

Abstract
Targeted deletion of two members of the FE65 family of adaptor proteins, FE65 and FE65L1, results in cortical dysplasia. Heterotopias resembling those found in cobblestone lissencephalies in which neuroepithelial cells migrate into superficial layers of the developing cortex, aberrant cortical projections and loss of infrapyramidal mossy fibers arise in FE65/FE65L1 compound null animals, but not in single gene knockouts. The disruption of pial basal membranes underlying the heterotopias and poor organization of fibrillar laminin by isolated meningeal fibroblasts from double knockouts suggests that FE65 proteins are involved in basement membrane assembly. A similar phenotype is observed in triple mutant mice lacking the APP family members APP, APLP1 and APLP2, all of which interact with FE65 proteins, suggesting that this phenotype may be caused by decreased transmission of an APP-dependent signal through the FE65 proteins. The defects observed in the double knockout may also involve the family of Ena/Vasp proteins, which participate in actin cytoskeleton remodeling and interact with the WW domains of FE65 proteins.
Keywords: APP, Alzheimer, axonal pathfinding, heterotopia, neuronal migration
Introduction
The mammalian FE65 protein family consists of FE65, FE65L1 and FE65L2. This class of scaffolding proteins has three structural domains, which include a WW and two phosphotyrosine binding domains (PID1/PTB1 and PID2/PTB2) that mediate protein–protein interactions. All FE65 protein family members bind members of the APP protein family (APP, APLP1 and APLP2) through the C-terminal PID2/PTB2 domain (King and Turner, 2004). The PID1/PTB1 domain of FE65 mediates binding to the low-density lipoprotein receptor-related protein, the histone acetyltransferase Tip60 and the transcription factor CP2/LSF/LBP1, whereas the WW domain of FE65 mediates its interaction with the Ena/Vasp proteins and the non-receptor tyrosine kinase Abl (Lambrechts et al, 2000; Cao and Sudhof, 2001; Zambrano et al, 2001; King and Turner, 2004).
APP processing can be regulated by the expression of FE65 family members in cultured cell lines. Higher levels of secreted Aβ, APPsα and APP C-terminal fragments including the γ-secretase-derived APP intracellular domain (AICD) have been reported upon overexpression of the FE65 proteins (King and Turner, 2004). FE65 also regulates gene transcription through interaction with APP protein family members (Cao and Sudhof, 2001; Bruni et al, 2002; Scheinfeld et al, 2002; Hass and Yankner, 2005). In addition, FE65 and APP may function in membrane extension and motility through their association with Mena; this possible function was proposed because MDCK cells overexpressing APP695 and FE65 showed accelerated wound healing (Sabo et al, 2001). Despite these findings, our understanding of the physiological importance of the molecular events that are regulated by the protein–protein interactions of the FE65 protein family is limited.
Recent studies have begun to address the in vivo function of the FE65 proteins using reverse genetics. A deletion allele of the single Caenorhabditis elegans FE65 gene (feh-1) causes developmental arrest at the late embryo or L1-larva stage and results in starvation due to increased pharyngeal pumping, which was recently shown to be associated with decreased acetylcholinesterase activity (Zambrano et al, 2002; Bimonte et al, 2004). Similarly, siRNA knockdowns of the single C. elegans APP gene (apl-1) produced a pharyngeal pumping defect (Zambrano et al, 2002). These data indicate that the FE65/APP interaction regulates an essential physiological process at the neuromuscular junction. Furthermore, an isoform-specific FE65 knockout has been generated in mice (Wang et al, 2004). These animals lack the 97 kDa isoform of FE65, but show a five-fold upregulation of the 60 kDa isoform of FE65 in mutant brains. No gross neuroanatomical brain abnormalities were observed, possibly due to functional compensation by the elevated levels of the 60 kDa isoform or by other FE65 family members (Wang et al, 2004).
In order to understand the function of the FE65 proteins in mammalian brain, we have generated mice that are deficient for two of the three FE65 protein family members, FE65 and FE65L1. Characterization of the single knockout mice revealed no abnormalities. However, the FE65−/−; FE65L1−/− mice displayed neuroanatomical abnormalities in the cortex and hippocampus that include the same types of marginal zone heterotopias, midline crossing defects and axonal pathfinding abnormalities occurring in animals lacking APP family members or for the latter two phenotypes in mice lacking Ena/Vasp proteins. Our findings suggest that FE65 family members cooperate with these proteins in controlling the development of the mammalian brain.
Results
Generation and characterization of mice lacking FE65 and FE65L1
FE65 is widely expressed in the adult mouse brain (King and Turner, 2004). However, little is known about the expression of FE65L1 and FE65L2 in brain. We performed in situ hybridization analyses of adult wild type (WT) mouse brain slices using antisense probes specific for FE65, FE65L1 and FE65L2, and found a similar spatial distribution in the brain for all three family members (Supplementary Figure S2), suggesting possible functional redundancy for the three FE65 proteins.
To better understand the role of the FE65 protein family in the central nervous system (CNS), we generated mice with targeted alleles for FE65 and FE65L1 (Supplementary Figure S1A). Mice lacking only FE65 or FE65L1 were indistinguishable from their WT littermates, viable and fertile, and histological examination of adult brains revealed no obvious neuroanatomical abnormalities (Supplementary Figure S1D).
However, live FE65−/−; FE65L1−/− progeny were found at a lower Mendelian frequency than expected at P21, whereas embryos (E12.5–E16.5) and perinatal pups (E18.5–P0) were closer to the expected frequency, indicating that death occurs between birth and weaning in the double mutants due to unknown causes (Table I). Viable FE65−/−; FE65L1−/− mice are smaller than their littermates (Table II) and often displayed bilateral circling. Routine histological staining of the major organs revealed no overt anatomical abnormalities (data not shown).
Table 1.
Expected and actual survival rates of FE65/FE65L1 compound null mutant mice
| Genotype | Expected (%) | P21 (%) | Perinatal pupsa (%) | Embryosb (%) |
|---|---|---|---|---|
| FE65−/− or FE65L1−/− | 25 | 30.9 (38/123) | 20.7 (6/29) | 29.4 (20/68) |
| FE65−/−; FE65L1+/− and FE65+/−; FE65L1−/− | 50 | 54.5 (67/123) | 48.3 (14/29) | 50.0 (34/68) |
| FE65−/−; FE65L1−/− | 25 | 14.6 (18/123) | 31 (9/29) | 20.6 (14/68) |
| FE65−/−; FE65L1+/− and FE65+/−; FE65L1−/− | 50 | 59.5 (44/74) | 52.0 (50/96) | 48.3 (28/58) |
| FE65−/−; FE65L1−/− |
50 |
40.4 (30/74) |
48.0 (46/96) |
51.7 (30/58) |
| aE18.5–P0. | ||||
| bE12.5–E16.5. | ||||
Table 2.
