

Abstract
The cholesterol-synthesizing enzyme seladin-1, encoded by the Dhcr24 gene, is a flavin adenine dinucleotide-dependent oxidoreductase and regulates responses to oncogenic and oxidative stimuli. It has a role in neuroprotection and is downregulated in affected neurons in Alzheimer's disease (AD). Here we show that seladin-1-deficient mouse brains had reduced levels of cholesterol and disorganized cholesterol-rich detergent-resistant membrane domains (DRMs). This was associated with inefficient plasminogen binding and plasmin activation, the displacement of β-secretase (BACE) from DRMs to APP-containing membrane fractions, increased β-cleavage of APP and high levels of Aβ peptides. In contrast, overexpression of seladin-1 increased both cholesterol and the recruitment of DRM components into DRM fractions, induced plasmin activation and reduced both BACE processing of APP and Aβ formation. These results establish a role of seladin-1 in the formation of DRMs and suggest that seladin-1-dependent cholesterol synthesis is involved in lowering Aβ levels. Pharmacological enhancement of seladin-1 activity may be a novel Aβ-lowering approach for the treatment of AD.
Keywords: Aβ, Alzheimer, cholesterol, neurodegeneration, therapy
Introduction
Accumulation of amyloid-β peptides (Aβ) in the CNS is an invariant feature of the pathology of Alzheimer's disease (AD), the most common form of dementia. Aβ peptides are derived from proteolytic cleavage of the amyloid precursor protein (APP), with the β-secretase (BACE) cleaving at the N-terminus and γ-secretase at the C-terminus of Aβ peptides. Genetic studies of familial AD cases have led to the identification of alterations in genes associated with the disease that result in increased production of Aβ peptide or to C-terminal extended forms that aggregate more readily (Capell et al, 1998; Hardy and Selkoe, 2002). These observations, together with studies on Aβ toxicity (Small et al, 2001), support the view of a key role of Aβ in AD pathophysiology (Huang et al, 1999; Chen et al, 2000; Janus et al, 2000; Morgan et al, 2000; Mucke et al, 2000; Small et al, 2001; Casas et al, 2004; Schmitz et al, 2004). Thus, concentrated effort has been focused on the identification of modifying factors (Corder et al, 1993) and regulatory mechanisms of the proteases that control APP cleavage and are involved in Aβ production, as well as on proteases with the capacity to degrade the peptide (Selkoe, 2001).
Seladin-1, encoded by a single gene (Dhcr24) on chromosome 1, is an evolutionarily conserved gene with homologies to a family of flavin adenine dinucleotide-dependent oxidoreductases (Greeve et al, 2000). It catalyzes the reduction of the Δ24 double bond of sterol intermediates of cholesterol metabolic pathway (Waterham et al, 2001). As an exemplar, seladin-1 reduces the Δ24 double bond of desmosterol, the immediate precursor of cholesterol, to form cholesterol (Waterham et al, 2001). Functional deficiency in the Dhcr24 gene causes, in humans, desmosterolosis, a severe autosomal recessive disorder characterized by gross developmental abnormalities and elevated desmosterol levels in plasma (Waterham et al, 2001). Moreover, seladin-1 is a key regulator of Ras-induced senescence, and cellular responses to oncogenic and oxidative stimuli (Wu et al, 2004). Greeve et al (2000) have previously shown the protective activity of seladin-1 against oxidative and Aβ-mediated toxicity and that seladin-1 levels are lower in affected neurons of AD brain.
