

Abstract
Actomyosin contractility regulates various cell biological processes including cytokinesis, adhesion and migration. While in lower eukaryotes, α-kinases control actomyosin relaxation, a similar role for mammalian α-kinases has yet to be established. Here, we examined whether TRPM7, a cation channel fused to an α-kinase, can affect actomyosin function. We demonstrate that activation of TRPM7 by bradykinin leads to a Ca2+- and kinase-dependent interaction with the actomyosin cytoskeleton. Moreover, TRPM7 phosphorylates the myosin IIA heavy chain. Accordingly, low overexpression of TRPM7 increases intracellular Ca2+ levels accompanied by cell spreading, adhesion and the formation of focal adhesions. Activation of TRPM7 induces the transformation of these focal adhesions into podosomes by a kinase-dependent mechanism, an effect that can be mimicked by pharmacological inhibition of myosin II. Collectively, our results demonstrate that regulation of cell adhesion by TRPM7 is the combined effect of kinase-dependent and -independent pathways on actomyosin contractility.
Keywords: cell adhesion, cytoskeleton, myosin, phosphorylation, TRPM7
Introduction
Actomyosin contractility in nonmuscle cells plays a fundamental role in regulating basic cellular functions such as cell shape, cytokinesis, adhesion and migration (De la Roche et al, 2002; Geiger and Bershadsky, 2002; Burridge and Wennerberg, 2004). Myosin II is the major motor protein driving contractility. Regulators of myosin II-based contractile responses include the Rho GTPase family and their effectors, myosin light chain kinases and phosphatases as well as myosin heavy chain (MHC) kinases (De la Roche et al, 2002; Burridge and Wennerberg, 2004). The Dictyostelium genome encodes several MHC kinases, which are essential for proper localization and assembly of myosin II during cell division and migration (Kolman et al, 1996; Rico and Egelhoff, 2003; Heid et al, 2004). Myosin II assembles into bipolar thick filaments that generate cortical tension by pulling together oppositely oriented actin filaments. MHC kinases inhibit myosin II function by phosphorylating the MHC tail on threonine residues, leading to filament disassembly and consequently a release in cortical tension (Egelhoff et al, 1993). The cell biological effects of overexpression or knockout of these MHC kinases are consistent with this paradigm. Defects in cytokinesis due to overexpression of MHC kinases are reversed by expressing an MHC mutant that cannot be phosphorylated and therefore forms stable myosin filaments (Rico and Egelhoff, 2003). Thus, MHC kinases play a key role in actomyosin remodeling in Dictyostelium.
Dictyostelium MHC kinases belong to a rare and novel class of kinases known as α-kinases that, with the exception of the ATP-binding site, show only limited sequence similarity with other members of the protein kinase superfamily (Ryazanov, 2002). Based on in vivo substrates identified to date, these kinases preferentially phosphorylate threonine residues presented within the context of an α-helix, hence the name α-kinases (Ryazanov, 2002). Whether mammalian α-kinases also regulate actomyosin contractility has yet to be established.
The mammalian genome encodes several α-kinases whose function, with the exception of elongation factor-2 kinase, is unknown (Drennan and Ryazanov, 2004). Notably, TRPM6 and TRPM7 are two bifunctional proteins encoding a TRP cationic channel fused to a COOH-terminal α-kinase domain. Electrophysiological characterization of TRPM7 has suggested that it forms part of the channel responsible for the magnesium-inhibited current and is implicated in cellular Mg2+ homeostasis (Nadler et al, 2001; Kozak and Cahalan, 2003; Schmitz et al, 2003). However, TRPM7 may also relay signals by phosphorylating downstream effector molecules. In line with a hypothesized role for TRPM7 as an active component in receptor-mediated signal transduction, it was reported that TRPM7 channels are highly permeable to Ca2+, that the TRPM7 COOH-terminus associates with phospholipase C (PLC) isoforms and that PLC activation regulates TRPM7 channel opening (Runnels et al, 2001, 2002; Langeslag et al, manuscript in preparation). We have previously found that bradykinin (BK), a Gq-PLC-coupled receptor agonist, induces Ca2+-dependent phosphorylation of the MHC and disassembly of myosin IIA at the cell periphery, which correlates with cell spreading and adhesion in mammalian cells (van Leeuwen et al, 1999). Thus, we hypothesized that the coupling of a cation channel to an α-kinase in a single polypeptide could explain the close relationship between Ca2+ signaling and Ca2+-dependent actomyosin remodeling. Here, we investigated whether TRPM7 could link receptor-mediated signaling to cytoskeletal contractility and cell adhesion.
Results
TRPM7 mediates bradykinin-induced calcium influx
To examine a role for TRPM7 in the regulation of actomyosin contractility, an HA-tagged TRPM7 cDNA encoding wild-type (TRPM7-WT) or a kinase-dead mutant (TRPM7-D1775A) was introduced into N1E-115 neuroblastoma cells by retroviral transduction (a technique that allows low, near-physiological expression of recombinant proteins; Supplementary Figure S1). Both TRPM7-WT and TRPM7-D1775A were equally expressed at 2–3 times the endogenous TRPM7 levels (Figure 1A and Supplementary Figure S2; Langeslag et al, manuscript in preparation). Both TRPM7-WT and TRPM7-D1775A run as doublets at 216 kDa due to differential glycosylation (Supplementary Figure S3). Moreover, proper localization of TRPM7-WT and TRPM7-D1775A to the plasma membrane was confirmed by fluorescence microscopy (Supplementary Figure S3; also see Figure 4).
Figure 1.
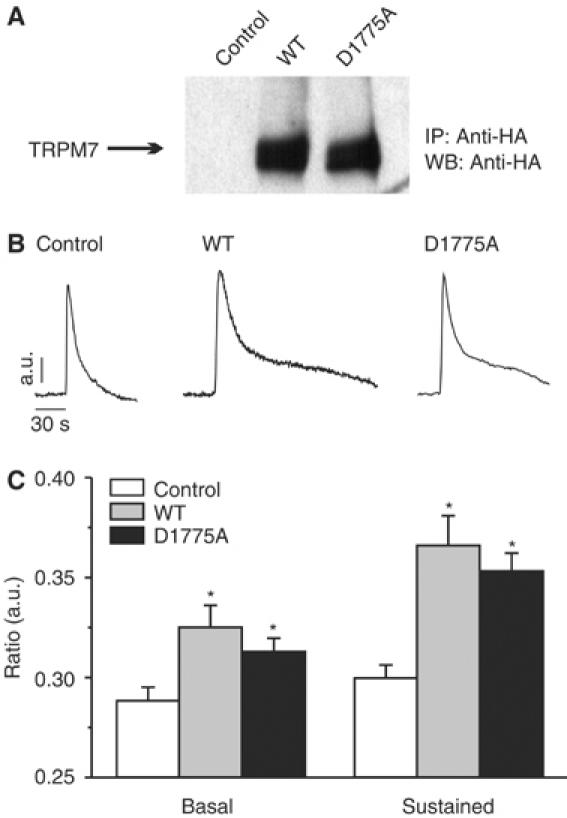
TRPM7 enhances receptor-operated calcium influx. (A) Expression of TRPM7-WT and TRPM7-D1775A in N1E-115 neuroblastoma cells was detected by IP–Western blot using antibodies against the HA tag. Cells containing the empty vector were used as control in all experiments. (B) TRPM7 expression causes sustained Ca2+ influx following BK stimulation independently of kinase activity. Left panel, typical time course of BK-induced Ca2+ changes in control N1E-115 cells; other panels, BK-induced Ca2+ mobilizations in N1E-115/TRPM7 cells. The sustained influx depends on Ca2+ influx, because it is acutely blocked by extracellular La3+ and Gd3+ (data not shown). (C) Quantification of Ca2+ data for control and TRPM7-overexpressing cells (mean±s.e.m. from at least 20 experiments for each condition). Significant differences (*) are P<0.01 from values obtained in control N1E-115 cells.
Figure 4.