Weight of mutant mice at P28
| Genotype | Weight (g) |
n | |
|---|---|---|---|
| Males | Females | ||
| WT | 17.19 | 14.76 | 37 |
| FE65−/− | 16.98 | 15.70 | 12 |
| FE65L1−/− | 17.36 | 15.86 | 35 |
| FE65−/−; FE65L1−/− | 13.06a | 13.14b | 13 |
| DKO littermatesc |
19.7 |
15.09 |
19 |
| at-Tests: DKO versus WT, P<0.05; DKO versus littermates, P<0.01. | |||
| bt-Tests: DKO versus WT, P<0.001; DKO versus littermates, P<0.02. | |||
| cFE65−/−; FE65L1+/− and FE65+/−; FE65L1−/−. | |||
Neuronal ectopias form during cortical development in FE65−/−; FE65L1−/− mice
Histological staining of adult FE65−/−; FE65L1−/− mouse brains revealed neuronal positioning defects in the cortex and significantly larger lateral ventricles than in WT brains. Focal heterotopias were observed in layer 1 of FE65−/−; FE65L1−/− adult cortices (Supplementary Figure S1E), and immunostaining for NeuN indicated that neurons were present in these heterotopias (data not shown). Heterotopias were found in frontal and parietal cortices, but were absent from the cerebellum and olfactory bulb.
The development of the cortical plate begins with neurogenesis and an initial wave of neuronal migration to form the preplate. Successive waves of newly differentiated neurons migrate from the ventricular zone to the preplate, causing the preplate to split, producing the marginal zone (layer 1 in the adult cortex) and subplate, and leading to the eventual assembly of the six layers that form the cortical plate (Marin and Rubenstein, 2003). To determine the stage at which focal heterotopias appear during FE65−/−; FE65L1−/− cortical development, we performed H&E staining on E12.5, E14.5, E16.5 and E18.5 FE65−/−; FE65L1−/− embryo brain slices. Clusters of misplaced cells on the neocortical surface in E18.5 FE65−/−; FE65L1−/− brains (Figure 1B), not present in control E18.5 brains (Figure 1A), indicated that heterotopias formed prior to birth. Cortical lamination appeared normal in the surrounding parenchyma, but heteretopias were often associated with a stream of cell bodies directed toward the ectopic cell foci (Figure 1D). Disruption of the pial membrane and occupation of the subarachnoid space by cell bodies (Figure 1D) was not detected in control neocortices (Figure 1A and C). Although the size of the foci increased incrementally with increases in brain volume, partial loss of viability for the FE65−/−; FE65L1−/− mice cannot be attributed to the size and location of heterotopias, as perinatal mice have heterotopias that are similar in relative size and location to those found in live adult mice. Heterotopias were also observed at earlier embryonic stages, indicating that they form during the period of neuronal migration that results in cortical layering. In the five adult and the 13 embryonic (four E18.5, five E16.5, four E14.5) FE65−/−; FE65L1−/− mice examined, all brains contained heterotopias, indicating 100% penetrance for this phenotype at E14.5 and onward. Furthermore, heterotopias were not observed in adult triple allele mutant (FE65+/−; FE65L1−/−, n=2; FE65−/−; FE65L1+/−, n=2) brains. To determine whether brain abnormalities could be identified at a stage of development occurring prior to the waves of neuronal migration that form the cortical plate, we performed H&E staining of E12.5 WT and FE65−/−; FE65L1−/− embryos. At this stage of neocortical development, differentiated neurons residing below the pial membrane form the preplate. We did not detect ectopic cells in the preplate of our E12.5 compound null mice by histological staining. However, a striking protrusion of ectopic cells was observed in the ventral pallium of E12.5 FE65−/−; FE65L1−/− brains (Figure 1F compare to Figure 1E). These data suggest that ectopia formation in these mice is not restricted to neuronal migration events occurring exclusively during the development of the cortical plate.
Figure 1.
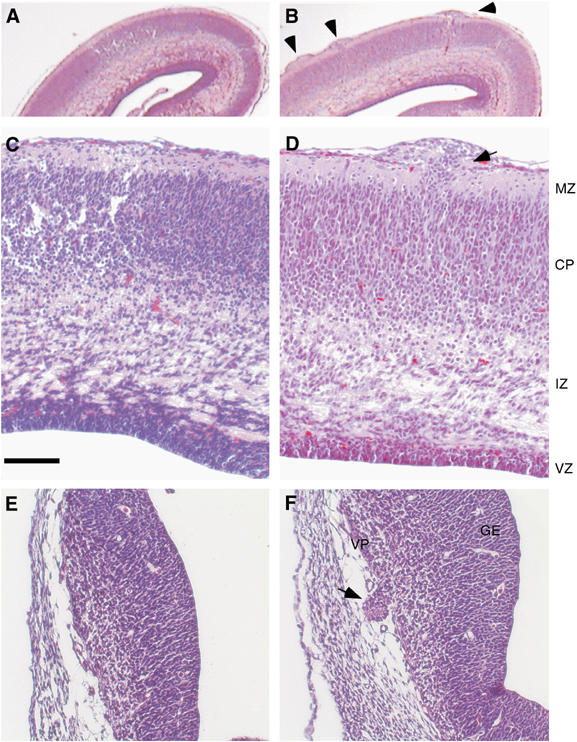
Cortical abnormalities in FE65−/−; FE65L1−/− mice. H&E-stained coronal sections of E18.5 (A–D) and E12.5 (E–F) mouse cortices. Control littermate FE65−/−; FE65L1+/− (A, C) and E12.5 WT (E) control sections are compared to FE65−/−; FE65L1−/− E18.5 (B, D) and E12.5 sections (F). Arrowheads point to ectopic cell nodules on the surface of the E18.5 cortex in the FE65−/−; FE65L1−/− brain (B) and arrows indicate ectopic neurons that have invaded the marginal zone and broken through the pial membrane of E18.5 and E12.5 cortex (D, F). The cranium is present in E12.5 sections. Scale bar, 100 μm (B–F). MZ, marginal zone; CP, cortical plate; IZ, intermediate zone; VZ, ventricular zone; VP, ventral pallium; GE, ganglionic eminence.
Localized displacement of reelin-positive neurons in FE65−/−; FE65L1−/− brains
Reelin, a large secreted protein essential for establishing the inside-out layering of the neocortex, is expressed by Cajal–Retzius (CR) neurons residing in the marginal zone of the developing embryonic cortex (Marin and Rubenstein, 2003). Signaling events mediated by the binding of reelin to its receptors, ApoER2 and VLDLR, are essential for normal cortical lamination and may act by altering the adhesive properties of neurons (Trommsdorff et al, 1999; Ballif et al, 2004). Loss or mislocalization of CR neurons has been associated with the formation of neuronal ectopias and disorganization of the surrounding cortical layers (Hartmann et al, 1999; Graus-Porta et al, 2001; Ligon et al, 2003; Herms et al, 2004). To determine whether CR cells are mispositioned in the cortex of FE65−/−; FE65L1−/− brains, cortical slices from E18.5, E14.5 and E12.5 mice were immunostained with an anti-reelin antibody. In WT embryos, staining of reelin and the large CR cells was observed as a uniform layer in the marginal zone (Figure 2A and C). By contrast, CR cells were displaced by clusters of ectopic neurons in E18.5 (Figure 2B) and E14.5 brains (Figure 2D). However, diffuse reelin staining, representing secreted reelin, could be detected in the marginal zone surrounding heterotopic neurons in E14.5 FE65−/−; FE65L1−/− brains (Figure 2D). Furthermore, reelin staining in the marginal zone of WT and compound mutant animals appeared similar. Thus, there is no striking loss of CR neurons or reelin throughout the marginal zone of mutant mice. In fact, in E12.5 brains, the ectopic cells express reelin (Figure 2I and J). CR cells are born between E10.5 and E12.5 at several focal sites including the ventral pallium from which they are redistributed to the marginal zone by tangential migration (Bielle et al, 2005). Thus, mislocalization of radially migrating neuroepithelial cells in the developing cortex (pallium) and tangentially migrating reelin-positive interneurons in the ventral pallium suggests that neuron mispositioning is due to defects in the local environment.
