

Abstract
Aggregation of the type 1 Fcɛ receptors (FcɛRI) on mast cells initiates a network of biochemical processes culminating in secretion of both granule-stored and de novo-synthesized inflammatory mediators. A strict control of this response is obviously a necessity; nevertheless, this regulation is hardly characterized. Here we report that a prototype inhibitory receptor, the mast cell function-associated antigen (MAFA), selectively regulates the FcɛRI stimulus–response coupling network and the subsequent de novo production and secretion of inflammatory mediators. Specifically, MAFA suppresses the PLC-γ2–[Ca2+]i, Raf-1–Erk1/2, and PKC–p38 coupling pathways, while the Fyn–Gab2-mediated activation of PKB and Jnk is essentially unaffected. Hence, the activities of several transcription/nuclear factors for inflammatory mediators (NF-κB, NFAT) are markedly reduced, while those of others (Jun, Fos, Fra, p90rsk) are unaltered. This results in a selective inhibition of gene transcription of cytokines including IL-1β, IL-4, IL-8, and IL-10, while that of TNF-α, MCP-1, IL-3, IL-5, or IL-13 remains unaffected. Taken together, these results illustrate the capacity of an immunoreceptor tyrosine-based inhibitory motif-containing receptor to cause tight and specific control of the production and secretion of inflammatory mediators by mast cells.
Keywords: cytokine, inhibition, mast cells
Introduction
It is only recently that the intricate and essential mechanisms controlling mast cells' secretory response to the FcɛRI stimulus are being addressed. Moreover, as this is a multiple phase process, resolving its regulatory elements is necessary.
FcɛRI aggregation on mast cells culminates in an immediate secretion of its granule-stored mediators followed by slower secretion of de novo-synthesized arachidonic acid (AA) metabolites and of more than a dozen different cytokines (IL-1–IL-6, IL-9–IL-14, IL-16, IL-18, MIF, TNF-α, GM-CSF, LIF, etc) and chemokines (IL-8, MCP-1–MCP-4, MIP-1, MIP-3, RANTES, etc) (Sayama et al, 2002). These molecules play a critical role in the vigorous immune response, affecting a wide range of tissues (Galli, 2000), as they stimulate, mobilize, and navigate immune cells and thereby initiate an early immune response (reviewed by Malaviya and Abraham, 2001). These mediators may also induce sustained inflammatory responses, such as type I IgE-dependent allergic reactions (Turner and Kinet, 1999). Therefore, the networks activated by FcɛRI or by other members of the multichain immune recognition receptors (MIRR) family are regulated by elaborate mechanisms. One of the most effective controls is exerted by receptor superfamily members, which contain one or more copies of an immunoreceptor tyrosine-based inhibitory motif (ITIM) essential for their function (Ravetch and Lanier, 2000).
The membrane glycoprotein named ‘mast cell function-associated antigen' (MAFA) has been shown to suppress the FcɛRI secretory response of rat mucosal type mast cells of the 2H3 line (RBL-2H3) (Ortega and Pecht, 1988). Expression cloning of MAFA's cDNA has shown that it is a 188-amino-acid-long molecule containing an ITIM sequence (SIYSTL) within its intracellular domain, classifying MAFA as an inhibitory receptor family member (Abramson et al, 2002). Unlike most other ITIM-containing receptors however, MAFA clustering alone (by its specific monoclonal antibody (mAb) G63), prior to the FcɛRI stimulation, suffices for suppressing the secretory response. Nevertheless, co-clustering of MAFA with FcɛRI has recently been shown to markedly increase the inhibition of mast cell degranulation. Analysis of the latter results has indicated that this enhancement could reach two orders of magnitude, probably by increasing the concentrations of inhibitory molecules in the proximity of the activating FcɛRI clusters (Licht et al, 2005).
The capacity of MAFA (and other ITIM-containing receptors) to regulate mast cells' late response of de novo synthesis and secretion of cytokines and fatty acid metabolites has so far not been characterized. Here we show that MAFA clustering or co-clustering with FcɛRI causes a selective suppression of FcɛRI-induced cytokine gene transcription and their subsequent secretion. The mechanism underlying this selective control is shown to be a selective interference with the FcɛRI-induced activation of several key enzymes and transcription factors. This yields a tight and specific control of gene expression and very selective inhibition of the production and secretion of inflammatory mediators.
Results
MAFA clustering inhibits the FcɛRI-induced de novo synthesis and secretion of leukotriene
We have first investigated whether MAFA suppresses the FcɛRI-induced de novo synthesis and secretion of AA-derived mediators. Enzyme-linked immunoassay kits specific for rat LTC4 and LTB4 were employed for monitoring their secretion by RBL-2H3 cells (primed with the DNP-specific IgE class mAb, A2IgE) that were either preincubated with mAb G63-F(ab′)2 for 5 min and/or challenged by antigen (BSA-DNP11) for 40 min. Co-clustering impact was assayed by cells' treatment (40 min) with either F(ab′)2 of mouse IgG derivatized with an average of three DNP (IgG-DNP3, which clusters FcɛRI only) or with a similarly derivatized F(ab′)2 fragment of mAb G63 (G63-DNP3, which co-clusters MAFA with FcɛRI), and determining the levels of secreted LTC4/LTB4. MAFA clustering prior to FcɛRI stimulation inhibited the FcɛRI-induced de novo synthesis and secretion of LTC4/LTB4 and this was further enhanced upon MAFA–FcɛRI co-clustering (Figure 1A).
Figure 1.
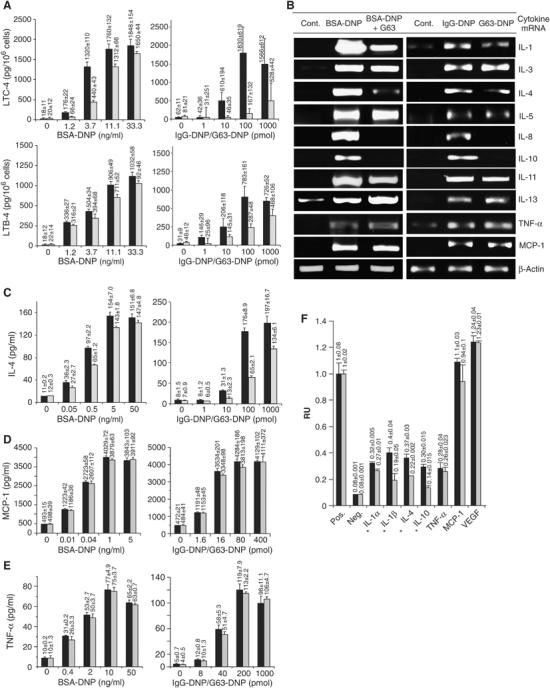
MAFA clustering selectively inhibits the FcɛR-induced de novo synthesis and secretion of proinflammatory mediators. (A) RBL-2H3 cells (1 × 105/well) were incubated with the indicated antigen (BSA-DNP11) concentrations in the absence (black) or presence (gray) of F(ab′)2 of mAb G63 added 5 min prior to antigen treatment (left panel). In a parallel set of experiments, the cells were treated with the indicated concentrations of IgG-DNP3 (black) or G63-DNP3 (gray) (right panel). After 30 min, the cells' supernatants were assayed for the secreted LTC4 (upper panel) or LTB4 (lower panel) by ELISA. (B) RBL-2H3 cells (1 × 107) were either left untreated as controls (Cont.) or FcɛRI stimulated for 3 h in the absence (BSA-DNP11) or presence of mAb G63-F(ab′)2 (BSA-DNP11+G63) (left panel). An additional set of cells was treated for 2 h with 1 nM (F(ab′)2) IgG-DNP3 (inducing FcɛRI–IgE clustering) or with 1 nM (F(ab′)2) G63-DNP3 (inducing FcɛRI–MAFA co-clustering) (right panel). The levels of respective cytokines were measured by RT–PCR. The results are of one representative set of at least three independent experiments. (C–E) RBL-2H3 cells were treated as in panel A. Cell supernatants were assayed by ELISA for secreted levels of (C) IL-4, 24 h post stimulation (PS); (D) MCP-1, 2 h PS; and (E) TNF-α, 12 h PS. The average value ±s.d. for each antigen concentration was calculated from triplicates; the plotted results are of one representative set out of three independent measurements (A, C–E). (F) RBL-2H3 cells (5 × 105 cells/well) were incubated with 0.5 nM IgG-DNP3 (black) or G63-DNP3 (gray) for 15 h. Their supernatants (100 μl) were then transferred onto cytokine array membranes and assayed for secretion of individual cytokines. The average intensities (duplicates ±s.d.) were plotted as relative values (where 1 is the intensity of a positive control). The results are of one set out of two independent experiments; values marked with ‘*' indicate results with a reproducible inhibition.