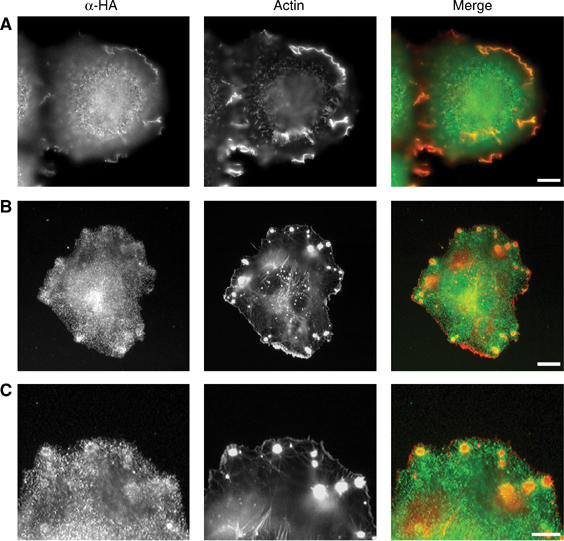
TRPM7 is present at the cell surface and localizes to cell adhesion structures. N1E-115/TRPM7-WT cells were stimulated with BK (10 nM, 30 min). Subsequently, TRPM7 was detected using anti-HA (3F10) antibody followed by alexa 488-conjugated anti-rat IgG. The actin cytoskeleton was visualized using phalloidin–Texas red. (A) TRPM7 is found in membrane ruffles. Scale bar=10 μm. (B) TRPM7 forms a ring surrounding the actin dense core of podosomes. Scale bar=10 μm. (C) High magnification of TRPM7 rings within podosomes. Scale bar=5 μm. A rat IgG control antibody did not show any specific staining (data not shown).
Initially, we investigated by Ca2+ fluorometry whether TRPM7 is activated following stimulation with BK, a peptide agonist that promotes actomyosin relaxation in N1E-115 cells (van Leeuwen et al, 1999). Low overexpression of TRPM7 in N1E-115 cells resulted in a moderate elevation of the resting Ca2+ levels (109±9 versus 85±4 nM). The addition of BK triggered a rapid increase in cytosolic Ca2+ from internal stores in both parental and TRPM7-transduced cells. Strikingly, in N1E-115/TRPM7 cells, this initial Ca2+ transient was followed by a sustained phase of elevated Ca2+ that lasted for several minutes before returning to basal levels and was not observed in the parental cells (Figure 1B). Removal of extracellular Ca2+ by BAPTA immediately terminated the plateau phase (data not shown). Patch-clamp studies and inhibitor profiling identified TRPM7 as the channel responsible for the Ca2+ influx (a detailed analysis of TRPM7 channel function will be published elsewhere; Langeslag et al, manuscript in preparation). Finally, channel activation of kinase-dead TRPM7-D1775A by BK was not altered in comparison to TRPM7-WT (Figure 1B and C), demonstrating that TRPM7 kinase activity is not required for BK-induced Ca2+ influx. From these results, we conclude that TRPM7 functions as a BK-regulated Ca2+ channel in N1E-115 cells.
TRPM7 expression promotes cell spreading and increases cell–matrix adhesion
Mouse N1E-115 neuroblastoma cells are an excellent model system to study the regulation of actomyosin contractility because signals that either activate contraction or promote relaxation are rapidly translated into cell rounding or cell spreading responses, respectively (Jalink et al, 1994; van Leeuwen et al, 1997, 1999). Consistent with a role for TRPM7 in actomyosin regulation, N1E-115/TRPM7 cells showed enhanced spreading when compared to the parental cells (Figure 2A and B). In addition to effects on cell spreading, low overexpression of TRPM7 increased cell–matrix adhesion of N1E-115 cells (Figure 2A and C). Both effects of TRPM7 were independent of kinase activity, suggesting that Ca2+-dependent mechanisms account for the morphological differences between parental and TRPM7-transduced cells.
Figure 2.
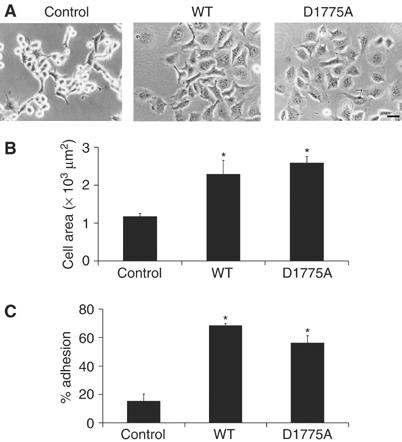
TRPM7 overexpression induces cell spreading and increases cell adhesion. (A) Phase-contrast images of control, TRPM7-WT and TRPM7-D1775A-expressing N1E-115 cells. Scale bar=50 μm. (B) Cell surface area covered by N1E-115 cells expressing WT and kinase-dead TRPM7. Cells were stained for F-actin and cell surface area was calculated by quantifying the amount of pixels that exceeded a threshold (n>5). (C) Quantification of cell adhesion of N1E-115 cells expressing WT and kinase-dead TRPM7 (n=3). Significant differences (*) are P<0.01 from values obtained in control N1E-115 cells.
TRPM7 activation leads to podosome formation
In light of the effects of TRPM7 on cell morphology and adhesion, we further investigated the effect of low TRPM7 overexpression and activation on the subcellular distribution of F-actin, myosin IIA and vinculin by confocal microscopy (Figure 3A). Control N1E-115 cells displayed a predominantly cortical distribution of F-actin and myosin IIA, with little or no focal adhesions nor stress fibers. Following BK-induced cell spreading, a part of this peripheral actomyosin was lost while small focal adhesions could now be observed at the cell periphery by vinculin staining. In N1E-115/TRPM7 cells, a profound redistribution of actomyosin was observed. In contrast to the parental cells, focal adhesions were already present in unstimulated cells, whereas myosin IIA was dispersedly staining the cytoplasm. BK stimulation caused further cell spreading and increased the number and size of adhesion complexes. These responses were noticeable within 2 min and peaked at approximately 10 min after BK stimulation (Figure 3B and Supplementary movie 1). The architecture and size (∼1 μm) of these adhesion structures were remarkably different from the focal adhesions observed in unstimulated cells and resembled those of podosomes (Linder and Aepfelbacher, 2003). Similar to podosomes, the TRPM7-induced adhesions, found at the interface of the cell and matrix, had a diameter of approximately 1 μm. Moreover, they consisted of an actin core surrounded by vinculin and conspicuous myosin lattices that were no longer associated with actin stress fibers (Figure 3C). We conclude therefore that TRPM7 activation promotes the formation of podosomes.
Figure 3.