Figure 2.
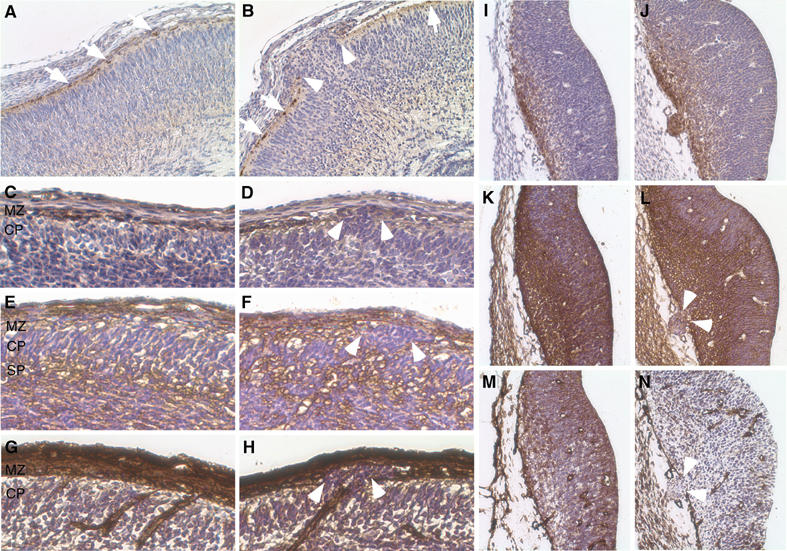
Reelin-positive neurons, reelin and ECM components in the developing cortical plate of FE65−/−; FE65L1−/− mice. IHC staining of coronal paraffin sections from E18.5 (A, B), E14.5 (C–H) and E12.5 (I–N) control (A, C, E, G, I, K, M) and FE65−/−; FE65L1−/− (B, D, F, H, J, L, N) mouse brains. In WT E18.5 and E14.5 cortices (A, C), G10 staining shows reelin-positive neurons and secreted reelin in the marginal zone. In the FE65−/−; FE65L1−/− mice (B, D), reelin-positive cells are displaced by ectopic neurons. In WT cortices, CSPGs (CS56 antibody) are present in the subplate and marginal zone (E), the latter being disrupted by heterotopic neurons in FE65−/−; FE65L1−/− mice (F). Laminin staining reveals blood vessels and the cortical basement membrane (G), which is discontinuous when neurons invade the marginal zone of FE65−/−; FE65L1−/− mouse brains (H). G10 staining shows reelin-positive cells in the ventral pallium of E12.5 WT brains (I), which have overmigrated in the E12.5 FE65−/−; FE65L1−/− mouse brain (J). CSPG (K, L) and laminin (M, N) staining in the ventral pallium of E12.5 brains is disrupted at the pial membrane by heterotopic cells in the FE65−/−; FE65L1−/−brains. Arrowheads in panels B, D, F, H, L and N show the breaks in the pial membrane associated with heterotopias. Arrows in panels A and B point to reelin-positive cells. MZ, marginal zone; CP, cortical plate; SP, subplate; IZ, intermediate zone.
Disruption of the pial–meningeal basement membrane in FE65−/−; FE65L1−/− brains
Mice lacking basement membrane constituents or proteins that play a prominent role in cell adhesion and adhesion-dependent signaling events display abnormal cortical development and focal marginal zone heterotopias (Blackshear et al, 1997; Georges-Labouesse et al, 1998; Graus-Porta et al, 2001; Halfter et al, 2002; Moore et al, 2002; Beggs et al, 2003). To examine the integrity of the basement membrane in the FE65−/−; FE65L1−/− mice, we stained mutant embryonic brains for chondroitin sulfate proteoglycans (CSPGs) and laminin. CSPGs are abundant in the marginal zone and subplate during cortical plate development, whereas laminin localizes to the pial basement membrane and cerebral vasculature (Bicknese et al, 1994). Both CSPG and laminin staining were reduced in regions of the marginal zone that contained heterotopic neurons (Figure 2F and H) when compared to E14.5 WT embryos (Figure 2E and G). However, CSPG staining of the subplate in FE65−/−; FE65L1−/− mouse brains did not differ significantly from control brains (Figure 2F and E). Furthermore, a dramatic disruption in both CSPG (Figure 2L) and laminin (Figure 2N) staining at sites of heterotopic neurons in E12.5 FE65−/−; FE65L1−/− mice was indicative of a discontinuity in the pial basement membrane when compared to control embryos (Figure 2K and M). Thus, foci of ectopic cells in the developing cortical plate and ventral pallium of FE65−/−; FE65L1−/− mice may be due to defects in the pial basement membrane.
FE65/FE65L1-deficient meningeal fibroblasts show altered laminin organization
Marginal zone heterotopias associated with a disruption in pial laminin develop in focal adhesion kinase (fak)-flox embryos derived from crosses with mice expressing Cre in neuroepithelial precursors and after injection of adenovirus bearing CMV-Cre to the brain outer surface (Beggs et al, 2003). In contrast, no heterotopias were identified in conditional knockouts obtained by crossing fak-flox and nexin-Cre mice, which produces a neuron-specific FAK knockout (Beggs et al, 2003). These data indicate that loss of the signaling response of meningeal fibroblasts to the ECM is sufficient to produce overmigration of cortical neurons. Furthermore, FAK-deficient meningeal fibroblasts show disorganized laminin (Beggs et al, 2003). To determine whether FE65−/−; FE65L1−/− meningeal fibroblasts also display altered laminin organization, we performed laminin immunocytochemistry on primary meningeal fibroblasts from WT and FE65−/−; FE65L1−/− brains. Meningeal fibroblasts from WT mice produced laminin that was arranged in a fibrillar and punctate pattern, whereas meningeal fibroblasts from FE65−/−; FE65L1−/− mice showed a reduction in punctate laminin and a lack of fibrillar laminin at the cell periphery (Figure 3). In addition, meningeal fibroblasts from FE65−/−; FE65L1−/− mice showed an increase in F-actin fiber thickness but fewer fibers compared to WT (Figure 3), possibly reflecting the response of these cells to loss of peripheral laminin. These data are consistent with heterotopia formation in the FE65−/−; FE65L1−/− being caused by a basement membrane defect at the pial surface resulting from meningeal cell dysfunction. Furthermore, in contrast to what has been observed in presenilin-1 (PS1)-deficient mice, which also display marginal zone heterotopias (Hartmann et al, 1999), fibrosis of the meninges does not occur in FE65−/−; FE65L1−/− mice (Figure 3B), nor was it reported for the APP/APLP1/APLP2 triple knockout (Herms et al, 2004).
Figure 3.