MAFA clustering suppresses the FcɛRI-induced de novo synthesis of only selected cytokines
MAFA's regulation of FcɛRI-induced de novo synthesis and secretion of cytokines and chemokines was first examined at mRNA levels of those inflammatory mediators known to be enhanced in response to FcɛRI stimulation. RBL-2H3 cells were either subjected to MAFA clustering (5 min) and/or stimulated with FcɛRI for 3 h. In addition, cells were either stimulated with IgG-DNP3 or subjected to MAFA–FcɛRI co-clustering by G63-DNP3, both for 2 h. The results of the reverse transcription–polymerase chain reaction (RT–PCR) analysis confirmed that the FcɛRI stimulation induces a marked increase in the mRNA levels of the following molecules: IL-1β, IL-3–IL-5, IL-8, IL-10, IL-11, IL-13, TNF-α, and MCP-1. MAFA clustering or co-clustering caused a selective decline in this response. Specifically, while no (or insignificant) change was observed in the FcɛRI-induced transcription of IL-3, IL-5, IL-11, IL-13, TNF-α, and MCP-1 genes (Figure 1B), mRNA levels of IL-1β, IL-4, IL-8, and IL-10 were significantly reduced. MAFA-mediated selective inhibition was then quantitatively analyzed and established by real-time PCR (Table I). To determine whether the above selective suppression of cytokine mRNA levels also results in a decrease of the subsequent protein synthesis and secretion, a cytokine assay array has been employed. Cells were subjected to either FcɛRI clustering or MAFA–FcɛRI co-clustering for 15 and 24 h and their supernatants' samples were transferred onto the array membranes and levels of the secreted cytokines were evaluated. The results (Figure 1F) show that while secretion of IL-1, IL-4, and IL-10 was notably reduced upon MAFA–FcɛRI co-clustering, that of TNF-α, MCP-1, and VEGF was unaffected (similarly after 24 h; not shown). Thus, the MAFA-mediated selective suppression of the cytokine mRNA levels also inhibits the synthesis and secretion of the respective proteins.
Table 1.
Real-time PCR analysis of cytokine mRNA levels induced by FcɛRI clustering or MAFA–FcɛRI co-clustering
| IL-1β | IL-3 | IL-4 | IL-5 | IL-8 | IL-10 | IL-11 | IL-13 | TNF-α | MCP1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| IgG-DNP | 19.7±2.1 | 278.7±28 | 52.8±6.9 | 45.3±0.6 | 7.3±.02 | 12714±46 | 9.9±1.4 | 10.6±0.9 | 17.3±2.1 | 82.3±9.3 |
| G63-DNP | 9.1±1.4 | 333.1±41 | 38.7±3.7 | 45.6±3.2 | 2.1±0.1 | 2655±27 | 9.9±2.1 | 10.1±1.2 | 20.7±2.3 | 100.6±18.1 |
| IgE-primed RBL-2H3 cells were treated for 3.5 h with 1 nM (F(ab′)2) IgG-DNP3 (inducing FcɛRI–IgE clustering) or with 1 nM (F(ab′)2) G63-DNP3 (inducing FcɛRI–MAFA co-clustering). Cells were then lysed, the total RNA was isolated and used for reverse transcription to produce cDNA. Levels of respective cytokines were measured as duplicates by light cycler using their specific primers. Values represent fold change in mRNA levels relative to control (untreated) cells. | ||||||||||
Secretion of IL-4, TNF-α, and MCP-1 was also quantitatively determined by enzyme-linked immunosorbent assay (ELISA), further confirming that MAFA clustering (or co-clustering) does indeed inhibit the de novo synthesis and secretion of IL-4 (Figure 1C), but not of MCP-1 (Figure 1D) and TNF-α (Figure 1E). Although our previous preliminary results of TNF-α bioassay suggested that MAFA clustering also affects this cytokine's secretion (Abramson et al, 2002), the above results of the more sensitive and reliable method demonstrated that this is not the case. Significantly, in all cases, the MAFA-induced inhibition was qualitatively identical to that caused by its co-clustering with FcɛRI, although amplitudes of the latter were much larger, suggesting that MAFA–FcɛRI co-clustering is able to bring more inhibitory molecules to the proximity of the activating FcɛRI clusters.
FcɛRI-induced tyrosine phosphorylation of multiple substrates is differentially affected by MAFA clustering
To resolve the mechanism of MAFA's selective inhibition process, the potential targets of its action in the FcɛRI coupling network were pursued. First, the effect of MAFA clustering (and MAFA–FcɛRI co-clustering) on the levels of FcɛRI-induced protein tyrosyl phosphorylation was investigated. RBL-2H3 cells were preincubated for 2 min with/without mAb G63 and then FcɛRI stimulated for the indicated times. Similarly, FcɛRI was clustered (with IgG-DNP3) or MAFA–FcɛRI co-clustered (with G63-DNP3) for the indicated time periods. FcɛRI clustering induced the known marked tyrosine phosphorylation of several proteins (Figure 2A and B, left upper panel). However, the level of tyrosine phosphorylation of several proteins was altered upon both MAFA clustering and MAFA–FcɛRI co-clustering (Figure 2A and B, right upper panel). To identify the molecules whose tyrosine phosphorylation was affected, the same membranes were sequentially probed with antibodies specific to molecules known to be involved in the FcɛRI stimulus-secretory response coupling. The sequential blotting revealed that MAFA clustering, as well as MAFA–FcɛRI co-clustering, markedly increased the tyrosine phosphorylation of proteins such as Dok-1, Dok-2, and SHIP relative to that caused by FcɛRI stimulation (Figure 2A and B). In contrast, tyrosine phosphorylation of PLC-γ2 was markedly reduced and that of Syk and LAT was only marginally affected. Noteworthy is that MAFA (co)-clustering did not alter the FcɛRI-induced tyrosine phosphorylation of its β- and γ-subunits or of the adaptor protein Gab2 (Figure 2A and B). These results therefore suggest that MAFA's inhibitory activity primarily affects PLC-γ2 activation, while it does not interfere (or only marginally interferes) with the Fyn–Gab2 and Lyn–Syk–LAT signaling paths.
Figure 2.