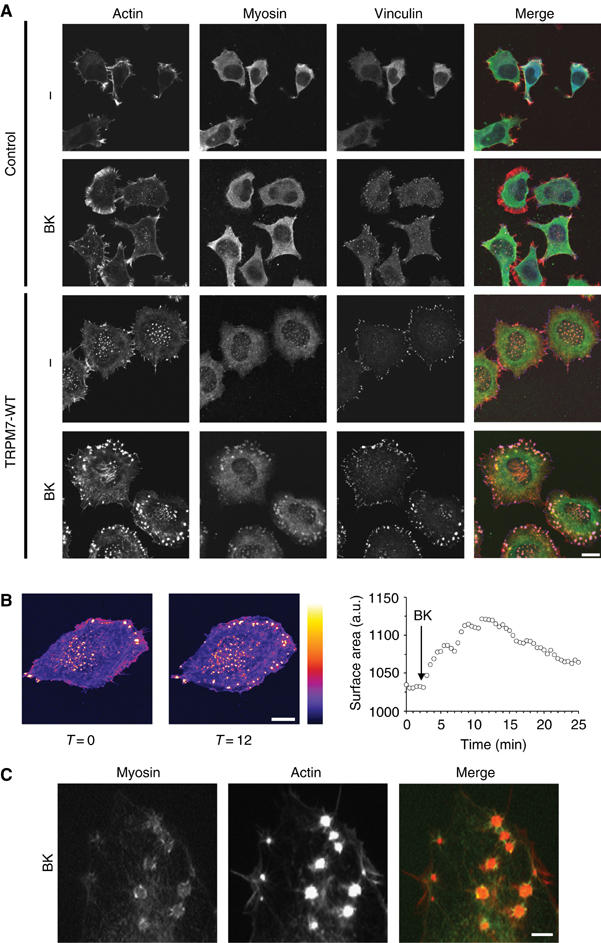
Activation of TRPM7 induces actomyosin reorganization in conjunction with podosome formation. (A) TRPM7 activation induces actomyosin reorganization and formation of podosomes in N1E-115/TRPM7 cells. Cells were either serum starved (0.1% FCS) or stimulated with BK (10 nM, 30 min) and stained for actin (red), myosin IIA heavy chain (green) and vinculin (blue). Cells were analyzed by confocal microscopy. Scale bar=20 μm. (B) Increase in cell surface area and production of podosomes occurs within minutes of BK stimulation in N1E-115/TRPM7 cells. N1E-115/TRPM7 cells expressing GFP-β-actin were followed by confocal microscopy before and after BK stimulation. Cell surface area was measured as described in Materials and methods. Gray values are displayed using the Fire LookUpTable (LUT) of the public domain software Image J (National Institutes of Health, Bethesda, MD, USA). Left panel, a resting cell; middle panel, the same cell 12 min post-BK stimulation; right panel, kinetics of BK-induced cell spreading. A movie is provided as Supplementary data. Scale bar=15 μm. (C) High magnification of myosin lattices found in N1E-115/TRPM7 cells following BK stimulation. N1E-115/TRPM7 cells were transfected with GFP-myosin, stimulated with BK and stained for actin. Scale bar=2 μm.
TRPM7 is present at the cell surface in proximity to cell adhesion structures
To examine whether TRPM7 may affect cell adhesion locally, we investigated the cellular distribution of TRPM7 by immunofluorescence. TRPM7 was clearly present within membrane ruffles, indicating that the protein is localized at the cell surface (Figure 4A). In addition, TRPM7 is enriched in cell adhesions where it is found in the podosome ring structure, together with myosin IIA (Figure 4B and C). Our results suggest that TRPM7 regulates cell adhesion by directly affecting components within these structures.
Induction of podosomes by TRPM7 is mediated by a kinase-dependent mechanism
Although the global effects of TRPM7 on cell spreading and adhesion appear to be independent of kinase activity, we tested whether the TRPM7 kinase domain affects the subcellular organization of the actomyosin cytoskeleton. By confocal microscopy, we compared N1E-115 cells expressing TRPM7-WT with cells expressing the kinase-dead TRPM7-D1775A mutant. No clear differences were noticed between unstimulated cells (data not shown). However, to our surprise, cells expressing kinase-dead TRPM7 failed to induce podosomes in response to BK stimulation (Figure 5). We conclude that TRPM7 activation results in a kinase-dependent remodeling of the actomyosin cytoskeleton, leading to the assembly of podosomes.
Figure 5.
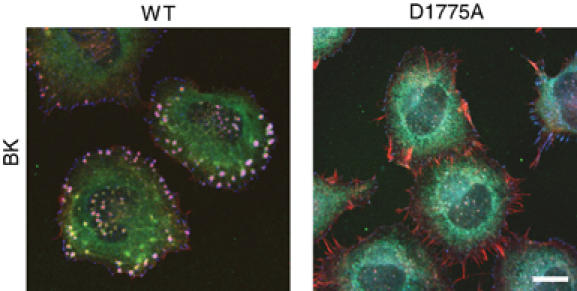
Regulation of podosome formation by TRPM7 is kinase dependent. N1E-115/TRPM7-WT and N1E-115/TRPM7-D1775A cells were stimulated with BK (10 nM, 30 min) and stained for actin (red), myosin IIA heavy chain (green) and vinculin (blue). Cells were analyzed by confocal microscopy. Scale bar=20 μm.
Inhibition of myosin II function leads to podosome formation both in cells expressing wild-type and kinase-dead TRPM7
Actomyosin contractility plays a central role in regulating the assembly and disassembly of adhesive contacts (Geiger and Bershadsky, 2002). Notably, the formation of podosomes requires a local inhibition of actomyosin contractility, suggesting that TRPM7 may regulate podosome assembly by promoting relaxation of the actomyosin cytoskeleton (Burgstaller and Gimona, 2004). To further test this model, we investigated the effect of directly inhibiting myosin II function on cell adhesion in N1E-115 cells expressing TRPM7-WT and TRPM7-D1775A. Treatment of cells with blebbistatin (a selective inhibitor of myosin II ATPase activity) led to the transformation of focal adhesions into podosomes independently of TRPM7 kinase activity (Figure 6A). These effects of blebbistatin do not require exogenous TRPM7, as parental N1E-115 cells also produce podosomes in response to myosin II inhibition (Supplementary Figure S4). Notably, the adhesive structures formed upon blebbistatin treatment are remarkably similar to those formed after BK stimulation of N1E-115/TRPM7-WT cells (Figure 6B). These findings indicate that the transformation of normal focal adhesions into podosomes by BK-mediated activation of TRPM7 is due to a kinase-dependent inhibition of myosin II function.
Figure 6.
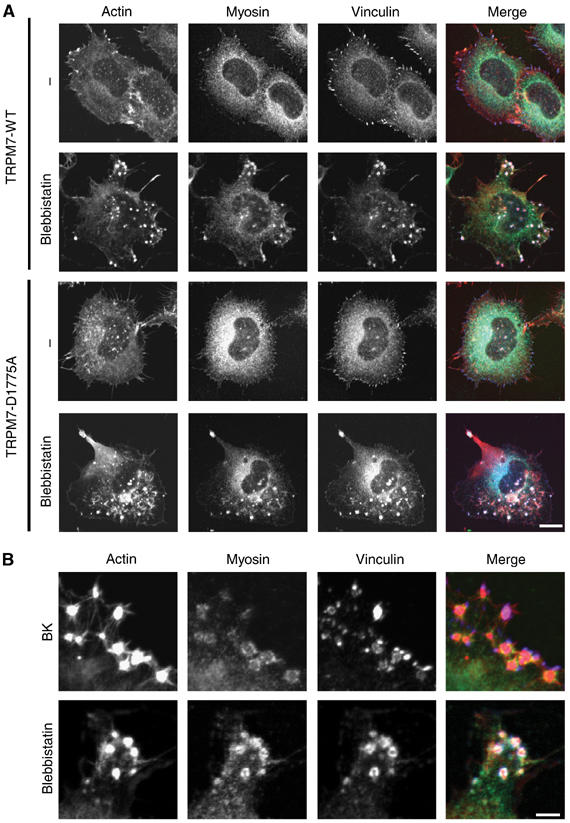
Inhibition of myosin II function in N1E-115 cells expressing TRPM7-WT and TRPM7-D1775A leads to podosome formation. (A) N1E-115/TRPM7-WT and N1E-115/TRPM7-D1775A cells were incubated in the presence of vehicle control or 50 μM blebbistatin for 30 min. Cells were stained for actin (red), myosin (green) and vinculin (blue) and visualized by confocal microscopy. Scale bar=15 μm. (B) High magnification view shows that the adhesion complexes induced by BK and blebbistatin in N1E-115/TRPM7-WT cells are architecturally similar, as they consist of an actin dense core surrounded by a ring of vinculin and myosin IIA. Scale bar=5 μm.
TRPM7 interacts with the actomyosin cytoskeleton
As TRPM7 is a member of the α-kinase family, we hypothesized that it may affect actomyosin remodeling and podosome assembly by directly coupling to and potentially phosphorylating components present within the actomyosin cytoskeleton. Therefore, we precipitated TRPM7 complexes with anti-HA antibodies and detected the presence of associated proteins by Western blotting. Both β-actin and the myosin IIA heavy chain were present in a complex with TRPM7 (Figure 7A). Importantly, endogenous TRPM7 also associates with the actomyosin cytoskeleton (Figure 7B). This interaction indeed suggests that TRPM7 can affect actomyosin function through a direct association with cytoskeletal proteins.