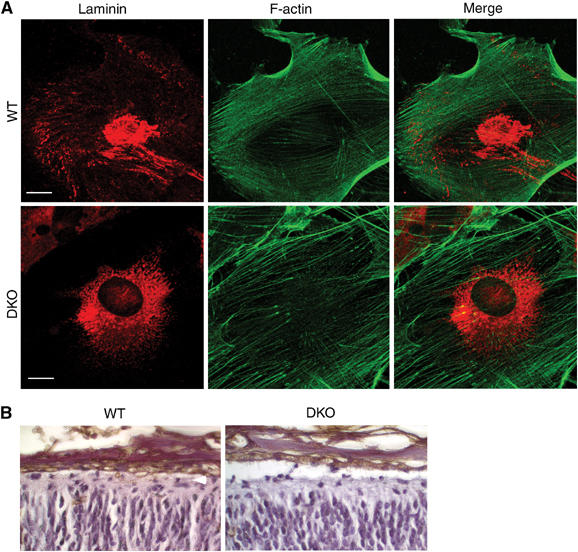
Defective laminin organization in FE65−/−; FE65L1−/− meningeal fibroblasts. (A) Primary meningeal fibroblasts from WT and FE65−/−; FE65L1−/− mice were double stained with a laminin antibody and phalloidin-AlexaFluor 488 to detect F-actin. Fibrillar and punctate laminin is observed at the periphery in WT meningeal fibroblasts, whereas it is absent from FE65−/−; FE65L1−/− meningeal fibroblasts. Scale bar, 20 μm. (B) Staining with the lectin RCA-1 shows no difference in the thickness of meninges for WT and FE65−/−; FE65L1−/− E18.5 mouse brains.
APP expression and processing changes do not account for heterotopia formation
As mice lacking all three APP family members develop marginal zone heterotopias (Herms et al, 2004), we performed IHC staining for APP and APLP2 using the A8717 antibody to determine whether overmigrating neurons expressed APP proteins. Our results indicate that neuronal overmigration is not due to loss of APP/APLP expression in the mispositioned neurons (Figure 4A). Similar results were obtained using the 22C11 antibody (data not shown). Furthermore, no decrease in APP/APLP2 steady-state levels were observed in whole brain lysates of FE65−/−; FE65L1−/− mice compared to WT and single knockout mice (Figure 4B). In addition, no changes in APP/APLP2α levels in brain lysates, cellular APP from primary cultured neurons and glial cells or secreted APP or CSPG-APPs levels from conditioned media were found (data not shown), thus precluding the simple explanation that common phenotypes between these two mouse mutants were due to loss of APP proteins or their α-secretase-derived products. Furthermore, APP/APLP2 localization did not differ in cultured WT and FE65−/−; FE65L1−/− cortical neurons (Supplementary Figure S3).
Figure 4.
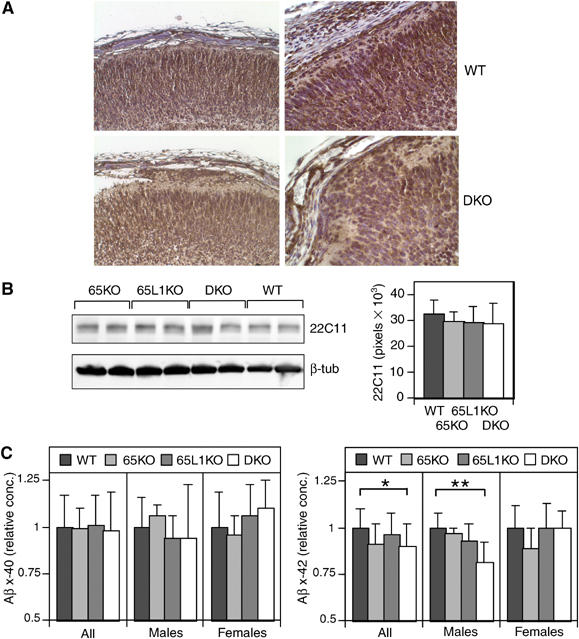
APP and Aβ levels in FE65−/−; FE65L1−/− brains. (A) IHC staining for APP (A8717) shows expression throughout the cortical plate in WT and FE65−/−; FE65L1−/− E18.5 brains and in heterotopic cells. (B) APP and APLP2 steady-state levels are unchanged in FE65−/−; FE65L1−/− adult mouse brains compared to WT. Western blot analyses using 22C11 and anti-β-tubulin (reprobe) on two mouse brain diethylamine extracts per genotype. Bar chart shows average APP steady-state levels that were normalized to β-tubulin (as load control), with standard error bars for WT (n=8), FE65−/− (n=7), FE65L1−/− (n=9), FE65−/−; FE65L1−/− (n=9) mouse brains. No significant difference was observed between genotypes. (C) Aβx-40 and Aβx-42 levels in diethylamine brain extracts of WT (n=5F, 7M), FE65−/−(n=7F, 4M), FE65L1−/− (n=10F, 4M) and FE65−/−; FE65L1−/− (n=6F, 6M) 14- to 21-week-old mice. Data have been normalized to the mean of the WT mice. Aβx-42 levels are significantly reduced in FE65−/−; FE65L1−/− males compared to WT. t-Test *P<0.05; **P<0.005.
Interestingly, a significant decrease in brain Aβx-42 levels was observed for adult FE65−/− mice (10.1%, P=0.03, n=11) and FE65−/−; FE65L1−/− mice (11.4%, P=0.022, n=12) when compared to WT (n=12), whereas no significant changes in Aβx-40 were observed (Figure 4C). When the comparison was restricted to animals of the same sex, no significant differences in Aβx-40 and Aβx-42 levels were observed between female mice, nor were any significant changes observed between WT and FE65−/− male mice. However, the reduction in Aβx-42 levels for FE65−/−; FE65L1−/− mice compared to WT males became more pronounced (23.4%, P⩽0.005) and a similar trend was observed for Aβx-40 (14.5% reduction) in these animals but it was not statistically significant. These data indicate subtle sex-dependent changes in Aβ levels in the FE65−/−; FE65L1−/− mice.
Axonal projection defects are varied in the FE65−/−; FE65L1−/− mice
Several axonal projection pathways are aberrant in FE65−/−; FE65L1−/− brains. Reduced size and medial shifting of the fimbria were observed in FE65−/−; FE65L1−/− mice when compared to WT coronal sections at similar rostro-caudal positions, possibly due to a reduction in the number of efferent fibers from the hippocampus (Figure 5A and B). Silver staining of coronal sections also revealed defects in the mossy fiber pathway formed by dentate granule cell axons. In WT hippocampi, mossy fiber bundles extend from dentate granule cells and run through the hilus of the dentate, above (suprapyramidal) and below (infrapyramidal) the pyramidal cell layer of the CA3 region (Figure 5C). By contrast, the infrapyramidal blade of the dentate gyrus in the FE65−/−; FE65L1−/− mice (n=4) is foreshortened, deflected toward the thalamus and lacks mossy fiber axons (Figure 5D). This does not occur in FE65+/−; FE65L1−/− or FE65−/−; FE65L1+/− hippocampi (data not shown). Interestingly, destruction of meningeal cells of the dentate anlage results in almost complete loss of the infrapyramidal blade of the dentate gyrus (Hartmann et al, 1992), suggesting that the defective infrapyramidal blade of the dentate gyrus and loss of the associated mossy fibers in the FE65−/−; FE65L1−/− mice may also be due to dysfunctional meningeal cells.
Figure 5.