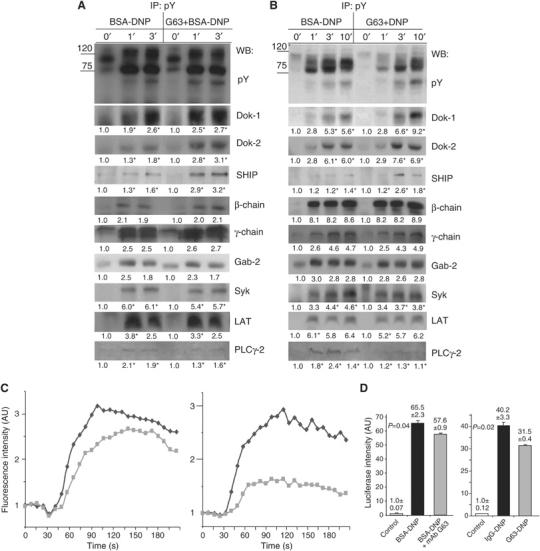
The impact of MAFA on the FcɛRI-induced multiple substrates' tyrosine phosphorylation, calcium mobilization, and NFAT transcriptional activity. (A) RBL-2H3 cells (1 × 107/sample) were either FcɛRI stimulated (BSA-DNP) or treated with mAb G63-F(ab′)2 for 2 min and then FcɛRI clustered (G63+BSA-DNP). Cells were lysed and immunoprecipitation (IP) was carried out with phosphotyrosine-specific antibodies (pY-99). Samples were then analyzed by sequential Western blotting (WB) with antibodies specific to phosphotyrosine (pY-99), Dok-1, Dok-2, SHIP, β-chain, γ-chain, Gab-2, Syk, LAT, and PLC-γ2. Detection was carried out by ECL and quantification of fold change (in AU) was carried out by densitometric analysis. Values highlighted with ‘*' indicate a change that was reproduced three times. The results are of one representative set of experiments out of four. (B) RBL-2H3 cells (1 × 107/sample) were treated for the indicated times with either 0.5 nM IgG-DNP3 or 0.5 nM G63-DNP3 at 37°C. The subsequent IP, WB, and evaluation were performed as in panel A. (C) RBL-2H3 cells were labeled with Fluo-3 (5 μg/1 × 106 cells in 1 ml) and then either stimulated with BSA-DNP11 (0.1 ng/ml) (black) or first treated by F(ab′)2 of 50 nM mAb G63 (2 min) and then stimulated with BSA-DNP11 (gray) (left panel). In parallel, the cells were subjected to either 0.1 nM IgG-DNP3 (black) or 0.1 nM G63-DNP3 (gray) (right panel). The respective increase in fluorescence intensity (reflecting the free [Ca2+]i mobilization) was monitored by flow cytometry for the indicated times in four independent experiments. (D) RBL-2H3 cells (5 × 106 cells in 0.5 ml) were transiently cotransfected with pNFAT-Luc reporter plasmid, NFAT-1 cDNA, and pRL-CMV Renilla plasmid by electroporation. The next day, the cells were either stimulated with BSA-DNP11 (30 ng/ml) (black) or first treated with 50 nM F(ab′)2 of mAb G63 (2 min) and then stimulated with BSA-DNP11 (gray) or left untreated (white) for 6 h (left panel). In parallel, cells were either stimulated with 1 nM IgG-DNP3 (black) or 1 nM G63-DNP3 (gray) or left untreated (white) for 6 h (right panel). Cells were lysed and their lysates were assayed for luciferase/Renilla activities. The normalized luciferase activities are expressed as fold change (average of three measurements+s.e.m.) compared to that in untreated cells. The P-value indicates the level of statistical significance. The plotted results are of one representative set out of three independent repetitions.
MAFA–FcɛRI (co)-clustering suppresses the FcɛRI-induced Ca2+ mobilization and NFAT transcriptional activity
Based on the above findings, the coupling events downstream of PLC-γ2 activity were examined by monitoring the effect of MAFA (co)-clustering on FcɛRI-induced transient rise in free intracellular Ca2+ ion concentrations ([Ca2+]i). Cells were labeled with Fluo-3 and either subjected to FcɛRI clustering (Figure 2C, black), MAFA/FcɛRI individual clustering (Figure 2C, left, gray), or to MAFA–FcɛRI co-clustering (Figure 2C, right, gray). The induced [Ca2+]i changes were monitored by flow cytometry. MAFA clustering (and even more so MAFA–FcɛRI co-clustering) considerably suppressed the FcɛRI-induced transient [Ca2+]i rise (Figure 2C). Moreover, experiments where the extracellular [Ca2+] was limited by EGTA demonstrated that this suppression already took place during the initial phase of Ca2+ release from internal stores (cf Supplementary data). These results therefore suggest that MAFA-mediated inhibition of PLC-γ2 phosphorylation lowers IP3 production and hence reduces the amplitude of the transient [Ca2+]i rise. As [Ca2+]i is the main activator of NFAT transcription factor family (Hutchinson and Mccloskey, 1995) (which is involved in the regulation of expression of several cytokine genes), the luciferase assay was employed to analyze the effect of MAFA (co)-clustering on NFAT transcriptional activity. Indeed, both MAFA clustering and, even more so, MAFA–FcɛRI co-clustering caused a significant (P=0.044 and 0.021, respectively) decline in NFAT-driven firefly luciferase production (Figure 2D). Although the latter decline appears to be relatively small (due to the known relatively low sensitivity of monitoring transcriptional inhibition by a luciferase assay), it is statistically significant and reproducible (n=3).
MAFA clustering increases Dok-1/2 binding to its intracellular tail as well as to RasGAP, Shc, and SHIP
The observed marked increase in the tyrosyl phosphorylation of Dok-1/2 upon MAFA–FcɛRI (co)-clustering suggests their possible role in mediating the inhibitory signals. Moreover, several reports demonstrated that Dok-1/2 are involved in the negative regulation of de novo cytokine synthesis by immunocytes (Nemorin et al, 2001; Abramson et al, 2003; Kepley et al, 2004). Therefore, Dok-1/2 function was further pursued, concentrating on the identification of molecules that bind them upon MAFA clustering. Dok-1/2 were isolated by IP from lysates of RBL-2H3 cells treated either with mAb G63-F(ab′)2 or with nonspecific F(ab′)2 fragments. Samples were then separated by SDS–PAGE and electrotransferred onto nitrocellulose membranes, which were then sequentially blotted with different specific antibodies. In this screening, we observed a time-dependent increase in Dok-1/2 association with the adaptor protein Shc, 5′-inositol phosphatase SHIP, and GTPase-activating protein RasGAP, suggesting the formation of an intermolecular complex (Figure 3A and B).
Figure 3.
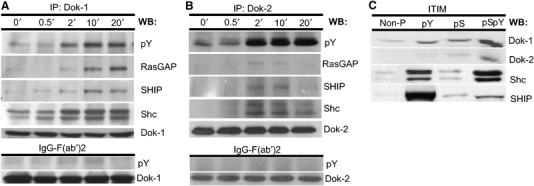
MAFA clustering leads to the formation of a multimolecular complex (Shc–SHIP–Dok–RasGAP), which is recruited to MAFA's intracellular tail. RBL-2H3 cells (1 × 107/sample) were subjected to MAFA clustering (upper panel) or were treated with F(ab′)2 fragments of nonspecific polyclonal mouse IgG (lower panel) for the indicated times. Cells were lysed and the IP was carried out with antibodies specific to either (A) Dok-1 or (B) Dok-2. Samples were analyzed by sequential WB with antibodies specific to phosphotyrosine, RasGAP, Shc, SHIP, Dok-1, and Dok-2. (C) RBL-2H3 cells (1 × 107/sample) were lysed and their lysates containing equal protein amounts were incubated with Sepharose beads conjugated with the following MAFA ITIM synthetic peptides: non-phosphorylated (non-P), tyrosyl-phosphorylated (pY), seryl-phosphorylated (pS) and seryl/tyrosyl-phosphorylated (pSpY). Bound proteins were eluted and analyzed by sequential WB using antibodies specific to Dok-1, Dok-2, Shc, and SHIP. Detection was carried out by ECL. Results are of one set out of three independent experiments (A–C).
A further in vitro examination of potential interactions between MAFA and these molecules was carried out using peptides with the sequence corresponding to MAFA's ITIM (YSTL), phosphorylated on its tyrosine, serine, or both residues that were conjugated to Sepharose beads. These beads were then used to isolate proteins from MAFA-clustered RBL-2H3 cell lysates. Figure 3C shows that while the nonphosphorylated ITIM peptide did not bind any of the tested proteins, the tyrosine phosphorylated one bound all four: SHIP, Shc, Dok-1, and Dok-2. Interestingly, the serine phosphorylated, and, even more so, the peptide phosphorylated on both serine and tyrosine residues also bound Dok-1 and Dok-2, suggesting that optimal recruitment of both proteins also requires phosphorylation of the MAFA ITIM serine. Furthermore, the results of surface plasmon resonance measurements demonstrated that it is only the SH2 domain of SHIP that binds MAFA's ITIM directly (cf Supplementary data). These results therefore suggest that MAFA clustering (or co-clustering) leads (via its phosphorylated ITIM) to the recruitment of SHIP. Subsequently, the MAFA-bound SHIP becomes tyrosine phosphorylated and may then serve as a docking site for other signaling molecules such as Dok, which thereafter becomes tyrosine phosphorylated as well. This may eventually lead to the formation and recruitment of a multimolecular complex (SHIP–Shc–Dok–RasGAP) in the proximity of the plasma membrane.