Figure 7.
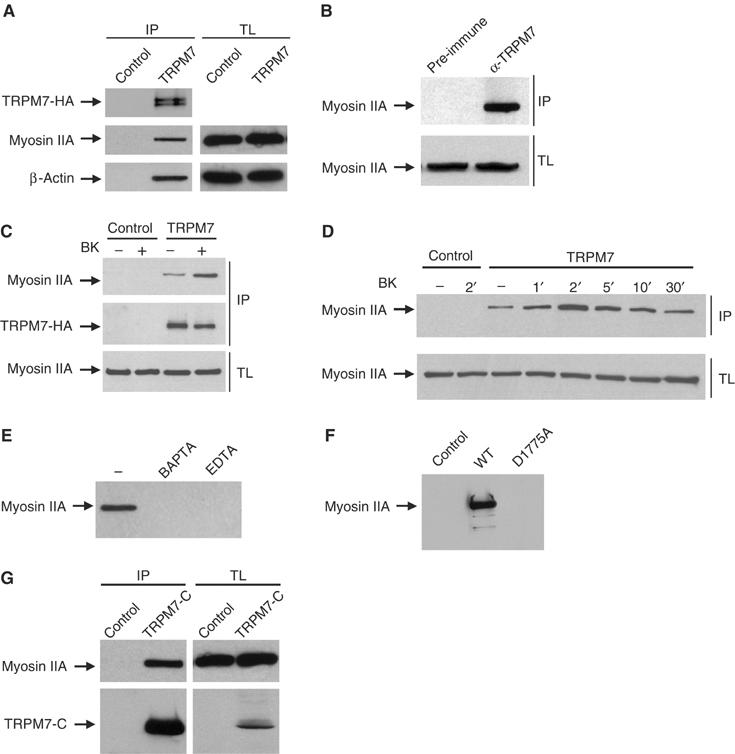
TRPM7 activation promotes its association via the COOH-terminus with the actomyosin cytoskeleton in a calcium- and kinase-dependent manner. In all experiments (except panel B), N1E-115 control or TRPM7-transduced cells were lysed and incubated with anti-HA-coupled protein G beads for 3 h at 4°C. Proteins in the complex were detected by Western blotting. (A) Co-immunoprecipitation of TRPM7 with β-actin and myosin IIA. Top, detection of TRPM7 with anti-HA antibodies; middle and bottom, detection of myosin IIA and β-actin, respectively, in the immune complexes (IP, left) and total lysates to control for protein levels (TL, right). (B) Endogenous TRPM7 associates with the actomyosin cytoskeleton. PC12 lysates were incubated with preimmune serum or anti-TRPM7 serum and protein complexes were isolated using protein G-Sepharose. The presence of myosin IIA was detected by Western blotting. Top, detection of associated myosin IIA heavy chain in immunoprecipitates; bottom, detection of associated myosin IIA heavy chain in total lysates. (C) Co-immunoprecipitation of TRPM7 with myosin IIA heavy chain pre- and post-BK stimulation (10 nM, 1 min). Top, detection of co-immunoprecipitated myosin IIA; middle, detection of TRPM7 with anti-HA antibodies; bottom, detection of myosin IIA heavy chain in total lysates to control for protein levels. (D) Kinetics of BK-induced TRPM7 association with myosin IIA heavy chain. Cells were stimulated with BK (10 nM) for the indicated times and TRPM7 was immunoprecipitated using anti-HA antibodies. Top, Western blot showing co-immunoprecipitated myosin IIA heavy chain; bottom, detection of myosin IIA heavy chain in total lysates. (E) Interaction between TRPM7 and myosin IIA heavy chain is Ca2+ dependent. N1E-115/TRPM7 cells were preincubated with 10 mM BAPTA or EDTA for 1 min prior to cell lysis and TRPM7 was immunoprecipitated using anti-HA antibodies. (F) TRPM7-D1775A does not interact with myosin IIA heavy chain. TRPM7-WT and -D1775A were immunoprecipitated with anti-HA antibodies and associating myosin IIA heavy chain was detected by Western blotting. (G) The COOH-terminus of TRPM7 interacts with myosin IIA. The soluble COOH-terminus of TRPM7 (aa 1158–1864) was immunoprecipitated from N1E-115/TRPM7-C cells with anti-HA antibodies and associated myosin IIA was detected by Western blotting. Top, detection of associated myosin IIA heavy chain; bottom, detection of TRPM7-C using anti-HA antibodies in immunoprecipitates (IP, left) and total lysates (TL, right).
Bradykinin causes calcium- and kinase-dependent association of the TRPM7 COOH-terminus with myosin IIA
As TRPM7 activation by BK results in Ca2+ influx and a kinase-dependent remodeling of the actomyosin cytoskeleton, we investigated whether the interaction between TRPM7 and the actomyosin cytoskeleton is subject to regulation. BK stimulation of N1E-115/TRPM7 cells led to a transient increase in the amount of TRPM7-associated myosin IIA (Figure 7C and D). Its kinetics closely correlate with those of calcium influx in response to TRPM7 activation (refer to Figure 1), with maximal association observed at about 2 min after agonist addition. Moreover, chelation of Ca2+, using BAPTA or EDTA, abrogated the association between TRPM7 and myosin IIA (Figure 7E). These results indicate that the interaction between TRPM7 and myosin IIA is strictly Ca2+ dependent, and suggest that TRPM7-mediated Ca2+ influx enhances the TRPM7/myosin IIA interaction. However, in addition to Ca2+, the association of TRPM7 with the cytoskeleton requires an active kinase domain, as the kinase-dead TRPM7-D1775A mutant did not interact with myosin IIA (Figure 7F). A soluble COOH-terminus variant containing the kinase domain of TRPM7 also interacted with myosin IIA (Figure 7G). We conclude therefore that the interaction between TRPM7 and myosin IIA is mediated by the COOH-terminus and requires an active kinase domain.
TRPM7 phosphorylates myosin IIA heavy chain
Hitherto, we have shown that TRPM7 associates with myosin IIA in a regulated manner and that both activation of TRPM7-WT (but not TRPM7-D1775A) and inhibition of myosin II function produce podosomes, suggesting that TRPM7 activation promotes actomyosin relaxation. How does TRPM7 mediate the inhibition of myosin II function? By analogy to regulation of Dictyostelium myosin II function, we have previously proposed that Ca2+-dependent MHC phosphorylation contributes to cytoskeletal relaxation (van Leeuwen et al, 1999). The finding that TRPM7 co-precipitates with myosin IIA, particularly after BK stimulation, enabled us to test whether TRPM7 can phosphorylate myosin IIA heavy chain. As TRPM7 and myosin IIA heavy chain co-migrate on SDS–PAGE gels, TRPM7 was immunoprecipitated both under conditions that favor association and dissociation (low versus high stringency) of the cytoskeletal complex, allowing us to distinguish between TRPM7 autophosphorylation and myosin phosphorylation. By in vitro kinase assays, TRPM7 underwent autophosphorylation, in agreement with earlier findings (Runnels et al, 2001; Schmitz et al, 2003; Ryazanova et al, 2004) (Figure 8A, bottom panel). Strikingly, clear phosphorylation of associated myosin IIA heavy chain (which runs slightly faster than TRPM7) was also observed in these kinase reactions (Figure 8A, bottom panel). In contrast, actin was not phosphorylated (data not shown).
Figure 8.