Abnormal axonal trajectories in FE65−/−; FE65L1−/− mouse cortex. Silver-stained coronal sections of 10-week-old WT (A, C), 12-week-old FE65−/−; FE65L1−/− (B, D) and 9-week-old (E) FE65−/−; FE65L1−/− mouse brain sections. An intact corpus callosum, well-developed fimbria and mossy fiber axons emanating from dentate granule cells are observed in the WT mouse brain (A). In the FE65−/−; FE65L1−/− mice, cortical projection neurons fail to cross the midline, forming Probst bundles, and fimbria are reduced in size and medially displaced (B). Higher magnification (boxed in A, B) reveals the absence of the infrapyramidal mossy fibers in FE65−/−; FE65L1−/− hippocampi (D) compared to WT (C). Aberrant fiber bundles are present in the cortical plate of adult (E) and E18.5 FE65−/−; FE65L1−/− mice (F). E18.5 brain slices were stained with Tuj1 (red) and counterstained with DAPI (blue) (F). Arrows indicate the position of fiber bundles and arrowheads indicate breaks in the marginal zone where DAPI-positive cells have invaded and migrated into the subarachnoid space. cc, corpus callosum; dg, dentate gyrus; f, fimbria; mf, mossy fibers; spmf, suprapyramidal mossy fibers; ipmf, infrapyramidal mossy fibers; P, Probst bundle. Scale bar, 250 μm (A).
The major axonal projection pathway joining the two hemispheres, the corpus callosum, is also disrupted in FE65−/−; FE65L1−/−mice (Figure 5A). Agenesis of the corpus callosum was observed in all adult (n=5) and E18.5 (n=3) FE65−/−; FE65L1−/− mouse brains that were examined for this defect (Figure 5B). However, no callosal abnormalities were detected in WT (n=6) mice, single knockout mice (FE65−/− n=2; FE65L1−/− n=3) or mice lacking three of the four FE65 and FE65L1 alleles (E18.5, n=2; adult, n=4). Probst bundles, which consist of callosal axonal projections that have grown toward but have failed to cross the midline (Ozaki and Wahlsten, 1993), were visualized in several FE65−/−; FE65L1−/− mouse brains (see Figure 5B). A variable frequency of callosal agenesis has been reported for the 129 inbred strain background, depending on the substrain (Magara et al, 1999). However, combined deficiency of FE65 and FE65L1 dramatically increased the frequency of callosal agenesis above that of the underlying genetic predisposition contributed by the 129SvEvBradley strain background and partial FE65/FE65L1 loss in our hybrid strain background (129SvEvBradley × C57BL/6).
Neural connections are established at the time of neuronal migration during cortical development. Ectopic axon bundles were observed in FE65−/−; FE65L1−/− adult (n=2) and embryonic (n=3) brains (Figure 5E and F, respectively). These axon bundles traverse the cortex in a radial orientation and appear to be directed toward heterotopic cell bodies (Figure 5F). Thalamocortical axons produce collaterals that begin to radially enter the cortex at E16 (Bicknese et al, 1994; Del Rio et al, 2000). To examine axonal projections during development, we stained E16.5 FE65−/−; FE65L1−/− brains with antibodies directed against the neural cell adhesion molecules L1 and TAG-1, which have been reported to label thalamocortical axons and cortical efferents, respectively (Fukuda et al, 1997). TAG-1 staining of E16.5 WT and FE65−/−; FE65L1−/− cortical fibers showed fewer fibers in the intermediate zone and fewer fibers leaving the intermediate zone to enter the internal capsule in the FE65−/−; FE65L1−/− brains compared to WT (Figure 6B and A). L1-positive fibers were fewer in the internal capsule of the FE65−/−; FE65L1−/− mouse brains. More importantly, L1-positive fibers projected in a radial orientation toward the marginal zone (Figure 6C and D), indicating that ectopic fibers observed in the cortical plate are of thalamic origin and suggesting that subplate abnormalities may exist in FE65−/−; FE65L1−/− mouse brains.
Figure 6.
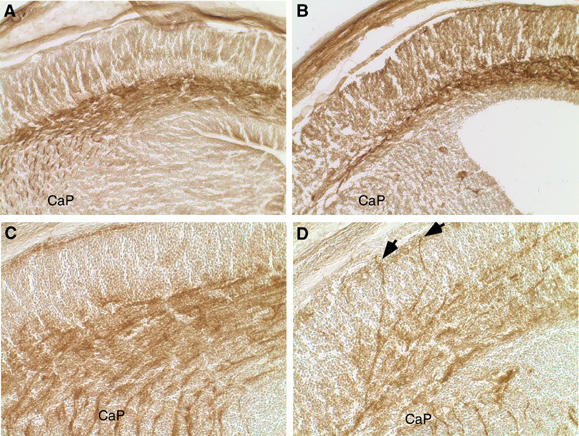
Aberrant Tag-1 corticofugal and L1-thalamocortical fiber projections in FE65−/−; FE65L1−/− mice. IHC staining of Tag-1 (A, B) and L1 (C, D) in coronal cryostat sections of E16.5 WT (A, C) and FE65−/−; FE65L1−/− (B, D) mouse brains. There is a reduction in Tag-1-positive fibers exiting the cortex in FE65−/− FE65L1−/− mice (B) compared to WT controls (A). Fewer L1-positive fibers enter the cortex from the caudate putamen (CaP) and are ectopically localized in the developing cortex of FE65−/−; FE65L1−/− mice (D) compared to the organized projections of L1-positive fibers in the WT control (C). Arrows identify L1-positive ectopic fibers that have reached the marginal zone.
Taken together, the ectopic thalamocortical fibers, loss of the infrapyramidal mossy fibers, reduced corticothalamic projections, reduced fimbria size and callosal agenesis observed in the FE65−/−; FE65L1−/− mouse brains suggest that FE65 protein deficiencies interfere with the establishment of axonal connections during development.
Discussion
In the present study, we examined the consequences of FE65 and FE65L1 deficiencies on mammalian brain development in mice. Until now, our understanding of the function of the FE65 family in vivo was limited to its role at the C. elegans neuromuscular junction (Zambrano et al, 2002; Bimonte et al, 2004). We report that the FE65 and FE65L1 proteins are required for integrity of the pial basement membrane, neuronal positioning and the establishment of normal axonal projections during cortical development. Furthermore, the phenotypes of the FE65 and FE65L1 double knockout share remarkable similarities with that reported for mutant mice lacking FE65-binding partners, the three members of the APP gene family, that is APP, APLP1 and APLP2 (Herms et al, 2004), and the Ena/Vasp protein family (Lanier et al, 1999).
Molecules implicated in neuronal migration such as those necessary for locomotion, nucleokinesis and adhesion of migrating neurons along radial glia contribute to cortical development (Marin and Rubenstein, 2003). Reelin, a key molecule for cortical lamination, has been proposed to act either as an attractant or a stop signal for neuronal positioning in the terminal phase of radial migration (Soriano and Del Rio, 2005). Premature loss of CR neurons from the marginal zone produces defects in neuronal positioning in the cortex, possibly resulting from defective reelin-dependent regulation of radial glia (Soriano and Del Rio, 2005). Disruption of the ‘limiting membrane', consisting of radial glia endfeet and basal lamina, is often associated with a phenotype that resembles the FE65−/−; FE65L1−/− heterotopias, resulting in the accumulation of ectopic neurons in the marginal zone and subarachnoid space (Lambert de Rouvroit and Goffinet, 2001; Marin and Rubenstein, 2003). PS1- or APP/APLP1/APLP2-deficient mice, which also develop focal heterotopias, are associated with reduced numbers of CR neurons throughout the marginal zone (Hartmann et al, 1999; Herms et al, 2004). Although a 37% reduction in marginal zone CR cells is observed in E18.5 but not E14.5 APP/APLP1/APLP2-deficient mice, it remains possible that cell dysfunction occurs at the onset of cortical layer formation, resulting in cell death at a later developmental stage. In the FE65−/−; FE65L1−/− mice, we cannot exclude the possibility that CR neuron function is suboptimal. However, neither the reelin-positive neurons nor reelin levels appeared to differ significantly in these mice.
