P7 (forward, 5′GCCCACCCTCGTGACCACCCTGACC-3′); P8 (reverse, 5′-CGAACTCCAGCAGGACCATGTGATC-3′); P9 (forward, 5′-ATGAGCAAGGGCGAGGAGCTGTTCAC-3′); P10 (reverse, 5′-CTTGAAGAAGTCGTGCTGCTTCATGTGGT-3′); P11 (forward, 5′GCGCCAGTCCTCCGATAGACTGCGT-3′); P12 (reverse, 5′AGTCCCTGGGACGTCTCCCAGGGTT-3′). For analysis of the methylation status of Oct4, primer pairs were used: site 1, forward, AGAGGGTGCAGTGCCAACAGGCTTTGT, reverse, CCCCAGGAAGGCCTTCATTTTCAACCTT; site 2, forward, AAGGTTGAAAATGAAGGCCTTCCTGGGG, reverse, TTTCACCTCTCCCTCCCCAATCCCAC. PCR reactions were carried out as follows: 5 min at 94°C, 35 cycles of 60 s at 94°C, 30 s at 60°C, and 60 s at 72°C, and finally 5 min at 72°C. The PCR products were electrophoresed on 1% agarose gels, stained with ethidium bromide, and photographed. For real-time PCR analysis, the ABI PRISM 7900 machine (Applied Biosystems) and Platinum SYBR Green qPCR SuperMix UDG (Invitrogen) were used. The PCR was initiated with one cycle of 50°C for 2 min, one cycle of 95°C for 5 min, followed by 45 cycles of 95°C for 30 s, 60°C for 30 s, 72°C for 30 s, and finally one cycle of 83°C for 10 s. The negative control without target was carried for each PCR. For every methylation-sensitive PCR, we adjusted the starting amount of template using undigested DNA in the PCR reaction before enzymatic digestion. The digested DNA was further controlled by using a primer set that amplified an area that did not contain methylation-sensitive restriction enzyme sites. To quantify the amount of methylation on the template using real-time PCR data, standard titration experiments for each template and each primer set were performed and linear regression equation and the calculation for the log copy number were established using Prism 3.0 software (GraphPad Software, Inc.) and Microsoft Excell.
Southern blot analysis
Genomic DNA was extracted from ES cells, digested with HpaII and MspI, separated by electrophoresis on 1% agarose gels, and blotted onto Nytran Plus membranes (Scheicher and Schuell). The blots were hybridized for 2 h at 65°C in Rapid-hyb buffer (Amersham) with 32P-labeled probes and washed twice in 2 × SSC/0.1% SDS at room temperature for 20 min and twice in 0.2 × SSC/0.1% SDS at 65°C for 20 min. The minor satellite repeat probe was a 66-mer oligonucleotide: 5′-GACTGAAAAACACATTCGTTGGAAACGGGATT TGTAGAACAGTGTATATCAATGAGTTACAATGAG- 3′.
Reverse transcription-PCR
For detection of GFP mRNA in ES cells transiently infected with pMSCV-hGFP (Jiang et al, 2004), the cells were harvested at 24, 48 and 72 h. Total RNA was prepared using RNeasy kit (Qiagen) and 1 μg of RNA was reverse transcribed using Omniscript Reverse Transcriptase (Qiagen). Omission of reverse transcriptase served as negative control. cDNA was amplified in a GeneAmp PCR system 9700 machine (Applied Biosystems) using Platinum PCR SuperMix (Invitrogen). The PCR was performed as follows: 5 min at 94°C, 30 cycles of 30 s at 94°C, 30 s at 60°C, and 30 s at 72°C, followed by one cycle 5 min at 72°C.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We are grateful for the kind gifts of pMSCV-hGFP from Dr Jonathan Keller, NCI, NIH, anti Chd4 Ab from Dr W Wang, NIH/NIA/IRP, and ES cells from Dr Lino Tessarolo, NCI, NIH. We also would like to thank Terry Stull and Rodney Wiles for excellent technical assistance. We would like to thank Dr Jiaqiang Huang for his kind support, Dr John Julias for technical assistance with real-time PCR, and Drs Xiaohu Zhang, Li Hua Wang and David Hodge for assistance of the fluorescence microscopy.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
This project has been funded in whole or part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. N01-C0-12400.