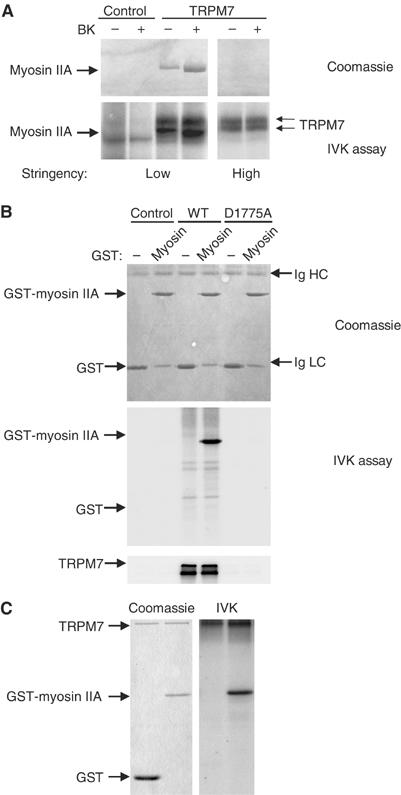
TRPM7 phosphorylates myosin IIA heavy chain. (A) In vitro kinase assay detecting the phosphorylation of associated myosin IIA heavy chain by TRPM7. TRPM7/myosin IIA heavy chain complexes were isolated before and after stimulation of N1E-115 control and N1E-115/TRPM7 cells with BK (10 nM, 2 min) under low (1% Triton X-100) and high (1% Triton X-100/0.5% deoxycholate/0.1% SDS) stringency conditions. Substrates of TRPM7 were detected by labeling proteins with [γ-32P]ATP, separating products of the in vitro kinase assay by SDS–PAGE (6% gel) followed by autoradiography. Top, Coomassie staining of precipitated proteins; bottom, autoradiogram of phosphorylated proteins. Note that myosin association is lost under high stringency conditions (1% Triton X-100/0.5% deoxycholate/0.1% SDS), whereas TRPM7 autophosphorylation is unaffected. (B) TRPM7 phosphorylates recombinant myosin IIA. TRPM7-WT and -D1775A were immunoaffinity purified from HEK293 cells using anti-HA antibodies under high stringency and mixed with 2 μg of GST or GST-myosin IIA in kinase buffer. To detect phosphorylated myosin IIA, the proteins were separated by SDS–PAGE (12% gel) followed by autoradiography. Top, Coomassie staining of GST-fusion proteins. Note that GST co-migrates with the antibody light chain (Ig LC). Middle, autoradiogram of phosphorylated GST-fusion proteins. Bottom, autoradiogram showing autophosphorylation of WT but not kinase-dead TRPM7. (C) Recombinant TRPM7 kinase phosphorylates myosin IIA. The TRPM7 kinase domain (aa 1403–1864) was produced in E. coli as a fusion with maltose-binding protein and purified on an amylose column. The purified kinase was incubated with 2 μg of GST or GST-myosin IIA in kinase buffer. The proteins were resolved on a 12% SDS–PAGE gel and detected by Coomassie staining. Phosphorylated proteins were visualized by autoradiography. Left, Coomassie staining of gel; right, autoradiogram.
To demonstrate that TRPM7 directly phosphorylates myosin IIA heavy chain, a myosin tail fragment was expressed as a GST-fusion protein in Escherichia coli to serve as substrate, whereas TRPM7 was immunoaffinity purified from mammalian cells or the COOH-terminus of TRPM7 was purified from E. coli. In in vitro kinase assays, immunoaffinity-purified WT but not kinase-dead TRPM7 efficiently phosphorylated recombinant myosin IIA (Figure 8B). Notably, GST was not phosphorylated by TRPM7 (Figure 8B). Similar results were obtained when using the soluble COOH-terminus of TRPM7 (data not shown). Importantly, contaminating actin in the purified TRPM7 fractions is below detection levels, and disruption of the actin cytoskeleton prior to TRPM7 purification had no effect on the level of myosin II phosphorylation, arguing against the presence of contaminating kinases (Supplementary Figure S5). Finally, recombinant TRPM7 purified from E. coli lysates also efficiently phosphorylated myosin IIA, in the absence of other sources of eukaryotic proteins (Figure 8C). Thus, TRPM7 itself and not an associated kinase is responsible for myosin IIA heavy chain phosphorylation. Collectively, our results demonstrate that TRPM7 associates with the actomyosin cytoskeleton in a Ca2+- and kinase-dependent manner to regulate myosin II activity and consequently actomyosin contractility. Moreover, our in vitro kinase data suggest that phosphorylation of the myosin IIA heavy chain by TRPM7 serves as a regulatory mechanism.
Discussion
In this study, we have tested the hypothesis that TRPM7, by analogy to its α-kinase family members from Dictyostelium, affects actomyosin contractility. We provide evidence that TRPM7 promotes relaxation of the actomyosin cytoskeleton via a kinase-dependent inhibition of myosin II, potentially involving myosin IIA heavy chain phosphorylation. The evidence includes (i) electrophysiological measurement of TRPM7 channel opening after BK stimulation, (ii) biochemical analyses of the TRPM7 complex showing that TRPM7 interacts in a Ca2+- and kinase-dependent manner with an actomyosin protein complex, (iii) in vitro kinase reactions demonstrating that TRPM7 phosphorylates myosin IIA heavy chain and (iv) cell biological studies revealing that TRPM7 promotes a loss of cortical tension leading to podosome formation by a kinase-dependent mechanism. Collectively, our data indicate that TRPM7 plays a role in linking receptor-mediated signals to actomyosin remodeling and cell adhesion. Furthermore, our observations reveal for the first time that TRP channels affect the cytoskeleton by directly associating with cytoskeletal proteins in a highly regulated manner.
What is the relationship between channel opening and the kinase domain? Several studies have suggested that kinase activity regulates TRPM7 channel opening (Runnels et al, 2001; Schmitz et al, 2003). However, in our model system, TRPM7 channel opening was independent of kinase activity, as the responses to BK stimulation were identical between cells expressing WT and kinase-dead TRPM7. Although TRPM7 kinase activity does not directly affect channel opening, it cannot be excluded that actomyosin remodeling serves to regulate TRPM7 function in an indirect manner as reported for other TRP channels (Lockwich et al, 2001; Itagaki et al, 2004). Based on our observations, rather than the kinase domain regulating TRPM7 channel function, the reverse relationship exists. In this model (Figure 9), phosphorylation of downstream targets (myosin II, annexin I; Dorovkov and Ryazanov, 2004) by the TRPM7 kinase domain is tightly regulated by ion influx (Ca2+, Mg2+) through the TRPM7 channel. In support of this model, we observed that Ca2+ is strictly required for the association of TRPM7 with the cytoskeleton, that increased Ca2+ influx through TRPM7 upon agonist-induced activation enhances its association with myosin IIA and that MHC phosphorylation is strictly Ca2+ dependent (van Leeuwen et al, 1999). Recently, Dorovkov and Ryazanov (2004) showed that phosphorylation of annexin I by TRPM7 is potentiated in vitro by the addition of Ca2+ and conversely is inhibited by chelation of Ca2+. Therefore, we propose that ion influx through the channel pore regulates the TRPM7 kinase domain by controlling the recruitment of its substrates.
Figure 9.