MAFA clustering leads to interference with the FcɛRI-induced Raf/MEK/Erk-1/2 activation yet does not affect the p90RSK activity
Recruitment of RasGAP to the plasma membrane may be important for the regulation of the cell's RasGTP/GDP balance and the subsequent activity of the Ras/Raf-1/MEK/Erk-1/2 signaling pathway (central to regulating cytokine gene transcription in lymphocytes). To examine whether MAFA does indeed interfere with the FcɛRI-induced activation of the above pathway, we have employed antibodies specific to the active forms of MEK-1/2 and Erk-1/2 and analyzed the effect of MAFA (co)-clustering on their activation. Figure 4A shows that while shorter periods of MAFA–FcɛRI co-clustering (1–5 min) had only a marginal effect on Erk-1/2 and MEK activation, longer co-clustering dramatically decreased the levels of their activated forms. This suggests that MAFA reduces the half-life of the activated MEK-1/Erk-1/2, rather than their early activation (Figure 4A).
Figure 4.
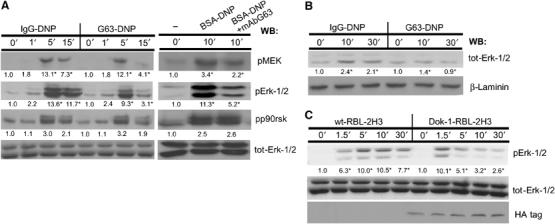
MAFA (co)-clustering interferes with the FcɛRI activation of the Erk-1/2 coupling pathway. (A) RBL-2H3 cells were treated for the indicated times with either IgG-DNP3 or G63-DNP3 (left panel). In an additional set of experiments, the cells were either FcɛRI stimulated (BSA-DNP) or first pretreated with 50 nM mAb G63 F(ab′)2 for 5 min and then FcɛRI stimulated (BSA-DNP+G63) (right panel). Cells were lysed and lysates' samples containing equal protein amounts were analyzed by WB using antibodies specific to the active forms of the following proteins: pMEK, pErk-1/2, pp90rsk, and total Erk-1/2. (B) RBL-2H3 cells (1 × 107) were treated for the indicated times with either IgG-DNP3 or G63-DNP3. Cells were lysed with Triton-based buffer and the cells' nuclei were separated and then resuspended with nuclear extract buffer. The nuclear supernatants were then separated by SDS–PAGE and WB was carried out as described above using antibodies specific to tot-Erk-1/2 and laminin. (C) Parental RBL-2H3 cells (wt-RBL-2H3) or their mutants overexpressing Dok-1 (Dok-1-RBL-2H3) were treated for the indicated times with 1 nM G63-DNP3 and lysed. Equal amounts of protein samples were taken from cell lysates, separated by SDS–PAGE, and electrotransferred onto nitrocellulose membranes. Erk-1/2 activation and the levels of proteins were monitored by sequential WB with antibodies specific to phosphorylated Erk-1/2 (pErk-1/2), total Erk-1/2 (Erk-1/2), or the HA tag (HA). Detection was carried out by ECL and quantification of fold change (in AU) was by densitometric analysis. The results represent one set out of three independent experiments; numbers highlighted with an ‘*' indicate a change that was reproduced three times (A–C).
Upon FcɛRI activation, cytosolic Erk translocates to the nucleus and activates the transcription process by phosphorylating different histones and ribosomal kinases such as MSK-1 and p90RSK, as well as transcription factors such as Elk-1, which then initiate the gene transcription process (Frodin and Gammeltoft, 1999). To test whether the observed decrease in Erk-1/2 activity results in suppression of downstream nuclear factor activity, the effect of MAFA (co)-clustering was examined on Erk-1/2 nuclear translocation and on the subsequent phosphorylation of the ribosomal kinase p90RSK. Figure 4A and B shows that while the FcɛRI-induced Erk-1/2 phosphorylation and translocation are markedly reduced upon MAFA–FcɛRI (co)-clustering, those of p90RSK were unaffected.
The role of Dok-1 in the MAFA-mediated inhibition of FcɛRI-induced Erk-1/2 activation was examined by its overexpression in RBL-2H3 cells using a retroviral expression system. As expected, Dok-1 overexpression dramatically decreased levels of p-Erk1/2 compared to wild-type cells upon G63-DNP3 treatment (Figure 4C).
MAFA clustering interferes with the FcɛRI-induced PKC/p38 MAPK activation, resulting in the suppression of NF-κB transcriptional activity
In addition to the calcium–NFAT and Erk1/2 signaling paths, the FcɛRI coupling network also involves other members of the MAPK family (Fukamachi et al, 1993; Turner and Kinet, 1999), that is, the c-Jun NH2-terminal kinase (Jnk) and p38 MAPK, as well as the protein S/T kinases PKB and PKC. When activated, these kinases induce several transcription factors (e.g. NF-κB, Jun, Fos), which initiate transcription of several cytokine genes. To determine whether MAFA also interferes with the above signaling pathways, cells were treated as described above and their lysates were first analyzed by WB with antibodies specific to phosphorylated forms of p38 MAPK, Jnk, PKB, and pan-PKC. The WB analysis demonstrated that phosphorylation of p38 MAPK and PKC (in particular, its 95 kDa isoform) (Figure 5A) is specifically suppressed upon MAFA (co)-clustering. In contrast, JNK phosphorylation was only marginally affected and that of PKB was unaffected (Figure 5A).
Figure 5.
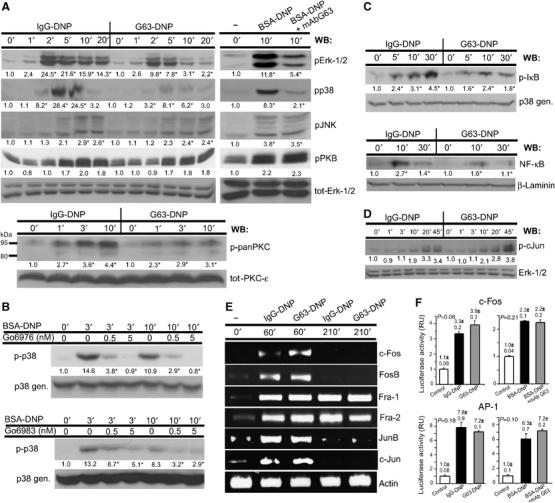
MAFA–FcɛRI co-clustering differentially regulates the activities of S/T kinases and several transcription factors. (A) Adherent IgE-primed RBL-2H3 cells were treated for the indicated times with either IgG-DNP3 or G63-DNP3 (left panel). In parallel, the cells were either stimulated by FcɛRI–IgE clustering (BSA-DNP) or first pretreated with G63 F(ab′)2 and then stimulated by an antigen (BSA-DNP+G63) for the indicated times (right panel). Cells were lysed and the samples were analyzed by WB using antibodies specific to the active forms of pErk-1/2, pp38, pJNK, pPKB, and p-pan-PKC. The loading of equal protein amounts was confirmed by WB using an antibody specific to either total Erk-1/2 or total PKC-ɛ. (B) RBL-2H3 cells were pretreated for 10 min with the indicated concentrations of two different PKC-specific inhibitors (Go6976 and Go6983). Cells were then stimulated by an antigen (BSA-DNP11) for the indicated times and lysed. Samples containing equal protein amounts were separated by SDS–PAGE and the activation of p38 MAPK was monitored by WB using an antibody specific to its active form. (C) RBL-2H3 cells (1 × 107) were treated for the indicated times with either IgG-DNP3 or G63-DNP3. Cells were lysed and samples analyzed by WB using an antibody specific to either the active form of IκB and general p38 MAPK (upper panel). Cells were treated as above and then lysed with Triton-based buffer. The cells' nuclei were separated and resuspended with nuclear extract buffer. The nuclear supernatants were collected and equal amounts of proteins were analyzed by WB with antibodies specific to NF-κB and laminin (lower panel). (D) RBL-2H3 cells were treated as in panel A and levels of active c-Jun were monitored by WB using an antibody specific to the phosphorylated form of c-Jun. The loading of equal protein amounts was confirmed by WB using an antibody specific to general p38 MAPK. Quantification in panels A–D was carried out by densitometric analysis, and fold change is expressed in AU; numbers highlighted with an ‘*' indicate a change that was reproduced three times. (E) Cells were treated for the indicated time periods with either 1 nM IgG-DNP3 or 1 nM G63-DNP3. Cells were then lysed and the total RNA was isolated. The mRNA levels of respective transcription factors were measured in four independent experiments by RT–PCR. The equal amounts of cDNA were confirmed by using primers specific to the actin gene. (F) RBL-2H3 cells (5 × 106cells in 0.5 ml) were transiently cotransfected by electroporation by pRL-CMV Renilla plasmids together with plasmids of cFos-Luc reporter (upper panel) or with 4XAP1 reporter (lower panel). The next day, the cells were primed with A2IgE and either stimulated with BSA-DNP11 (black) or first MAFA clustered by mAb G63 F(ab′)2 and stimulated with BSA-DNP11 (gray) or left untreated (white) for 6 h (left panel). In parallel, cells were stimulated with either IgG-DNP3 (black) or G63-DNP3 (gray) or left untreated (white) for 6 h (right panel). Cells were then lysed and their lysates were assayed for luciferase/Renilla activities. Normalized luciferase activities are expressed as fold change (average of three measurements±s.e.m.) compared to that in untreated cells. The P-value indicates the level of statistical significance. The plotted results are of one set out of three independent experiments.