Cell adhesion plays a central role in neuronal migration and positioning during cortical plate formation. The ECM proteins of the basal lamina are secreted by meningeal fibroblasts located at the interface of the cortical plate and subarachnoid space. Focal marginal zone heterotopias have been observed in mice deficient for molecules that regulate cell adhesion, integrins, dystroglycan and FAK (Georges-Labouesse et al, 1998; Graus-Porta et al, 2001; Moore et al, 2002; Beggs et al, 2003; Niewmierzycka et al, 2005) or in ECM molecules that are components of the pial basement membrane such as laminin γ1 and perlecan (Costell et al, 1999; Halfter et al, 2002). In these mutants, CR neurons were either found at deeper levels in the cortical plate or were pushed aside by ectopic neurons and were associated with local disruption of cortical layers, similar to what we observed for the FE65−/−; FE65L1−/− mice. Pial basement membrane disruption is not always observed when neurons overmigrate (Ligon et al, 2003). This finding and our observation that a defective pial membrane is associated with ectopic neurons as early as E12.5 in the FE65−/−; FE65L1−/− mice supports the hypothesis that pial basement membrane disruption is the primary defect in these mice. Our findings that FE65−/−; FE65L1−/− primary meningeal fibroblasts fail to organize fibrillar laminin and have altered laminin distribution compared to WT fibroblasts further support this hypothesis and suggest defects in meningeal cells that may extend to the production and secretion of neuronal growth cone inhibitory CSPG molecules (Shearer et al, 2003). Inappropriate positional cues from the ECM to migrating neurons, regardless of their mode of migration (tangential or radial), may lead to aberrant positioning of neurons in FE65−/−; FE65L1−/− mice. A role for the APP protein family in cell adhesion is supported by binding of APP to type 1 collagen, laminin, fasciclin II, the heparan sulfate proteoglycan glypican-1, and disruption of the epithelial cell layers in wings of APP transgenic Drosophila (Kibbey et al, 1993; Williamson et al, 1996; Coulson et al, 2000; Ashley et al, 2005). Furthermore, colocalization of FE65, APP and β1-integrin has been reported in adhesion sites of dynamic membranes known as focal complexes (Sabo et al, 2001). Taken together, these data suggest that defective ECM deposition and neuronal adhesion may be responsible for cortical neuron positioning defects in both the APP/APLP1/APLP2 and the FE65/FE65L1 compound null mice. Our results suggest that disruption of FE65 family members alters laminin distribution through its effects on APP/APLP function.
As the FE65 proteins bind APP family members and have been shown to modulate APP processing in cell culture, it is tempting to speculate that modulation of APP/APLP processing is responsible for the shared cortical phenotypes for these knockout mice. Our data do not support this hypothesis, as Aβ levels were marginally decreased only in male FE65−/−; FE65L1−/− brains, whereas heterotopias are detected in both male and female mice. The reasons for this sex-specific decrease in brain Aβ are presently unclear. Importantly, however, our data confirm that FE65 proteins participate in the modulation of APP processing in vivo and our findings are consistent with the reported decrease in Aβ in p97−/−/APP transgenic cortical neuronal cultures (Wang et al, 2004).
FE65 was shown to play a role in AICD-dependent transcriptional activation (Cao and Sudhof, 2001), but, unlike FE65, the FE65L1 and FE65L2 interactions with APP do not activate APP-dependent transcription (Tanahashi and Tabira, 2002; Chang et al, 2003). As we only observed cortical phenotypes in mice lacking both FE65 and FE65L1, there is insufficient evidence to implicate transcriptional dysregulation as an underlying cause for neuronal overmigration in FE65−/−; FE65L1−/− knockout mice. Nevertheless, the similarity of phenotypes in both these mouse mutants suggests that FE65-dependent molecular events may occur downstream of APP in response to events at the cell surface. In this regard, a recent study showing that APP and FE65 contribute to notch signaling (Fischer et al, 2005) suggests that impairment of notch-dependent interactions between neurons and glia may contribute to the similar phenotype of compound APP and FE65 family defects, respectively.
Our results show that axons from thalamocortical neurons are misrouted from the CSPG-rich subplate into the developing cortical plate of FE65−/−; FE65L1−/− mice. Thalamocortical axon outgrowth occurs in the subplate before production of axon collaterals that begin to radially enter the cortex at E16 (Bicknese et al, 1994; Del Rio et al, 2000). In reeler mice, the trajectory of thalamic fibers is normal until they reach and enter the cortex in diagonal fascicles, where they extend throughout the entire thickness of the cortex to accumulate in the superplate, which contains the cells normally found in the subplate of WT brains (Molnar et al, 1998). Ectopic fibers expressing the neural cell adhesion molecule, L1, were found to extend into the cortex in a radial orientation in developing E16.5 FE65−/−; FE65L1−/− mouse embryos, suggesting that some subplate cells may be mislocalized in the FE65−/−; FE65L1−/− mouse brains. Intriguingly, aberrant fiber projections from the intermediate zone through the cortex toward ectopic neurons residing in the marginal zone were reported in laminin-deficient mice (Halfter et al, 2002; Moore et al, 2002; Beggs et al, 2003). Thus, the presence of two distinct phenotypic manifestations that can arise from laminin defects in the FE65−/−; FE65L1−/− mice provides strong evidence for the role of these scaffolding proteins in modulating neuronal cell positioning through their indirect effects on neuronal cell adhesion.
FE65 also interacts with Mena and Evl, proteins involved in actin cytoskeleton remodeling of neuronal filopodia and fibroblast lamellipodia (Ermekova et al, 1997; Lambrechts et al, 2000; Bear et al, 2002; Lebrand et al, 2004). Callosal agenesis in Mena-deficient mice resulted from axonal projection failure across the midline, and increased rates of neuronal migration, resulting in neurons that are localized to more superficial layers of the mouse cortex, were observed for neurons in which the Ena/Vasp proteins were neutralized (Lanier et al, 1999; Goh et al, 2002). Thus, Ena/Vasp deficiency results in phenotypes resembling those observed in the FE65−/−; FE65L1−/− mice and suggests that the interactions between these two protein families, which involve the WW domain in the FE65 proteins, may be important for axon guidance and neuronal positioning in the developing brain. Our observation that meningeal fibroblasts lacking FE65 and FE65L1 have altered laminin and actin filament organization may be due to the loss of FE65/Ena/Vasp interactions. Thus, interactions of the FE65 proteins with the Ena/Vasp protein family and/or the APP protein family may be responsible for producing some if not all of the phenotypes observed in the FE65−/−; FE65L1−/− mice.