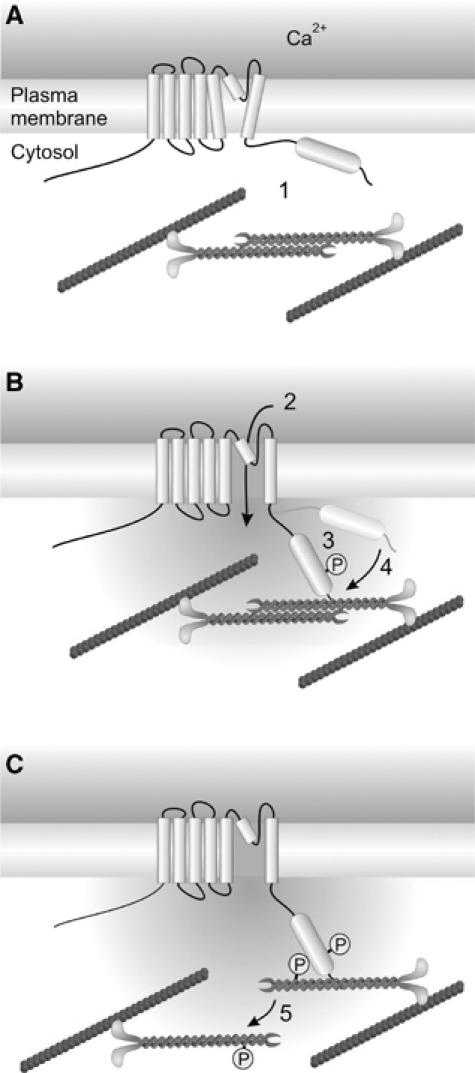
Regulation of actomyosin contractility by TRPM7. (A) In the resting state, TRPM7 is not associated with the actomyosin cytoskeleton (1). (B) Stimulation of cells with PLC-activating agonists induces TRPM7-mediated Ca2+ influx (2) as well as TRPM7 kinase activity. Autophosphorylation (3) promotes a conformational change within TRPM7, allowing for Ca2+-dependent association with myosin IIA (4). (C) Subsequently, TRPM7 phosphorylates the myosin IIA heavy chain, presumably leading to dissociation of myosin filaments and cytoskeletal remodeling (5).
Kinases are maintained in an active state by intramolecular and intermolecular protein–protein or protein–lipid interactions (Van Etten, 2003; Roskoski, 2004). Activation of kinases generally involves autophosphorylation or transphosphorylation at regulatory sites inducing conformational remodeling to expose the catalytic domain to its substrate. We postulate that TRPM7 is similarly regulated, as its kinase activity appears to be essential for its association with the actomyosin cytoskeleton. Autophosphorylation of TRPM7, which occurs on serine and threonine residues (Schmitz et al, 2003; Ryazanova et al, 2004), may release a cryptic binding site for myosin IIA heavy chain (Figure 9). Thus, autophosphorylation of TRPM7 may serve as an important regulatory mechanism to modulate the interactions between TRPM7 and its substrates.
A functional consequence of TRPM7 activation is an increase in cell adhesion and spreading, which involves both kinase-dependent and -independent pathways. Expression of a TRPM7 kinase-dead mutant still promotes cell adhesion and spreading, but does not lead to the formation of podosomes upon BK stimulation. The fact that the global effects of TRPM7 on cell morphology are independent of kinase activity or its association with the cytoskeleton may reflect the increase in cytosolic Ca2+ concentrations observed in TRPM7-transduced cells. Indeed, reducing the internal Ca2+ concentration with BAPTA-AM impaired cell spreading (data not shown). Ca2+ may act directly at the level of integrin activation or affect the actomyosin cytoskeleton. The activation of integrins can lead to the remodeling of the actomyosin cytoskeleton to promote cell spreading via outside-in signaling pathways (DeMali et al, 2003). Alternatively, Ca2+ is an important second messenger in actin remodeling including polymerization, severing of filaments and F-actin–membrane interactions (Forscher, 1989; Sun et al, 1999). Moreover, as TRPM7 can directly interact with PLC isoforms, it may influence local concentrations of PIP2, and thus affect actin polymerization (van Rheenen and Jalink, 2002).
The kinase-dependent effects of TRPM7 on cell adhesion are consistent with a role in regulating myosin IIA-dependent contractile responses. Notably, both BK stimulation and myosin II inhibition of N1E-115/TRPM7 cells induce the formation of large podosomes. Spatial and temporal regulation of actomyosin contractility is central to modulating the assembly and disassembly of focal adhesions and podosomes (Geiger and Bershadsky, 2002; DeMali et al, 2003; Linder and Aepfelbacher, 2003; Burgstaller and Gimona, 2004; Burridge and Wennerberg, 2004). Although local regulation of contractility appears to be controlled by Rho GTPases (Burgstaller and Gimona, 2004), α-kinases may also fulfill a role in spatial and temporal regulation of myosin II activity. Indeed, TRPM7, which is expressed at the cell surface, was found enriched in cell adhesion structures. By similarity, Dictyostelium myosin II function is precisely regulated by a family of MHC kinases, with each member possessing a unique spatial distribution (Liang et al, 2002). Therefore, we postulate that an increase in TRPM7-mediated MHC phosphorylation contributes to local relaxation of the cortical cytoskeleton causing the transformation of focal adhesions into podosomes. Ablation of TRPM7 kinase activity maintains myosin-based cytoskeletal contractility preventing the reorganization of focal adhesions. Hence, TRPM7 regulates cell adhesion by modulating actomyosin contractility in a kinase-dependent manner, and it is tempting to speculate that this phenomenon operates through MHC phosphorylation.
How to reconcile the small increase in TRPM7 expression (two-fold) with dramatic changes in cell morphology and response to BK? Although the kinase activity of TRPM7 is a linear relationship with concentration in vitro, it is most likely not linear in vivo as the protein will be subject to regulation. In support of this hypothesis, we have found that TRPM7 channels have increased activity as measured by patch-clamp and Ca2+ fluorometric experiments in N1E-115/TRPM7 cells. It should be noted that the basal Ca2+ levels in the cells are significantly higher upon TRPM7 overexpression, which could influence phosphorylation of its substrates as reported by Dorovkov and Ryazanov (2004). Moreover, TRPM7-mediated Ca2+ influx affects the recruitment of substrates for the kinase, by associating with the actomyosin cytoskeleton. Therefore, it can be expected that the activity of the TRPM7 complex in the N1E-115/TRPM7 cells in vivo is significantly more than the two-fold increase in kinase activity that is observed in vitro. Finally, if TRPM7 is targeted to a particular part of the cytoskeleton including cell adhesion structures, it is likely that the local activity of TRPM7 kinase increases significantly more than two-fold in those areas of the cell.
BK stimulation of N1E-115 cells leads to cell spreading and focal adhesion formation, whereas it promotes the transformation of focal adhesions into podosomes in N1E-115/TRPM7 cells. Although N1E-115 cells express endogenous TRPM7, these cells do not produce podosomes in response to BK. We believe that these results reflect the contractile state of the cell and that it is necessary to overexpress TRPM7 to achieve complete relaxation of the actomyosin cytoskeleton required for BK-mediated podosome formation (see Supplementary Figure S6). In parental N1E-115 cells, myosin II is very active, leading to a contractile phenotype. BK stimulation of these cells relaxes the cytoskeleton but since focal adhesions are formed, myosin II must be at least partly active as tension is required for focal adhesion biogenesis (Geiger and Bershadsky, 2002). Overexpression of TRPM7 mimics these effects and BK stimulation of these cells leads to further inhibition of myosin II and relaxation of the actomyosin cytoskeleton, which allows for the formation of podosomes (adhesion structures, which require an inhibition of myosin II for their formation; Burgstaller and Gimona, 2004). In support of this model, inhibition of myosin II by blebbistatin in parental N1E-115 cells similarly induces podosomes (Supplementary Figure S4). Thus, since TRPM7 has a basal activity, its expression levels dictate myosin II activity, and thereby cellular phenotype.
Although mammalian nonmuscle actomyosin contractility appears to be primarily regulated by phosphorylation of the myosin regulatory light chain, a role for MHC phosphorylation is emerging. Several studies have demonstrated that the MHC tail is phosphorylated on threonine residues upon stimulation of mammalian cells (Wilson et al, 1998; van Leeuwen et al, 1999). Based on homology to Dictyostelium MHC kinases, we hypothesized that TRPM7 may regulate myosin II function by phosphorylating the MHC α-helical tail. Indeed, in vitro kinase assays demonstrated that WT but not kinase-dead TRPM7 directly phosphorylates the MHC tail, indicating broad evolutionary conservation with respect to both substrate and function, an unexpected result since other α-kinases, α-kinase 1 and EF2K, regulate protein transport and protein synthesis, respectively (Ryazanov, 2002; Heine et al, 2005).