The cholesterol synthesizing activity of seladin-1 suggests a possible role in the formation of the cholesterol-rich detergent-resistant membrane domains (DRMs or rafts), but this activity has not been studied to date. DRMs are operationally defined as membrane domains resistant to solubilization by nonionic detergent at 4°C (Ciana et al, 2005). The in vivo counterpart of the DRMs is under intense investigation. Experimental evidence has revealed the disorganization of DRMs in AD brains, possibly because of low cholesterol content (Ledesma et al, 2003a). In a recent study, it has been described that DRMs participate in the segregation of APP from BACE, therefore reducing APP β-cleavage and Aβ production in cultured primary neurons and CHO cells, respectively (Abad-Rodriguez et al, 2004). Furthermore, disruption of DRMs results in diminished activity of the Aβ-degrading enzyme plasmin, because of decreased membrane plasminogen binding to the plasma membrane (Ledesma et al, 2000, 2003a, 2003b). Altogether, these data support a possible link between AD and brain cholesterol loss. In contrast, the use of statins to reduce cholesterol led to the reduction of Aβ production, favoring the view of high cholesterol as a risk factor for AD (Simons et al, 1998; Fassbender et al, 2001).
To determine the role of seladin-1 in APP processing and Aβ production, we analyzed seladin-1-deficient mice and we overexpressed seladin-1 in cultured human neuroblastoma cells. We provide here evidence for a key role of seladin-1 in the regulation of brain cholesterol levels, DRM formation, plasmin activation, APP processing and Aβ levels in vivo and in vitro.
Results
Seladin-1 influences the membrane and cellular cholesterol levels in mouse brains and in cultured human neuroblastoma cells
To determine to which extent the levels of brain cholesterol depend on seladin-1, we first analyzed the effects of genetic seladin-1 depletion on steady-state cholesterol levels in vivo. The analysis of seladin-1 mRNA levels in the brain of wild-type mice, heterozygous mice with depletion of one (heterozygous) or knockout mice with depletion of both (homozygous) seladin-1 alleles (Wechsler et al, 2003) revealed a gene-dose-dependent reduction in the heterozygous mice compared to their wild-type littermates (43.8±3.3%, P=0.009). There was no seladin-1 mRNA expression detectable in the homozygous mice (Figure 1A). The brains of seladin-1 heterozygous mice exhibited an average reduction of 29% in membrane (P=0.02; Figure 1B) and 15% in total cellular cholesterol (P=0.042; Figure 1C) compared to wild-type brains. Furthermore, cholesterol amount was undetectable in the homozygous mouse brains (Figure 1B and C). To characterize the effect of seladin-1 expression on cholesterol metabolism, we analyzed the effects of seladin-1 depletion on the levels of the cholesterol precursor, desmosterol, and of the major degradation product of cholesterol in the brain, 24OH-cholesterol. Desmosterol was 5.7-fold (P=0.012) and 50-fold (P=0.00006) increased in heterozygous and homozygous mouse brains, respectively (Figure 1D), indicating that desmosterol accumulates upon seladin-1 deficiency. Moreover, decreased seladin-1 expression led to a significant reduction in the catabolic product 24OH-cholesterol in heterozygous (24.6±4.3%, P=0.017) and knockout (97.6±0.47%, P=0.0004) compared to control brains. Altogether, these results unveil for the first time the key role of seladin-1 in the regulation of brain cholesterol metabolism. Consistent with an essential role of cholesterol in vertebrate cell viability, homozygous mice did not survive beyond the first month.
Figure 1.