Marginal zone heterotopias and leptomeningeal glioneuronal heterotopias are common neuropathological abnormalities found in human brains with cortical dysplasia (Hirano et al, 1992; Mischel et al, 1995). Cobblestone or type II lissencephalies are characterized by leptomeningeal heterotopias that are often associated with other CNS malformations and are found in congenital syndromes such as Fukumaya congenital muscular dystrophy, muscle-eye brain disease and Walker–Warburg syndrome (Ross and Walsh, 2001). Although we have exclusively focused on the roles of FE65 and FE65L1 in the CNS, it should nevertheless be noted that defects at neuromuscular junctions have been reported in both FE65- and APP-deficient C. elegans (Zambrano et al, 2002; Bimonte et al, 2004) and in the APP/APLP2 double knockout mice (Wang et al, 2005). Taken together, these findings suggest that the FE65 protein family may mediate multiple developmental and tissue-specific functions through a molecular pathway that participates in cortical neuronal development as well as in the regulation of muscle function, a shared feature of many of the type II lissencephalies.
Materials and methods
Generation of FE65−/−, FE65L1−/− and FE65−/−; FE65L1−/− mice
See Supplementary data for methods describing the generation of the FE65−/−, FE65L1−/− and FE65−/−; FE65L1−/− mice.
Antibodies
The pan-FE65 antibody generated against a glutathione S-transferase-FE65L1 fusion protein was previously described (Chang et al, 2003). The Tuj1 (Covance), anti-NeuN (Chemicon Int.), GFAP (Sigma-Aldrich) and CD-45 (Serotec) antibodies were used to stain immature neurons, postmitotic neuronal nuclei, glia and microglia, respectively. Antibodies directed against EHS-laminin and chondroitin sulfate modifications (both from Sigma-Aldrich) were used to detect extracellular matrix (ECM) proteins and reelin antibodies (G10, a gift from A Goffinet, University of Louvain, Belgium) detected secreted reelin and reelin-positive cells (de Bergeyck et al, 1998). APP antibodies included 22C11 (Chemicon Int.) and A8717 (Sigma-Aldrich). Anti-TAG-1 (Developmental Studies Hybridoma Bank) and anti-L1 antibodies (Chemicon Int.) were used to stain corticofugal and thalamocortical neurons, respectively.
Histology and immunohistochemistry
Mice were deeply anesthetized and perfused with PBS and 4% paraformaldehyde (PFA). Brains were dissected and immersed in 4% PFA for 4–16 h. Embryos were immersed in 4% PFA/PBS for fixation. Paraffin-embedded coronal sections were used for hematoxylin and eosin (H&E) staining, silver staining (Fink and Heimer, 1967) and some immunohistochemical (IHC) staining. Frontal and parietal cortex embryo brain sections were collected and a single slice representative of a group of at least 10 serially sliced sections was stained with H&E to localize foci of heterotopic neurons. Adjacent slices were used for IHC staining of CSPGs, laminin, reelin, APP (A8717) and NeuN. Coronal cryostat sections obtained from tissue submerged in 30% sucrose overnight at 4°C and embedded in OCT (Electron Microscopy Sci.) were used for IHC staining of Tag-1, L1 and β-tubulin. Primary antibody detection for IHC staining was performed with biotinylated goat anti-rat, rabbit or mouse secondary antibodies, ABC solution and diaminobenzidine using Vectastain and MOM kits (Vector Labs) and counterstained with hematoxylin or with fluorescent secondary Alexa Fluor 546 goat antibodies (Molecular Probes) and counterstained with DAPI (Sigma-Aldrich). The biotinylated lectin from Ricinus communis, RCA-1 (Vector Labs), was used to stain leptomeningeal fibroblasts and endothelia (Hartmann et al, 1999).
Immunocytochemistry of primary meningeal fibroblasts
Meningeal fibroblasts were isolated from WT and FE65−/−; FE65L1−/− E16.5 embryonic cortex. After reaching confluency, fibroblasts were passaged and cultured for 3 days before fixation in 4% PFA for immunocytochemistry and WT cells were extracted for total protein. Glial contamination was undetectable in these cultures by immunocytochemistry using anti-GFAP and anti-CD45 antibodies. Double staining of WT and FE65−/−; FE65L1−/− fibroblasts was performed with an anti-laminin antibody followed by Alexa Fluor 546 goat anti-rabbit antibody and phalloidin-conjugated Alexa Fluor 488. Images were captured using a confocal microscope (Zeiss Axiovert 200).
Aβ measurements
Mouse brains were homogenized in 9 volumes of 0.2% diethylamine in 50 mM NaCl using a Potter-Elvehjem homogenizer. Homogenates were centrifuged at 100 000 g for 1 h and the supernatant was neutralized with a 1/10 volume of 50 mM Tris–HCl (pH 6.8). Aβx-40 and Aβx-42 ELISAs were performed using the BNT-77/BA-27 (Aβx-40) and BNT-77/BC-05 (Aβx-42) system (Duff et al, 1996).
Supplementary Material
Supplementary Materials and Methods
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We thank Uwe Beffert, John Shelton, Kai Zurhove, Charo Gonzalez-Agosti and Giuseppina Tesco for their scientific expertise; Angela Robak, Loren Lindsley and Liz Lummus for technical assistance; Lindee Goh and Frank Gertler for mouse strain rederivations. This work was supported by the Alzheimer Association, the JD French Alzheimer's and the Wolfgang Paul Program of the Humboldt Foundation, MADRC and the NIH: AG15903, HL20948, HL63762 and NS43408.