Myosin II forms bipolar thick filaments through electrostatic interactions of the MHC tail (Hostetter et al, 2004). In Dictyostelium, phosphorylation of the α-helical MHC tail leads to filament disassembly thereby releasing cortical tension (Egelhoff et al, 1993). Whether phosphorylation of mammalian myosin II heavy chain by TRPM7 will have a similar effect on filament assembly remains to be determined (Figure 9C). Importantly, the region of myosin II phosphorylated by TRPM7 corresponds to the domain responsible for regulating myosin filament assembly (Nakasawa et al, 2005). Moreover, phosphorylation by PKC and casein kinase as well as point mutations affecting electrostatic charges (E1841K) within this domain affect myosin II function (Dulyaninova et al, 2005; Franke et al, 2005). Therefore, we propose that phosphorylation of this region of the myosin IIA tail affects filament formation rather than ATPase activity of myosin II. Future investigations aimed at mapping the TRPM7 target residues within the myosin IIA heavy chain and defining the physiological relevance of MHC phosphorylation will provide further insight into the mechanisms underlying actomyosin contractility in mammalian cells.
In conclusion, we identified TRPM7 as a novel regulator of actomyosin contractility and cell adhesion in response to agonist stimulation. Moreover, we have demonstrated that TRP channels can interact with the actomyosin cytoskeleton to affect cell adhesion. Future experiments will be aimed at defining the role of TRPM7-containing protein assemblies in regulating actomyosin function, and establishing how these cytoskeletal changes affect cell adhesion and/or TRPM7 function.
Materials and methods
Constructs
Mouse TRPM7 in pTracer-CMV2 and GFP-myosin IIA in pEGFP-C3 were kind gifts from David Clapham (Harvard, Boston, MA, USA) and Robert Adelstein (NIH, Bethesda, MD, USA), respectively. An HA tag has been inserted at the COOH-terminus of TRPM7 (Runnels et al, 2001). To generate retroviral expression vectors, the TRPM7 cDNA was inserted as an XhoI–NotI fragment into the LZRS-ires-neomycin vector. The kinase-dead mutant TRPM7-D1775A was produced by site-directed mutagenesis using the Quickchange kit (Stratagene). The soluble TRPM7 COOH-terminus construct encodes for aa 1158–1864 and contains an HA tag at the NH2-terminus. The cDNA was amplified by PCR and inserted as an XhoI–NotI fragment into the LZRS-ires-neomycin vector. GFP-β-actin cDNA was inserted as an EcoRI–NotI fragment into the LZRS-ires-neomycin vector. Mouse myosin IIA heavy chain tail (aa 1795–1960) was amplified by RT–PCR from N1E-115 cells and cloned in-frame in pGEX-1N using BamHI–EcoRI sites. The TRPM7 kinase domain (aa 1403–1864) in pMAL-p2x was described previously (Ryazanova et al, 2004). All constructs were verified by DNA sequencing.
Cell culture
Mouse N1E-115, HEK293 and Phoenix packaging cells were cultured in DMEM medium with 10% FCS, whereas PC12 cells were grown in RPMI medium with 10% horse serum and 5% FCS. Stable cell lines expressing the various TRPM7 constructs and GFP-β-actin were generated by retroviral transduction (van Leeuwen et al, 1999). N1E-115 cells transduced with the empty vector served as a control for all experiments. Cells were selected by the addition of 0.8 mg/ml G418 to the media and the selection was complete within 7 days. For transient expression, cells were transfected using Fugene 6 (Roche) according to the manufacturer's recommendations.
Intracellular calcium determinations
Calcium measurements were performed by confocal ratiometric imaging of cells loaded with Oregon Green 488 BAPTA-1 AM and Fura Red AM (Molecular Probes) essentially as described (Schild et al, 1994). Experiments were performed at 37°C in a HEPES/bicarbonate-buffered saline under 5% CO2, pH 7.3 (140 mM NaCl, 23 mM NaHCO3, 5 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 10 mM HEPES, 10 mM glucose), and calibration was with ionomycin (Calbiochem). Representative traces from experiments performed at least in 10-fold are shown; data are mean±s.e.m.
Adhesion assay
Cells (5 × 105) were seeded in a T25 flask and allowed to adhere for 16 h. To quantitate cell adhesion, non-adherent cells were washed away with PBS. The adherent cells were fixed in 3.7% formaldehyde in PBS for 10 min, washed with PBS and subsequently stained with 2% crystal violet for 50 min. Excess stain was removed by extensive washing in PBS. Cells were lysed in PBS containing 1% NP-40 and the absorbance at 570 nm of the lysate was measured. Maximal adhesion as measured on poly-L-lysine-coated dishes was set to 100%. Values reported are representative of two independent experiments performed in triplicate.
Microscopy
Cells seeded on glass coverslips were serum starved (0.1% FCS) overnight prior to stimulation with BK or blebbistatin (Calbiochem). For fixed specimens, cells were stained as previously described (van Leeuwen et al, 1997). Antibodies used for immunofluorescence were rat anti-HA (3F10; 1:100; Roche), mouse anti-vinculin (1:400; Sigma), rabbit anti-myosin IIA (1:100; BTI), alexa 488-conjugated anti-rabbit and anti-rat IgG and alexa 647-conjugated anti-mouse IgG (1:200; Molecular Probes). F-actin was detected using either Texas red or alexa 546-labelled phalloidin (1:50; Molecular Probes). Cells were viewed using either a Zeiss-LSM 510-meta microscope equipped with a Plan-Apochromat × 63, 1.4 NA oil immersion lens or a Leica-DMRA microscope with a × 63, 1.32 NA oil immersion lens. For video microscopy, cells were transferred to a bicarbonate-buffered medium as described above and maintained at 37°C on a temperature- and CO2-controlled stage. Cells were imaged with a DM-IRE2 inverted microscope fitted with a TCS-SP2 scanhead and a × 63, 1.32 NA oil immersion lens (Leica). Images were collected at 30 s time intervals and surface area was calculated by quantifying the amount of pixels under a digital mask constructed by using the binary-fill operation after a threshold step.
Generation of anti-TRPM7 antibodies
To generate anti-TRPM7 antibodies, a GST-fusion protein encoding aa 1447–1555 of TRPM7 was expressed in E. coli and purified on a glutathione-Sepharose column. Rabbits were injected with the antigen mixed with Freund's adjuvant and serum was collected 10 days after every vaccination. Serum collected prior to immunization (preimmune) was used as a negative control in all experiments.
Immunoprecipitations
Cells were serum starved (0.1% FCS) overnight prior to stimulation, as indicated in the figure legends. After stimulation, cells were lysed on ice for 20 min in lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 0.5 mM DTT, 1.5 mM MgCl2, 0.2 mM EDTA, 1% Triton X-100 supplemented with protease inhibitors) and the extract was cleared by centrifugation. For immunoprecipitation of exogenously expressed TRPM7, protein G-Sepharose beads, which were blocked with 0.5% BSA and precoupled with anti-HA antibody (clone 12CA5), were added to the lysate of N1E-115/empty vector (control) or N1E-115/TRPM7 cells. The samples were incubated at 4°C for 3 h. Endogenous TRPM7 was immunoprecipitated by incubating cellular lysates with anti-TRPM7 antibodies at 4°C for 3 h followed by the addition of protein G-Sepharose beads at 4°C for 15 min. Subsequently, the beads were washed three times with lysis buffer, protein complexes were solubilized in Laemmli sample buffer and separated by SDS–PAGE. Proteins were detected by immunoblotting using the following antibodies: anti-HA (clone 3F10; 1:1000), anti-myosin IIA (1:500) and anti-β-actin (1:20 000; Sigma).
In vitro kinase assays
Recombinant TRPM7 kinase domain (aa 1402–1864) and myosin tail fragment (aa 1795–1960) were expressed, respectively, as a maltose-binding protein- and GST-fusion protein in E. coli and purified by standard methods. Immunocomplexes containing TRPM7 with or without associated myosin or TRPM7 mixed with 2 μg of GST-myosin were solubilized in kinase buffer (50 mM HEPES pH 7.0, 4 mM MnCl2, 5 mM DTT) without ATP. The kinase reaction was initiated by adding 0.1 mM ATP in combination with 5 μCi [γ-32P]ATP and proceeded for 30 min at 30°C. The products of the kinase reaction were resolved by SDS–PAGE and detected by autoradiography.