Animal care was provided in accordance with the procedures outlined in the ‘Guide for the Care and Use of Laboratory Animals' (NIH Publication No. 86-23, 1985).
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188 [DOI] [PubMed] [Google Scholar]
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG (2000) Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 10: 687–692 [DOI] [PubMed] [Google Scholar]
- Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 [DOI] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH (2004) Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431: 96–99 [DOI] [PubMed] [Google Scholar]
- Chen T, Li E (2004) Structure and function of eukaryotic DNA methyltransferases. Curr Top Dev Biol 60: 55–89 [DOI] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Dodge JE, Wang Z, Li E (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23: 5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Ueda Y, Xie S, Li E (2002) A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J Biol Chem 277: 38746–38754 [DOI] [PubMed] [Google Scholar]
- Chen W, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, Karp JE, Gokgoz N, Wunder JS, Andrulis IL, Levine AJ, Mankowski JL, Baylin SB (2004) Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 6: 387–398 [DOI] [PubMed] [Google Scholar]
- Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF (1997) Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277: 1996–2000 [DOI] [PubMed] [Google Scholar]
- Dennis K, Fan T, Geiman TM, Yan QS, Muegge K (2001) Lsh, a member of the SNF2 family, is required for genome wide methylation. Genes Dev 15: 2940–294411711429 [Google Scholar]
- Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300: 455. [DOI] [PubMed] [Google Scholar]
- Einav Y, Shistik E, Shenfeld M, Simons AH, Melton DW, Canaani D (2003) Replication and episomal maintenance of Epstein–Barr virus-based vectors in mouse embryonal fibroblasts enable synthetic lethality screens. Mol Cancer Ther 2: 1121–1128 [PubMed] [Google Scholar]
- Fan T, Hagan JP, Kozlov SV, Stewart CL, Muegge K (2005) Lsh controls silencing of the imprinted Cdkn1c gene. Development 132: 635–644 [DOI] [PubMed] [Google Scholar]
- Fan T, Yan Q, Huang J, Austin S, Cho E, Ferris D, Muegge K (2003) Lsh deficient murine embryonal fibroblasts show reduced proliferation with signs of abnormal mitosis. Cancer Res 63: 4677–4683 [PubMed] [Google Scholar]
- Fyodorov DV, Kadonaga JT (2001) The many faces of chromatin remodeling: SWItching beyond transcription. Cell 106: 523–525 [DOI] [PubMed] [Google Scholar]
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R (2003) Induction of tumors in mice by genomic hypomethylation. Science 300: 489–492 [DOI] [PubMed] [Google Scholar]
- Geiman TM, Durum SK, Muegge K (1998) Characterization of gene expression, genomic structure, and chromosomal localization of Hells (Lsh). Genomics 54: 477–483 [DOI] [PubMed] [Google Scholar]
- Geiman TM, Muegge K (2000) Lsh, an SNF2/helicase family member, is required for proliferation of mature T lymphocytes. Proc Natl Acad Sci USA 97: 4772–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiman TM, Tessarollo L, Anver MR, Kopp JB, Ward JM, Muegge K (2001) Lsh, a SNF2 family member, is required for normal murine development. Biochim Biophys Acta 1526: 211–220 [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR (2000) Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 24: 368–371 [DOI] [PubMed] [Google Scholar]
- Gidekel S, Bergman Y (2002) A unique developmental pattern of Oct-3/4 DNA methylation is controlled by a cis-demodification element. J Biol Chem 277: 34521–34530 [DOI] [PubMed] [Google Scholar]
- Goll MG, Bestor TH (2005) Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74: 481–514 [DOI] [PubMed] [Google Scholar]
- Hata K, Okano M, Lei H, Li E (2000) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129: 1983–1993 [DOI] [PubMed] [Google Scholar]