The mechanisms underlying activation of p38 MAPK in FcɛRI-stimulated mast cells are not yet fully resolved. It was however demonstrated that p38 activity can be regulated by members of the PKC family (Hashimoto et al, 1998). To analyze whether p38 indeed acts downstream of PKC, we have used several isotype-specific PKC inhibitors and monitored their effect on FcɛRI-induced p38 phosphorylation. Cells were treated with two different concentrations of either Go6976 (inhibitor of α and β) or Go6983 (inhibits α, β, γ, δ, ζ) and the FcɛRI–IgE was clustered. Figure 5B shows that Go6976 at 5 nM concentration led to an essentially complete and at 0.5 nM concentration to a considerable suppression of the FcɛRI-mediated p38 MAPK phosphorylation. A similar, although less marked, decline was observed using Go6983 (Figure 5B, lower panel), suggesting that the reduced p38 activity is due to MAFA-induced suppression of the calcium-dependent isoforms of PKC.
FcɛRI-induced PKC activation was recently shown to be essential for regulating the transcriptional activity of NF-κB (Kalesnikoff et al, 2002; Peng et al, 2005). Therefore, the phosphorylation/nuclear translocation of components of the NF-κB signaling path were explored. MAFA–FcɛRI co-clustering led to a marked reduction in IκB phosphorylation, which was followed by a decreased NF-κB translocation to the nucleus compared to that induced by FcɛRI clustering alone (Figure 5C). These results therefore suggest that the selective inhibition of gene transcription is, at least partially, caused by the MAFA-mediated negative regulation of PKC, which reduces NF-κB transcriptional activity.
MAFA does not inhibit the FcɛRI-induced activation of AP-1 transcription factor components: Jun, Fos, and Fra
The above results suggest that the MAFA-mediated selective inhibition may be due to MAFA's lack of interference with FcɛRI-induced PKB and Jnk activation. These two pathways regulate expression of several FcɛRI-induced inflammatory mediators by controlling several transcription factors, in particular, members of the activator protein (AP-1) family (Karin, 1995; Hata et al, 1998; Kitaura et al, 2000). The activation of AP-1 proteins is regulated by either rapid induction of their de novo synthesis (Fos/Jun family) or by their (subsequent) phosphorylation mediated by S/T kinases (Jun family).
Therefore, the effect of MAFA–FcɛRI co-clustering on mRNA induction of individual AP-1 components was analyzed by RT–PCR. Figure 5E shows that the FcɛRI-induced mRNA levels of c-Jun, Jun-B, c-Fos, FosB, Fra-1, and Fra-2 were not affected by MAFA–FcɛRI co-clustering, suggesting that MAFA does not interfere with their rapid de novo synthesis. Moreover, WB analysis using antibodies specific to phosphorylated c-Jun demonstrated that its phosphorylation (i.e. activation) is also unaffected by MAFA–FcɛRI co-clustering (Figure 5D). Thus, MAFA's inability to inhibit transcription of certain cytokines (e.g. IL-3, IL-5, IL-13, TNF-α) may partially be a result of its failure to suppress the rapid de novo synthesis and phosphorylation of AP-1 components. In order to directly monitor the AP-1 transcriptional activity, luciferase assays of AP-1 and c-Fos were carried out. Cells were transiently transfected by either 4xAP-1 or c-Fos luciferase reporter plasmids and their transcriptional activities were monitored upon FcɛRI stimulation and MAFA–FcɛRI co-clustering. The results demonstrated that MAFA–FcɛRI co-clustering does not suppress, but rather slightly increases, c-Fos activity compared to FcɛRI clustering (Figure 5F). This fully agrees with the results shown in Figure 5E, demonstrating that the c-Fos mRNA levels are slightly increased upon FcɛRI–MAFA co-clustering compared to those induced by FcɛRI stimulation. Monitoring the effect of MAFA–FcɛRI co-clustering on AP-1 transcriptional activity demonstrated rather insignificant changes compared to FcɛRI-stimulated cells, suggesting that MAFA does not interfere with AP-1-controlled cytokine gene transcription.
Discussion
The mechanisms by which ITIM-bearing receptors regulate the MIRR-induced gene transcription and subsequent de novo protein synthesis are still poorly understood. This is particularly true for FcɛRI, which induces de novo synthesis of over a dozen different cytokines and chemokines. Resolving these mechanisms is of major importance, as these powerful mediators affect a wide range of body functions and orchestrate the immune/inflammatory responses (Galli, 2000). The present results show that an inhibitory receptor can differentially regulate several specific paths of the FcɛRI coupling network, resulting in a selective suppression of the de novo synthesis and secretion of inflammatory mediators. Furthermore, molecular details of the mechanisms underlying this regulation have now been examined and identified.
Each cytokine is known to have a very specific activity regulating different physiological functions, notably those of the immune system. The observed MAFA-mediated selective inhibition of the FcɛRI-induced cytokines' de novo synthesis therefore indicates a capacity for tight and specific modulation of the immune response. IL-1β (suppressed by MAFA) is, for example, the key trigger of the acute-phase immune response (e.g. by stimulating hepatocytes to synthesize acute-phase proteins) and can also serve as an endogenous pyrogen (Ramadori et al, 1985). In contrast, other mast cell-derived cytokines such as IL-3 and IL-5 (both unaffected by MAFA) induce growth, maturation, and survival of eosinophils (Shakoory et al, 2004), cells that play an essential role in the development of inflammatory response. Mast cell-derived TNF-α (unaffected by MAFA) plays a central role in inducing T-cell migration to draining lymph nodes upon bacterial infection (McLachlan et al, 2003). MCP-1 (unaffected by MAFA) is known to affect monocytes (but not neutrophils), while IL-8 (suppressed by MAFA) predominantly mediates the recruitment of neutrophils and basophils (but not monocytes) to sites of inflammation (Leonard, 1990). Interestingly, IL-4, which induces IgE production by B cells and promotes preferential differentiation of T cells into the TH2 subset (Lebman and Coffman, 1988) is modestly downregulated upon MAFA (co)-clustering. Conversely, the same treatment does not suppress IL-13 gene transcription (which has a very similar function as IL-4), suggesting that even cytokines with similar functions can be differentially regulated by MAFA. Therefore, the present findings indicate that inhibitory receptors, as exemplified by MAFA, may selectively regulate only certain aspects of MIRR-induced responses rather than causing a general inhibition. Hence, only specific components of the immune system might be modulated while others may stay unaffected. Such a tight regulation might be particularly important for orchestrating and achieving the required accurate and balanced immune response.
The FcɛRI-induced transcription of the above genes is regulated by several transcription factors that are, upon activation, shuttled to the nucleus where they provide their positive or negative input (Escoubet-Lozach et al, 2002). These factors are activated by multiple (separate and alternative) coupling pathways that are however often linked at specific points and thus exhibit considerable crosstalk with each other (Johnson and Lapadat, 2002). These pathways involve members of MAPK family such as Erk-1/2, p38, and Jnk-1/2. FcɛRI-induced gene transcription is also regulated by the activities of other enzymes such as PKB, PKC, or the calcium-dependent S/T phosphatase calcineurin.