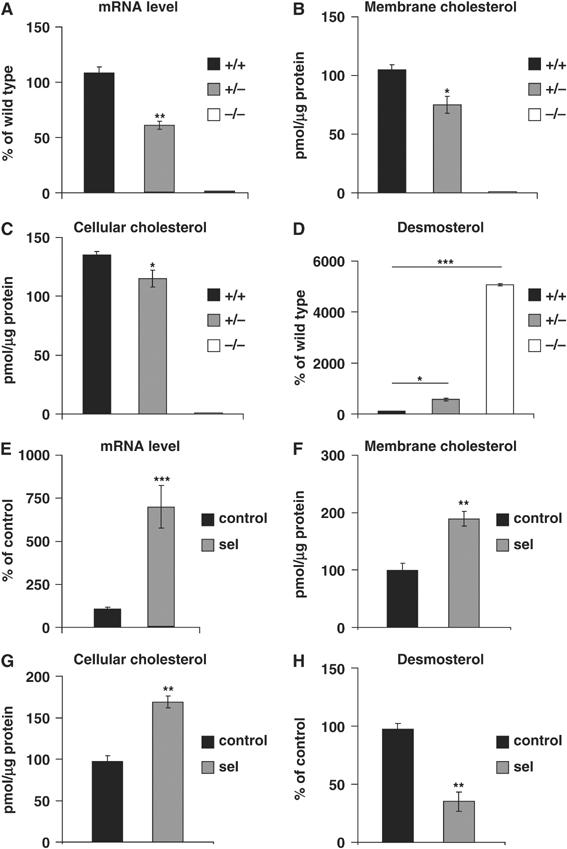
The level of cholesterol is dependent on seladin-1 expression in vivo and in vitro. Seladin-1 mRNA expression level was 44% lower in heterozygous (+/−) compared to wild-type mouse brains (+/+), whereas no seladin-1 mRNA was detectable in the brains of seladin-1 knockout mice (−/−) (A). In brain extracts of seladin-1 heterozygous (+/−) mice, membrane cholesterol (B) and cellular cholesterol (C) were significantly reduced by 29 and 15%, respectively, compared to wild-type (+/+) littermates. In knockout (−/−) mice, membrane and cellular cholesterol were undetectable (B, C). Desmosterol was 5.7- and 50-fold increased in heterozygous (+/−) and in knockout (−/−) brains, respectively, when compared to wild-type littermates (D). In seladin-1-overexpressing SH-SY5Y human neuroblastoma cells, seladin-1 mRNA expression level was 680% higher compared to control cells (E), and membrane cholesterol (F) and cellular cholesterol (G) were significantly increased by 1.9- and 1.7-fold, respectively. Desmosterol was significantly lower (two-fold) in seladin-1-overexpressing cells compared to control cultures (H). Values in panels A–D and E–H are expressed as a percentage change in the corresponding values of wild-type mouse brains or control cells, respectively, that were considered as 100%. The graphs show the average and standard error from three different mouse brains for each condition, and from three independent seladin-1-overexpressing and control SH-SY5Y cultures. Statistical significance is indicated by asterisks: *P<0.05, **P<0.009, ***P<0.0002.
Having demonstrated the consequences of seladin-1 deficiency in cholesterol metabolism, we next tested whether increasing seladin-1 levels would cause the opposite effects. To this aim, we generated human neuroblastoma SH-SY5Y cells constitutively expressing seladin-1. Analysis of seladin-1 mRNA revealed a significant increase (684±121%, P=0.0001) in the seladin-1-overexpressing cells (Figure 1E). Significant increases of 1.9-fold in membrane cholesterol (P=0.003; Figure 1F) and 1.7-fold in total cellular cholesterol (P=0.001; Figure 1G) were found in seladin-1-overexpressing cells when compared to the control cultures, confirming the bioactivity of the transgene. Consistent with the in vivo data, desmosterol levels were dramatically reduced in cells overexpressing seladin-1 (43±5.2%, P=0.001) (Figure 1H), whereas 24OH-cholesterol amounts were increased (172±2.3%, P=0.00001) when compared to control cultures.