- Hirt B (1967) Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26: 365–369 [DOI] [PubMed] [Google Scholar]
- Hsieh CL (1999) In vivo activity of murine de novo methyltransferases, Dnmt3a and Dnmt3b. Mol Cell Biol 19: 8211–8218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Fan T, Yan Q, Zhu H, Fox S, Issaq HJ, Best L, Gangi L, Munroe D, Muegge K (2004) Lsh, an epigenetic guardian of repetitive elements. Nucleic Acids Res 32: 5019–5028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis CD, Geiman T, Vila-Storm MP, Osipovich O, Akella U, Candéias S, Nathan I, Durum SK, Muegge K (1996) A novel putative helicase produced in early murine thymocytes. Gene 169: 203–207 [DOI] [PubMed] [Google Scholar]
- Jiang Q, Li WQ, Hofmeister RR, Young HA, Hodge DR, Keller JR, Khaled AR, Durum SK (2004) Distinct regions of the interleukin-7 receptor regulate different Bcl2 family members. Mol Cell Biol 24: 6501–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Laird PW (1999) Cancer epigenetics comes of age. Nat Genet 21: 163–167 [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428 [DOI] [PubMed] [Google Scholar]
- Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, Sasaki H (2004) Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429: 900–903 [DOI] [PubMed] [Google Scholar]
- Langst G, Becker PB (2004) Nucleosome remodeling: one mechanism, many phenomena? Biochim Biophys Acta 1677: 58–63 [DOI] [PubMed] [Google Scholar]
- Lee JH, Hart SRL, Skalnik DG (2004) Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis 38: 32–38 [DOI] [PubMed] [Google Scholar]
- Leonhardt H, Page AW, Weier HU, Bestor TH (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71: 865–873 [DOI] [PubMed] [Google Scholar]
- Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3: 662–673 [DOI] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R (1993) Role for DNA methylation in genomic imprinting. Nature 366: 362–365 [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926 [DOI] [PubMed] [Google Scholar]
- Muegge K (2005) Lsh, a guardian of heterochromatin at repeat elements. Biochem Cell Biol 83: 548–554 [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19: 219–220 [DOI] [PubMed] [Google Scholar]
- Pradhan S, Bacolla A, Wells RD, Roberts RJ (1999) Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J Biol Chem 274: 33002–33010 [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293: 1089–1093 [DOI] [PubMed] [Google Scholar]
- Sakatani T, Kaneda A, Iacobuzio-Donahue CA, Carter MG, de Boom Witzel S, Okano H, Ko MS, Ohlsson R, Longo DL, Feinberg AP (2005) Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science 307: 1976–1978 [DOI] [PubMed] [Google Scholar]
- Sun LQ, Lee DW, Zhang Q, Xiao W, Raabe EH, Meeker A, Miao D, Huso DL, Arceci RJ (2004) Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev 18: 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Yang XY, Zhang X, Huang J, Hou J, Li J, Xiong H, Mihalic K, Zhu H, Xiao W, Farrar WL (2004) Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 20: 205–218 [DOI] [PubMed] [Google Scholar]
- Yan Q, Cho E, Lockett S, Muegge K (2003a) Association of Lsh, a regulator of DNA methylation, with pericentromeric heterochromatin is dependent on intact heterochromatin. Mol Cell Biol 23: 8416–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Huang J, Fan T, Zhu H, Muegge K (2003b) Lsh, a modulator of CpG methylation, is crucial for normal histone methylation. EMBO J 22: 5154–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Liu C, Vandeusen J, Becknell B, Dai Z, Wu YZ, Raval A, Liu TH, Ding W, Mao C, Liu S, Smith LT, Lee S, Rassenti L, Marcucci G, Byrd J, Caligiuri MA, Plass C (2005) Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet 37: 265–274 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3