A transcription factor that plays a pivotal role in the FcɛRI-induced response is the NF-κB (Marquardt and Walker, 2000). In unperturbed mast cells, NF-κB is associated with the inhibitory protein IκB, which sequesters it in the cells' cytoplasm by masking the NF-κB nuclear localization signal. Upon FcɛRI stimulation, the IκB is phosphorylated by the inhibitory κB protein kinase (IKK), causing its dissociation from NF-κB. The released NF-κB rapidly enters the nucleus where it binds to promoters of several genes coding for inflammatory molecules. Our findings demonstrate that MAFA (co)-clustering interferes with the FcɛRI-induced NF-κB activation (Figure 5B), which is followed by suppressing production of several NF-κB-dependent cytokines (e.g. IL-1β and IL-8) (Figure 1B). Furthermore, recent studies demonstrated that in FcɛRI-stimulated mast cells, NF-κB activity is regulated by PKC (Kalesnikoff et al, 2002; Peng et al, 2005). This agrees with the present results showing that MAFA (co)-clustering suppresses the FcɛRI-induced phosphorylation (and thereby activation) of PKC (specifically its ∼95 kDa isoform) (Figure 5A) as well as it markedly reduces [Ca2+]i transients (Figure 2C) essential for activation of Ca2+-dependent PKC isoforms. Moreover, the reduced [Ca2+]i levels resulted in a significantly suppressed activity of NFAT (Figure 2D), another transcription factor involved in the regulation of the expression of a broad range of inflammatory mediators. The above data therefore suggest that the observed selective inhibition of certain inflammatory mediators may be due to MAFA-mediated interference with the PKC–NF-κB and the PLC-γ2–[Ca2+]i–NFAT signaling pathways.
Further experiments revealed that MAFA also interferes with FcɛRI-induced p38 and Erk-1/2 MAPK activation (Figures 4A and 5A). The immediate decrease in p38 activity is apparently also mediated by MAFA's suppression of the [Ca2+]i transient, which then results in a decline in the activities of calcium-dependent PKC isoforms. These isoforms seem to be responsible for the activation of p38 (Figure 5B). In contrast, suppression of the ERK signaling pathway was somewhat delayed and was found to depend on a different process, that is, the MAFA-induced tyrosine phosphorylation of Dok-1. The present results demonstrated that this adaptor becomes a scaffold for multimolecular complexes composed of RasGAP, Shc, and the 5′-inositol phosphatase SHIP (Figure 3). We have previously shown that SHIP's SH2 domain binds with a high affinity to MAFA's tyrosine phosphorylated ITIM (Xu et al, 2001), suggesting that it thereby mediates the recruitment of the (Shc–SHIP–Dok–RasGAP) complex to the plasma membrane. There, RasGAP downregulates the Ras-induced Raf-1/MEK/ERK signaling pathway by decreasing the RasGTP levels. This may eventually lead to a decline in gene transcription and in synthesis of cytokines regulated by Erk-1/2. Moreover, the regulatory role of Dok-1 in this process is supported by the results of experiments using cells overexpressing Dok-1. These exhibited lower Erk-1/2 activation upon FcɛRI–MAFA co-clustering compared to wild-type cells (Figure 4C). A similar involvement of Dok-1 was recently reported in the FcγRIIB-mediated inhibition of FcɛRI-induced signaling (Ott et al, 2002; Kepley et al, 2004). Taken together, these results suggest that MAFA-mediated inhibition of Erk signaling is another distinct mechanism that can partly account for the observed selective suppression of mediators' synthesis.
In contrast to MAFA's interference with the above FcɛRI-induced coupling processes, MAFA was not found to affect (or only to marginally affect) pathways dependent on the activities of PKB and Jnk-1/2 (Figure 5A). Both these pathways were recently shown to depend on tyrosine phosphorylation of the adaptor Gab2 by the PTK Fyn (Parravicini et al, 2002). Gab2 phosphorylation is essential for the recruitment and activation of phosphatidyl inositol-3 kinase (PI3K), which generates PI-3,4-P2, a second messenger essential for the recruitment and activation of PKB (Franke et al, 1997). Studies using either Fyn or Gab2 null mast cells demonstrated that these deficiencies abolished the activation of PKB and considerably suppressed that of Jnk-1/2, while they had no (or only marginal) effect on Erk-1/2 and p38 activation respectively (Parravicini et al, 2002; Gu et al, 2003). This agrees with our results demonstrating that FcɛRI-induced Gab2 tyrosine phosphorylation is also unaffected by MAFA (co)-clustering (Figure 2A and B). Our findings therefore suggest that MAFA does not interfere with the FcɛRI-induced Fyn–Gab2–PI3K signaling path, which is essential for PKB and central for Jnk pathway(s) activation. These two pathways were reported to control several transcription factors including members of AP-1 family (Kitaura et al, 2000).
AP-1 is a hetero- or homodimeric complex composed of members of the Fos and/or Jun families of transcription factors (Macian et al, 2001). Jun proteins are activated upon phosphorylation by S/T kinases (e.g. Jnk-1/2, Erk-1/2), while the activity of Fos family members is regulated by a rapid induction of their de novo synthesis. Both the increased synthesis of Fos and phosphorylation of Jun lead to the full activation of AP-1 complex. We found that MAFA does not reduce the FcɛRI-induced mRNA levels of c-Fos, FosB, Fra-1, Fra-2, JunB, and c-Jun (Figure 5E), nor does it suppress c-Jun phosphorylation. Moreover, the results of the luciferase assay of c-Fos transcriptional activity confirmed that MAFA–FcɛRI co-clustering does not suppress, but rather slightly increase its activity compared to that caused by FcɛRI clustering (Figure 5F). These results therefore suggest that MAFA's failure to inhibit transcription of certain cytokines (e.g. IL-3, IL-5, IL-13, or TNF-α) may partly be due to its inability to suppress the de novo synthesis/phosphorylation of certain AP-1 components. This is further supported by a body of evidence demonstrating that in FcɛRI-stimulated mast cells AP-1 is essential for transcription of most of the genes that are unaffected by MAFA (i.e. TNF-α, IL-3 or IL-5 and IL-13) (Macian et al, 2001; Masuda et al, 2004).
Thus, inhibitory receptors, as exemplified by MAFA, may not cause a general suppression of the coupling networks induced by their activating counterparts but rather exert a selective regulation (Figure 6). Such a combinatorial process provides the required tight and specific control of gene expression resulting in a selective inhibition of mediators' production, which is essential for orchestrating and achieving the required physiological, primarily immune response.
Figure 6.
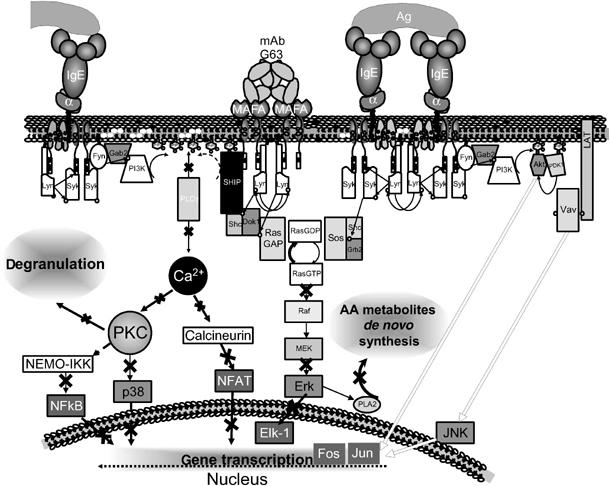
The proposed tentative mechanism underlying MAFA-mediated selective inhibition of FcɛRI-induced cytokine gene transcription. MAFA clustering leads to a rapid tyrosine phosphorylation of its ITIM sequence, probably by the PTK Lyn, which is associated with MAFA even in the unperturbed cells. Once phosphorylated, the ITIM directly binds several SH2 domain-containing molecules, such as SHIP and Lyn itself. When recruited to the proximity of the plasma membrane, SHIP dephosphorylates PI-3,4,5-P3 and PI-4,5-P2 to PI(3,4)P2 and PI-4-P, respectively, and thereby interferes with the FcɛRI-induced recruitment and activation of PLCγ via PI-3,4,5-P3, as well as with the production of IP3 and DAG from the PLCγ substrate PI-4,5-P2. This in turn leads to inhibition of downstream coupling events including the transient rise of [Ca2+]i and the subsequent activation of PKC, p38 MAPK, NF-κB, and NFAT, which are all involved in the regulation of cytokine gene transcription. In parallel, phosphorylated SHIP recruits the adaptor molecule Dok-1, which then binds the Ras GTPase-activating protein (RasGAP). RasGAP is a negative regulator of the Ras signaling pathway and its recruitment to the plasma membrane inhibits activation of Erk-1/2 signaling and the cytokine gene transcription regulated by this pathway. In contrast, MAFA clustering (or co-clustering) was found to only marginally affect tyrosine phosphorylation of several key coupling elements such as PTK Syk and adaptor proteins LAT and Gab2. As a result, the PKB and Jnk signaling and the subsequent activation of AP-1 components (Jun and Fos) stay essentially unaffected, leading to unaltered transcription of several downstream inflammatory genes. The white arrows indicate signaling pathways unaffected by MAFA clustering; black strikes on black arrows indicate MAFA-mediated suppression.