Statistical analysis
All data are representative of at least three independent experiments. Quantitative data are presented as the mean±s.e.m. Statistical significance of differences between experimental groups was assessed with Student's t-test. Differences in means were considered significant if P<0.05.
Supplementary Material
Supplementary Movie
Supplementary Movie
Supplementary data- Figure S1
Supplementary data- Figure S2
Supplementary data- Figure S3
Supplementary data- Figure S4
Supplementary data- Figure S5
Supplementary data- Figure S6
Acknowledgments
We are grateful to David Clapham, Loren Runnels and Robert Adelstein for providing DNA constructs. We thank Nannette Teunissen for technical assistance and Frank de Lange for help with confocal microscopy. We also thank Wiljan Hendriks and Erik Danen for critically reading the manuscript. This work was supported by grants from the Dutch Cancer Society (WHM, FL, CF; grant NKB 2002-2593).
References
- Burgstaller G, Gimona M (2004) Actin cytoskeleton remodelling via local inhibition of contractility at discrete microdomains. J Cell Sci 117: 223–231 [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K (2004) Rho and Rac take center stage. Cell 116: 167–179 [DOI] [PubMed] [Google Scholar]
- De la Roche MA, Smith JL, Betapudi V, Egelhoff TT, Cote GP (2002) Signaling pathways regulating Dictyostelium myosin II. J Muscle Res Cell Motil 23: 703–718 [DOI] [PubMed] [Google Scholar]
- DeMali KA, Wennerberg K, Burridge K (2003) Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol 15: 572–582 [DOI] [PubMed] [Google Scholar]
- Dorovkov MV, Ryazanov AG (2004) Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem 279: 50643–50646 [DOI] [PubMed] [Google Scholar]
- Drennan D, Ryazanov AG (2004) Alpha-kinases: analysis of the family and comparison with conventional protein kinases. Prog Biophys Mol Biol 85: 1–32 [DOI] [PubMed] [Google Scholar]
- Dulyaninova NG, Malashkevich VN, Almo SC, Bresnick AR (2005) Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry 44: 6867–6876 [DOI] [PubMed] [Google Scholar]
- Egelhoff TT, Lee RJ, Spudich JA (1993) Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell 75: 363–371 [DOI] [PubMed] [Google Scholar]
- Forscher P (1989) Calcium and polyphosphoinositide control of cytoskeletal dynamics. Trends Neurosci 12: 468–474 [DOI] [PubMed] [Google Scholar]
- Franke JD, Dong F, Rickoll WL, Kelley MJ, Kiehart DP (2005) Rod mutations associated with MYH9-related disorders disrupt nonmuscle myosin-IIA assembly. Blood 105: 161–169 [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A (2002) Exploring the neighborhood: adhesion-coupled cell mechanosensors. Cell 110: 139–142 [DOI] [PubMed] [Google Scholar]
- Heid PJ, Wessels D, Daniels KJ, Gibson DP, Zhang H, Voss E, Soll DR (2004) The role of myosin heavy chain phosphorylation in Dictyostelium motility, chemotaxis and F-actin localization. J Cell Sci 117: 4819–4835 [DOI] [PubMed] [Google Scholar]
- Heine M, Cramm-Behrens CI, Ansari A, Chu HP, Ryazanov AG, Naim HY, Jacob R (2005) Alpha-kinase 1, a new component in apical protein transport. J Biol Chem 280: 25637–25643 [DOI] [PubMed] [Google Scholar]
- Hostetter D, Rice S, Dean S, Altman D, McMahon PM, Sutton S, Tripathy A, Spudich JA (2004) Dictyostelium myosin bipolar thick filament formation: importance of charge and specific domains of the myosin rod. PLoS Biol 2: 1880–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki K, Kannan KB, Singh BB, Hauser CJ (2004) Cytoskeletal reorganization internalizes multiple transient receptor potential channels and blocks calcium entry into human neutrophils. J Immunol 172: 601–607 [DOI] [PubMed] [Google Scholar]
- Jalink K, van Corven EJ, Hengeveld T, Morii N, Narumiya S, Moolenaar WH (1994) Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J Cell Biol 126: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolman MF, Futey LM, Egelhoff TT (1996) Dictyostelium myosin heavy chain kinase A regulates myosin localization during growth and development. J Cell Biol 132: 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak JA, Cahalan MD (2003) MIC channels are inhibited by internal divalent cations but not ATP. Biophys J 84: 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W, Licate L, Warrick H, Spudich J, Egelhoff T (2002) Differential localization in cells of myosin II heavy chain kinases during cytokinesis and polarized migration. BMC Cell Biol 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Aepfelbacher M (2003) Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol 13: 376–385 [DOI] [PubMed] [Google Scholar]
- Lockwich T, Singh BB, Liu X, Ambudkar IS (2001) Stabilization of cortical actin induces internalization of transient receptor potential 3 (Trp3)-associated caveolar Ca2+ signaling complex and loss of Ca2+ influx without disruption of Trp3-inositol trisphosphate receptor association. J Biol Chem 276: 42401–42408 [DOI] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A (2001) LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411: 590–595 [DOI] [PubMed] [Google Scholar]
- Nakasawa T, Takahashi M, Matsuzawa F, Aikawa S, Togashi Y, Saitoh T, Yamagishi A, Yazawa M (2005) Critical regions for assembly of vertebrate nonmuscle myosin II. Biochemistry 44: 174–183 [DOI] [PubMed] [Google Scholar]
- Rico M, Egelhoff TT (2003) Myosin heavy chain kinase B participates in the regulation of myosin assembly into the cytoskeleton. J Cell Biochem 88: 521–532 [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2004) Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun 324: 1155–1164 [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE (2001) TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291: 1043–1047 [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE (2002) The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol 4: 329–336 [DOI] [PubMed] [Google Scholar]
- Ryazanov AG (2002) Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett 514: 26–29 [DOI] [PubMed] [Google Scholar]
- Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG (2004) Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to TRP ion channel. J Biol Chem 279: 3708–3716 [DOI] [PubMed] [Google Scholar]
- Schild D, Jung A, Schultens HA (1994) Localization of calcium entry through calcium channels in olfactory receptor neurones using a laser scanning microscope and the calcium indicator dyes Fluo-3 and Fura-Red. Cell Calcium 15: 341–348 [DOI] [PubMed] [Google Scholar]
- Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM (2003) Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114: 191–200 [DOI] [PubMed] [Google Scholar]
- Sun HQ, Yamamoto M, Mejillano M, Yin HL (1999) Gelsolin, a multifunctional actin regulatory protein. J Biol Chem 274: 33179–33182 [DOI] [PubMed] [Google Scholar]
- Van Etten RA (2003) c-Abl regulation: a tail of two lipids. Curr Biol 13: R608–R610 [DOI] [PubMed] [Google Scholar]
- van Leeuwen FN, Kain HE, Kammen RA, Michiels F, Kranenburg OW, Collard JG (1997) The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol 139: 797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG (1999) Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat Cell Biol 1: 242–248 [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Jalink K (2002) Agonist-induced PIP(2) hydrolysis inhibits cortical actin dynamics: regulation at a global but not at a micrometer scale. Mol Biol Cell 13: 3257–3267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JR, Ludowyke RI, Biden TJ (1998) Nutrient stimulation results in a rapid Ca2+-dependent threonine phosphorylation of myosin heavy chain in rat pancreatic islets and RINm5F cells. J Biol Chem 273: 22729–22737 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie
Supplementary Movie
Supplementary data- Figure S1
Supplementary data- Figure S2
Supplementary data- Figure S3
Supplementary data- Figure S4
Supplementary data- Figure S5
Supplementary data- Figure S6