Seladin-1 contributes to the specific recruitment of DRM proteins and lipids into DRMs
Given the above results and that cholesterol is a main component of DRMs and affects DRM functions (Simons et al, 1998; Simons and Toomre, 2000), we first tested whether changes in seladin-1 expression alter these membrane domains in vivo. Therefore, DRM protein and lipid composition was analyzed in the brains of wild-type and seladin-1 heterozygous mice that, in contrast to the seladin-1 homozygous mice, develop normally and survive to adulthood without major health problems but present a moderate, still significant, reduction of cholesterol (see above). DRMs were isolated upon cold-detergent extraction of total brain extracts and gradient centrifugation. In the seladin-1 heterozygous brains, significantly lower protein levels were found in the light fractions 4–6 of the gradient, which correspond to detergent-resistant membranes (Figure 2A). To determine whether this reduction of protein content was the consequence of general or DRM-specific protein loss, the flotation profiles of the DRM markers flotillin 1 and the cellular prion protein (PrPc) as well as of the non-DRM marker transferrin receptor (TfR) were analyzed. In agreement with a DRM-specific shortage, reduction of brain seladin-1 levels resulted in the displacement of DRM markers, flotillin 1 (Figure 2B and C) and PrPc (Figure 2D and E), away from the DRM fractions. Thus, while in wild-type mice 20.2±1.45% of total protein, 12.3±1.21% of total flotillin 1 and 40.5±4.68% of total PrPc were present in DRM fractions 4–6, these percentages decreased to 13.8±1.56, 5.1±2.19 and 12.5±5.32%, respectively, in seladin-1 heterozygous mouse brains. All the differences found were statistically significant (P<0.03). In contrast, no change was observed in the flotation profile of the non-DRM protein TfR (Figure 2F and G).
Figure 2.
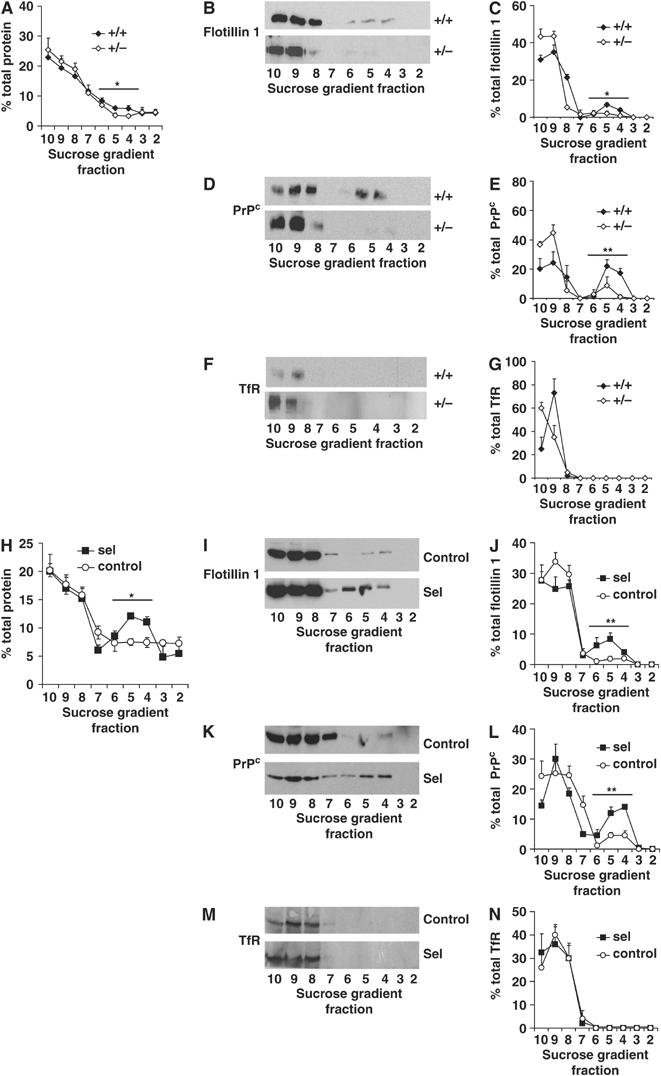
Seladin-1 modulates DRM protein composition. Homogenates from wild-type (+/+) and seladin-1 heterozygous (+/−) mouse brains and from control and seladin-1-overexpressing SH-SY5Y cells were extracted and centrifuged in a sucrose gradient. Fractions were collected and numbered from the lightest to the heaviest (2 to 10). The amount of protein in each gradient fraction is expressed as a percentage of the total protein along the gradient (A, H). Representative Western blots of the gradient fractions using antibodies against the DRM marker proteins flotillin 1 (B, I) and PrPc (D, K), and the non-DRM protein TfR (F, M) are shown. Graphs on the right show the distribution of flotillin 1 (C, J), PrPc (E, L) and TfR (G, N) in each fraction as a percentage of the total amount of the respective proteins along the entire gradient. Seladin-1 deficiency in mouse brains led to a significant decrease of the protein amount in fractions 4–6 corresponding to DRMs (A) and resulted in significantly lower amounts of DRM markers in these fractions (B–E), whereas the distribution of TfR did not differ between wild-type (+/+) and heterozygous (+/−) mouse brains (F, G). In contrast, overexpression of seladin-1 in SH-SY5Y cells led to an increased amount of protein in fractions 4–6 compared to control cells (H) and to significantly higher levels of the DRM markers flotillin 1 (I, J) and PrPc (K, L), whereas the distribution of the non-DRM protein TfR remained unchanged (M, N). The graphs show the average and standard error from three different mouse brains for each condition and from three independent seladin-1-overexpressing and control SH-SY5Y cultures. Asterisks show statistical significance of the difference in the total amount of respective proteins in the DRM fractions 4–6. *P<0.02, **P<0.008.