As our efforts toward identifying the physiological MAFA ligand have failed so far, the (co)-clustering of MAFA and FcɛRI has been induced by the MAFA-specific mAb. Use of mAbs as substitutes for natural ligands is common, although it does not allow an unambiguous extrapolation to the physiological case. The increasing insights now available for other ITIM-containing receptors, where both specific mAbs and natural ligands were used in parallel, however indicate that both types of reagents yield qualitatively similar responses (Katz, 2002). Although this may rationalize the use of mAb G63 in the above studies, we do continue our efforts toward identifying MAFA's ligand and hope to eventually use it in probing the validity of the above conclusions.
Materials and methods
Reagents and antibodies
See Supplementary data 4.
Cell culture and stimulation
The rat mucosal-type mast cells (RBL-2H3 line) were cultured as described by Xu et al, 2001; Supplementary data 4).
FcɛRI clustering: RBL-2H3 cells were incubated with either DNP11-BSA (10–30 ng/ml), or F(ab′)2 of mouse polyclonal nonspecific IgG derivatized with an average of three DNP (IgG-DNP3; 1 nM); MAFA/FcɛRI clustering: cells were first preincubated with 50 nM MAFA-specific mAb G63-F(ab′)2 for an indicated time and then treated with DNP11-BSA (10–30 ng/ml); MAFA–FcɛRI co-clustering: MAFA co-clustering with FcɛRI was carried out by means of F(ab′)2 of mAb G63 derivatized with an average of three DNP (G63-DNP3; 1 nM); MAFA (co)-clustering: refers to all experiments where a set of RBL-2H3 cells was subjected to MAFA/FcɛRI clustering and another set to MAFA–FcɛRI co-clustering.
Immunoprecipitation and immunoblotting
This was carried out according to a protocol described elsewhere (Xu et al, 2001; Supplementary data 4).
Affinity isolation by ITIM peptides
MAFA ITIM peptides are described elsewhere (Xu et al, 2001; Supplementary data 4).
Generation of RBL-2H3 cells overexpressing Dok-1 variants
This was carried out by retrovirus infection as described elsewhere (Abramson et al, 2003).
Cytokine and leukotriene assays
RBL-2H3 cells were seeded into 96-well plates (1 × 105 cells/well) and incubated with various concentrations of either IgG-DNP3/G63-DNP3 or BSA-DNP11 (with or without 5 min pretreatment with 50 nM G63 F(ab′)2) for 30 min (LTC4), 2 h (MCP-1), 12 h (TNF-α), or 24 h (IL-4) (at 37°C in DMEM). Their supernatants (100 μl) were then transferred into new 96-well plates and the levels of secreted mediators were assayed by ELISA kits according to the manufacturers' instructions. For the cytokine array analysis, RBL-2H3 cells were seeded into 96-well plates (5 × 105 cells/well) and incubated with 0.5 nM IgG-DNP3 or G63-DNP3 for 15 and 24 h. Supernatants (100 μl) were then transferred onto cytokine array membranes and assayed according to the manufacturer's instructions. Intensities of individual dots on the array membranes were determined by using Scion Image software (Scion Corporations, Frederick, MD, USA).
RNA extraction and RT–PCR
IgE-primed RBL-2H3 cells (10 × 106) were first cultured in tissue dishes of 10 cm diameter and then either treated for 3 h with BSA-DNP11 (30 ng/ml) or G63 (50 nM)+BSA-DNP11 (30 ng/ml) or left untreated as controls. In addition, cells were treated for 2 h with 1 nM (F(ab′)2) IgG-DNP3 (inducing FcɛRI–IgE clustering) or with 1 nM (F(ab′)2) G63-DNP3 (inducing FcɛRI–MAFA co-clustering). Total cellular RNA was isolated from adherent monolayers and used for single-strand cDNA synthesis (each sample contained 2 μg of total RNA extract in 20 μl of reaction mixture; after reaction, the volumes were adjusted to 100 μl). PCR was performed in a total volume of 50 μl, containing 5 μl of RT product. The amplification reaction was carried out for 38 cycles (94°C for 40 s, 60°C for 40 s, 72°C for 50 s) in a Perkin-Elmer Cetus Thermal Cycler. PCR products were resolved on agarose gels and stained with ethidium bromide. Real-time PCR was carried out by using specific primers, real-time master mix, and a Light Cycler (Roche, Mannheim, Germany) apparatus under the following conditions: initial activation at 95°C for 15 min and then followed by 35–55 cycles of 94°C/15 s, 60°C/20 s, and 72°C/20 s. A list of primers specific for individual rat chemokines is given in Supplementary data 1.
[Ca2+]i measurements
RBL-2H3 cells (1 × 106/sample) were harvested and incubated with A2IgE (200 ng/ml) for 1 h at 37°C, washed and resuspended in 500 μl of DMEM (containing 5% FCS), and then transferred into round-bottom FACS tubes. To each sample, 5 μl of Fluo3 solution (1 μg Fluo3+2 ng pluronic acid per 1 μl DMSO) was added, followed by 30 min incubation at 37°C. Cell samples were then transferred into 14 ml polystyrene round-bottom tubes, and after the addition of 10 ml of DMEM, incubated for an additional 30 min. Samples were centrifuged, resuspended in DMEM, and transferred into FACS tubes (final concentration 0.5 × 106 cells/ml). [Ca2+]i was measured at 37°C by flow cytometry using Facscan flow cytometer. Samples were first run for 20 s to set up the baseline, acquisition was then paused, and samples were stimulated by the addition of 10 μl of stimulating reagent (final concentrations: IgG-DNP3/G63-DNP3, 0.1 nM; BSA-DNP11, 0.1 ng/ml; mAb G63, 50 nM). Tubes were mixed and acquisition continued for a total of 200 s and was analyzed with the CellQuest software (Becton-Dickinson, San Jose, CA).
Nuclear translocation assay
RBL-2H3 cells (1 × 107) were treated either with 1 nM IgG-DNP3 or G63-DNP3, washed with ice-cold PBS, and lysed with 0.3 ml buffer A (10 mM HEPES, pH 7.5, 1.5 mM MgCl2, 1 mM DTT, 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 0.05% Nonidet P-40). Samples were incubated for 20 min on ice. The lysates were centrifuged at 3000 g for 5 min and the pellets were resuspended with 150 μl of nuclear extract buffer (300 mM HEPES, pH 7.5, 450 mM NaCl, 12 mM MgCl2, 0.5 mM EDTA, 6 mM DTT, 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 25% glycerol). The suspension was incubated for 30 min on ice. After centrifugation (14 000 g, 15 min), the supernatants were collected and equal protein amounts were separated by SDS–PAGE. WB was carried out as described above using antibodies specific to tot-Erk-1/2, NF-κB, and laminin.
Cell transfection and luciferase assay
Parental RBL-2H3 (5 × 106 cells in 0.5 ml) or their mutants overexpressing NFAT-1 were transiently cotransfected with 5 μg of either pNFAT-Luc, c-Fos-Luc, or 4xAP-1-Luc reporter plasmids and 0.5 μg of pRL-CMV Renilla plasmid by electroporation (in DMEM containing 20 mM PIPES, 128 mM potassium glutamate, 10 μM calcium acetate, and 2 mM magnesium acetate; final pH 7) using Bio-Rad Gene Pulsar (Hercules, CA) set at 400 V and 960 μF. The following day, cells were stimulated with 0.5 nM of either IgG-DNP3 or G63-DNP3 for 6 h. Reactions were stopped by washing cells with ice-cold PBS and the addition of 150 μl of passive lysis buffer. Aliquots (20 μl) of lysates were transferred into black 96-well plate and assayed for luciferase/Renilla activities using the luciferase and Renilla reporter assay systems and Luminoskan instrument and Ascent Software (ThermoLabsystems, Helsinki, Finland). Luciferase activities were then normalized to Renilla luciferase activity. Statistical analysis of groups treated with IgG-DNP3 and G63-DNP3 F(ab)′2 fragments was carried out by t-test (results with P<0.05 were considered statistically significant).