References
- Ashley J, Packard M, Ataman B, Budnik V (2005) Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci 25: 5943–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA (2004) Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol 14: 606–610 [DOI] [PubMed] [Google Scholar]
- Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB (2002) Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell 109: 509–521 [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF (2003) FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron 40: 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicknese AR, Sheppard AM, O'Leary DD, Pearlman AL (1994) Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci 14: 3500–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielle F, Griveau A, Narboux-Neme N, Vigneau S, Sigrist M, Arber S, Wassef M, Pierani A (2005) Multiple origins of Cajal–Retzius cells at the borders of the developing pallium. Nat Neurosci 8: 1002–1012 [DOI] [PubMed] [Google Scholar]
- Bimonte M, Gianni D, Allegra D, Russo T, Zambrano N (2004) Mutation of the feh-1 gene, the Caenorhabditis elegans orthologue of mammalian Fe65, decreases the expression of two acetylcholinesterase genes. Eur J Neurosci 20: 1483–1488 [DOI] [PubMed] [Google Scholar]
- Blackshear PJ, Silver J, Nairn AC, Sulik KK, Squier MV, Stumpo DJ, Tuttle JS (1997) Widespread neuronal ectopia associated with secondary defects in cerebrocortical chondroitin sulfate proteoglycans and basal lamina in MARCKS-deficient mice. Exp Neurol 145: 46–61 [DOI] [PubMed] [Google Scholar]
- Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T (2002) Fe65, a ligand of the Alzheimer's beta-amyloid precursor protein, blocks cell cycle progression by down-regulating thymidylate synthase expression. J Biol Chem 277: 35481–35488 [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC (2001) A transcriptionally (correction of transcriptively) active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293: 115–120 [DOI] [PubMed] [Google Scholar]
- Chang Y, Tesco G, Jeong WJ, Lindsley L, Eckman EA, Eckman CB, Tanzi RE, Guenette SY (2003) Generation of the beta-amyloid peptide and the amyloid precursor protein C-terminal fragment gamma are potentiated by FE65L1. J Biol Chem 278: 51100–51107 [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R (1999) Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol 147: 1109–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson EJ, Paliga K, Beyreuther K, Masters CL (2000) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int 36: 175–184 [DOI] [PubMed] [Google Scholar]
- de Bergeyck V, Naerhuyzen B, Goffinet AM, Lambert de Rouvroit C (1998) A panel of monoclonal antibodies against reelin, the extracellular matrix protein defective in reeler mutant mice. J Neurosci Methods 82: 17–24 [DOI] [PubMed] [Google Scholar]
- Del Rio JA, Martinez A, Auladell C, Soriano E (2000) Developmental history of the subplate and developing white matter in the murine neocortex. Neuronal organization and relationship with the main afferent systems at embryonic and perinatal stages. Cereb Cortex 10: 784–801 [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature 383: 710–713 [DOI] [PubMed] [Google Scholar]
- Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T, Sudol M (1997) The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J Biol Chem 272: 32869–32877 [DOI] [PubMed] [Google Scholar]
- Fink RP, Heimer L (1967) Two methods for selective silver impregnation of degenerating axons and their synaptic endings in the central nervous system. Brain Res 4: 369–374 [DOI] [PubMed] [Google Scholar]
- Fischer DF, van Dijk R, Sluijs JA, Nair SM, Racchi M, Levelt CN, van Leeuwen FW, Hol EM (2005) Activation of the Notch pathway in Down syndrome: cross-talk of Notch and APP. FASEB J 19: 1451–1458 [DOI] [PubMed] [Google Scholar]
- Fukuda T, Kawano H, Ohyama K, Li HP, Takeda Y, Oohira A, Kawamura K (1997) Immunohistochemical localization of neurocan and L1 in the formation of thalamocortical pathway of developing rats. J Comp Neurol 382: 141–152 [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A (1998) Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol 8: 983–986 [DOI] [PubMed] [Google Scholar]
- Goh KL, Cai L, Cepko CL, Gertler FB (2002) Ena/VASP proteins regulate cortical neuronal positioning. Curr Biol 12: 565–569 [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U (2001) Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367–379 [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U (2002) A critical function of the pial basement membrane in cortical histogenesis. J Neurosci 22: 6029–6040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann D, De Strooper B, Saftig P (1999) Presenilin-1 deficiency leads to loss of Cajal–Retzius neurons and cortical dysplasia similar to human type 2 lissencephaly. Curr Biol 9: 719–727 [DOI] [PubMed] [Google Scholar]
- Hartmann D, Sievers J, Pehlemann FW, Berry M (1992) Destruction of meningeal cells over the medial cerebral hemisphere of newborn hamsters prevents the formation of the infrapyramidal blade of the dentate gyrus. J Comp Neurol 320: 33–61 [DOI] [PubMed] [Google Scholar]
- Hass MR, Yankner BA (2005) A {gamma}-secretase-independent mechanism of signal transduction by the amyloid precursor protein. J Biol Chem 280: 36895–36904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J 23: 4106–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Houdou S, Hasegawa M, Kamei A, Takashima S (1992) Clinicopathologic studies on leptomeningeal glioneuronal heterotopia in congenital anomalies. Pediatr Neurol 8: 441–444 [DOI] [PubMed] [Google Scholar]
- Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE, Kleinman HK (1993) beta-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc Natl Acad Sci USA 90: 10150–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Turner RS (2004) Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol 185: 208–219 [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM (2001) Neuronal migration. Mech Dev 105: 47–56 [DOI] [PubMed] [Google Scholar]
- Lambrechts A, Kwiatkowski AV, Lanier LM, Bear JE, Vandekerckhove J, Ampe C, Gertler FB (2000) cAMP-dependent protein kinase phosphorylation of EVL, a Mena/VASP relative, regulates its interaction with actin and SH3 domains. J Biol Chem 275: 36143–36151 [DOI] [PubMed] [Google Scholar]
- Lanier LM, Gates MA, Witke W, Menzies AS, Wehman AM, Macklis JD, Kwiatkowski D, Soriano P, Gertler FB (1999) Mena is required for neurulation and commissure formation. Neuron 22: 313–325 [DOI] [PubMed] [Google Scholar]
- Lebrand C, Dent EW, Strasser GA, Lanier LM, Krause M, Svitkina TM, Borisy GG, Gertler FB (2004) Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron 42: 37–49 [DOI] [PubMed] [Google Scholar]
- Ligon KL, Echelard Y, Assimacopoulos S, Danielian PS, Kaing S, Grove EA, McMahon AP, Rowitch DH (2003) Loss of Emx2 function leads to ectopic expression of Wnt1 in the developing telencephalon and cortical dysplasia. Development 130: 2275–2287 [DOI] [PubMed] [Google Scholar]
- Magara F, Muller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, Wolfer DP (1999) Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc Natl Acad Sci USA 96: 4656–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL (2003) Cell migration in the forebrain. Annu Rev Neurosci 26: 441–483 [DOI] [PubMed] [Google Scholar]
- Mischel PS, Nguyen LP, Vinters HV (1995) Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54: 137–153 [DOI] [PubMed] [Google Scholar]
- Molnar Z, Adams R, Goffinet AM, Blakemore C (1998) The role of the first postmitotic cortical cells in the development of thalamocortical innervation in the reeler mouse. J Neurosci 18: 5746–5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP (2002) Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 418: 422–425 [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF (2005) Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci 25: 7022–7031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki HS, Wahlsten D (1993) Cortical axon trajectories and growth cone morphologies in fetuses of acallosal mouse strains. J Comp Neurol 336: 595–604 [DOI] [PubMed] [Google Scholar]
- Ross ME, Walsh CA (2001) Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci 24: 1041–1070 [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol 153: 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D'Adamio L (2002) Processing of beta-amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J Biol Chem 277: 44195–44201 [DOI] [PubMed] [Google Scholar]
- Shearer MC, Niclou SP, Brown D, Asher RA, Holtmaat AJ, Levine JM, Verhaagen J, Fawcett JW (2003) The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci 24: 913–925 [DOI] [PubMed] [Google Scholar]
- Soriano E, Del Rio JA (2005) The cells of Cajal–Retzius: still a mystery one century after. Neuron 46: 389–394 [DOI] [PubMed] [Google Scholar]
- Tanahashi H, Tabira T (2002) Characterization of an amyloid precursor protein-binding protein Fe65L2 and its novel isoforms lacking phosphotyrosine-interaction domains. Biochem J 367: 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97: 689–701 [DOI] [PubMed] [Google Scholar]
- Wang B, Hu Q, Hearn MG, Shimizu K, Ware CB, Liggitt DH, Jin LW, Cool BH, Storm DR, Martin GM (2004) Isoform-specific knockout of FE65 leads to impaired learning and memory. J Neurosci Res 75: 12–24 [DOI] [PubMed] [Google Scholar]
- Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H (2005) Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J Neurosci 25: 1219–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TG, Mok SS, Henry A, Cappai R, Lander AD, Nurcombe V, Beyreuther K, Masters CL, Small DH (1996) Secreted glypican binds to the amyloid precursor protein of Alzheimer's disease (APP) and inhibits APP-induced neurite outgrowth. J Biol Chem 271: 31215–31221 [DOI] [PubMed] [Google Scholar]
- Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P (2002) feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J Cell Sci 115: 1411–1422 [DOI] [PubMed] [Google Scholar]
- Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T (2001) The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem 276: 19787–19792 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials and Methods
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3