Supplementary Material
Supplementary information 1
Supplementary information 2
Supplementary information 3
Supplementary information 4
Acknowledgments
We are deeply indebted to Mr D Medgyesi, Ms J Coppens and Ms A Barbu for their helpful comments and discussions.
References
- Abramson J, Rozenblum G, Pecht I (2003) Dok protein family members are involved in signaling mediated by the type 1 Fc ɛ receptor. Eur J Immunol 33: 85–91 [DOI] [PubMed] [Google Scholar]
- Abramson J, Xu R, Pecht I (2002) An unusual inhibitory receptor-the mast cell function-associated antigen (MAFA). Mol Immunol 38: 1307. [DOI] [PubMed] [Google Scholar]
- Escoubet-Lozach L, Glass CK, Wasserman SI (2002) The role of transcription factors in allergic inflammation. J Allergy Clin Immunol 110: 553–564 [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275: 665–668 [DOI] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65–77 [DOI] [PubMed] [Google Scholar]
- Fukamachi H, Takei M, Kawakami T (1993) Activation of multiple protein kinases including a MAP kinase upon Fc ɛ RI cross-linking. Int Arch Allergy Immunol 102: 15–25 [DOI] [PubMed] [Google Scholar]
- Galli SJ (2000) Mast cells and basophils. Curr Opin Hematol 7: 32–39 [DOI] [PubMed] [Google Scholar]
- Gu HH, Botelho RJ, Yu M, Grinstein S, Neel BG (2003) Critical role for scaffolding adapter Gab2 in Fc γ R-mediated phagocytosis. J Cell Biol 161: 1151–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, Kurosaki T (1998) Involvement of guanosine triphosphatases and phospholipase C-γ 2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J Exp Med 188: 1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata D, Kitaura J, Hartman SE, Kawakami Y, Yokota T, Kawakami T (1998) Bruton's tyrosine kinase-mediated interleukin-2 gene activation in mast cells—dependence on the c-Jun N-terminal kinase activation pathway. J Biol Chem 273: 10979–10987 [DOI] [PubMed] [Google Scholar]
- Hutchinson LE, Mccloskey MA (1995) Fc-epsilon-Ri-mediated induction of nuclear factor of activated T-cells. J Biol Chem 270: 16333–16338 [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911–1912 [DOI] [PubMed] [Google Scholar]
- Kalesnikoff J, Baur N, Leitges M, Hughes MR, Damen JE, Huber M, Krystal G (2002) SHIP negatively regulates IgE+antigen-induced IL-6 production in mast cells by inhibiting NF-κB activity. J Immunol 168: 4737–4746 [DOI] [PubMed] [Google Scholar]
- Karin M (1995) The Regulation of Ap-1 Activity by Mitogen-Activated Protein-Kinases. J Biol Chem 270: 16483–16486 [DOI] [PubMed] [Google Scholar]
- Katz HR (2002) Inhibitory receptors and allergy. Curr Opin Immunol 14: 698–704 [DOI] [PubMed] [Google Scholar]
- Kepley CL, Taghavi S, Mackay G, Zhu DC, Morel PA, Zhang K, Ryan JJ, Satin LS, Zhang M, Pandolfi PP, Saxon A (2004) Co-aggregation of Fc γ RII with Fc ɛ RI on human mast cells inhibits antigen-induced secretion and involves SHIP–Grb2–Dok complexes. J Biol Chem 279: 35139–35149 [DOI] [PubMed] [Google Scholar]
- Kitaura J, Asai K, Maeda-Yamamoto M, Kawakami Y, Kikkawa U, Kawakami T (2000) Akt-dependent cytokine production in mast cells. J Exp Med 192: 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebman DA, Coffman RL (1988) Interleukin-4 causes isotype switching to IgE in T-cell-stimulated clonal B-cell cultures. J Exp Med 168: 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard EJ (1990) Nap-1 (Il-8). Immunol Today 11: 223–224 [DOI] [PubMed] [Google Scholar]
- Licht A, Pecht I, Schweitzer-Stenner R (2005) Regulation of mast cells' secretory response by co-clustering the Type 1 Fcɛ receptor with the mast cell function-associated antigen. Eur J Immunol 35: 1621–1633 [DOI] [PubMed] [Google Scholar]
- Macian F, Lopez-Rodriguez C, Rao A (2001) Partners in transcription: NFAT and AP-1. Oncogene 20: 2476–2489 [DOI] [PubMed] [Google Scholar]
- Malaviya R, Abraham SN (2001) Mast cell modulation of immune responses to bacteria. Immunol Rev 179: 16–24 [DOI] [PubMed] [Google Scholar]
- Marquardt DL, Walker LL (2000) Dependence of mast cell IgE-mediated cytokine production on nuclear factor-kappa B activity. J Allergy Clin Immunol 105: 500–505 [DOI] [PubMed] [Google Scholar]
- Masuda A, Yoshikai Y, Kume H, Matsuguchi T (2004) The interaction between GATA proteins and activator protein-1 promotes the transcription of IL-13 in mast cells. J Immunol 173: 5564–5573 [DOI] [PubMed] [Google Scholar]
- McLachlan JB, Hart JP, Pizzo SV, Shelburne CP, Staats HF, Gunn MD, Abraham SN (2003) Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol 4: 1199–1205 [DOI] [PubMed] [Google Scholar]
- Nemorin JG, Laporte P, Berube G, Duplay P (2001) p62dok negatively regulates CD2 signaling in Jurkat cells. J Immunol 166: 4408–4415 [DOI] [PubMed] [Google Scholar]
- Ortega SE, Pecht I (1988) A monoclonal antibody that inhibits secretion from rat basophilic leukemia cells and binds to a novel membrane component. J Immunol 141: 4324–4332 [PubMed] [Google Scholar]
- Ott VL, Tamir I, Niki M, Pandolfi PP, Cambier JC (2002) Downstream of kinase, p62(dok), is a mediator of Fc gamma RIIB inhibition of Fc ɛ RI signaling. J Immunol 168: 4430–4439 [DOI] [PubMed] [Google Scholar]
- Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalez-Espinosa C, Furumoto Y, Saitoh S, Samelson LE, O'Shea JJ, Rivera J (2002) Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat Immunol 3: 741–748 [DOI] [PubMed] [Google Scholar]
- Peng Y, Power MR, Li B, Lin TJ (2005) Inhibition of IKK down-regulates antigen+IgE-induced TNF production by mast cells: a role for the IKK-I{κ}B–NF-{κ}B pathway in IgE-dependent mast cell activation. J Leukoc Biol 77: 975–983 [DOI] [PubMed] [Google Scholar]
- Ramadori G, Sipe JD, Dinarello CA, Mizel SB, Colten HR (1985) Pretranslational modulation of acute phase hepatic protein-synthesis by murine recombinant interleukin-1 (Il-1) and purified human Il-1. J Exp Med 162: 930–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetch JV, Lanier LL (2000) Immune inhibitory receptors. Science 290: 84–89 [DOI] [PubMed] [Google Scholar]
- Sayama K, Diehn M, Matsuda K, Lunderius C, Tsai M, Tam SY, Botstein D, Brown PO, Galli SJ (2002) Transcriptional response of human mast cells stimulated via the Fc(ɛ)RI and identification of mast cells as a source of IL-11. BMC Immunol 3: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G (2004) The role of human mast cell-derived cytokines in eosinophil biology. J Interferon Cytokine Res 24: 271–281 [DOI] [PubMed] [Google Scholar]
- Turner H, Kinet JP (1999) Signalling through the high-affinity IgE receptor FcɛRI. Nature 402: 24–30 [DOI] [PubMed] [Google Scholar]
- Xu R, Abramson J, Fridkin M, Pecht I (2001) SH2 domain-containing inositol polyphosphate 5′-phosphatase is the main mediator of the inhibitory action of the mast cell function-associated antigen. J Immunol 167: 6394–6402 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information 1
Supplementary information 2
Supplementary information 3
Supplementary information 4