Conversely, significantly higher protein levels were found in the light fractions 4–6 of the seladin-1-overexpressing cell extracts compared to control cells (31.6±2.02 and 22.2±2.02%, respectively, P=0.01; Figure 2H). Consistent with a recruitment of DRM-specific proteins and not to a general protein contribution, flotillin 1 (Figure 2I and J) and PrPc (Figure 2K and L) were enriched in fractions 4–6 of seladin-1-overexpressing cell gradients when compared to control cells, whereas TfR remained always in detergent-soluble heavy fractions (8–10) (Figure 2M and N). Quantitative analysis showed that, 4.8±1.15% of total flotillin 1 and 10.4±1.73% of total PrPc float to DRMs in control cells, which increased to 18.7±2.66 and 30.5±2.89%, respectively, in seladin-1-overexpressing cells (P=0.004 and 0.001).
The analysis of the distribution of several lipids in the flotation gradient fractions revealed that the presence of cholesterol, sphingomyelin and the ganglioside GM1 was also modulated by seladin-1 levels, whereas no changes in the amounts of desmosterol and total phospholipids were detected in the DRM fractions in vivo and in vitro (Supplementary Figures 1 and 2).
Altogether, these results indicate that seladin-1 is required for the specific recruitment of DRM proteins and lipids into detergent-insoluble membrane domains and that its deficiency results in DRM disorganization, evident by displacement of DRM-specific lipids and proteins.
Seladin-1 affects DRM-dependent plasminogen binding and activation
Given the influence of seladin-1 on DRM composition, we reasoned that changes in its expression would also affect DRM-dependent functions. Plasminogen binding to the membrane leads to the activation of the Aβ-degrading enzyme plasmin in DRMs (Ledesma et al, 2003a). Therefore, we first investigated whether changes in seladin-1 expression affect plasminogen binding and plasmin activity in vivo. Endogenous plasmin activity was reduced in heterozygous brains when compared to controls (Figure 3A). To analyze plasminogen binding, exogenous plasminogen was added to isolated membranes and plasmin activity was measured. Seladin-1 deficiency resulted in reduced plasmin activity, indicating a diminished ability of heterozygous seladin-1 brain membranes to bind plasminogen (Figure 3B). Accordingly, the amount of endogenous plasminogen bound to the membrane was clearly reduced in seladin-1 heterozygous mouse brains (42.9±10.9% of that in wild-type brains, P=0.005; Figure 3C and E), whereas the endogenous levels of total plasminogen were similar in all mouse brains (Figure 3D and F). Endogenous plasmin activity was also measured in membranes derived from control and seladin-1-overexpressing cells. The latter exhibited a clear increase in plasmin activity (Figure 3G). Furthermore, plasmin activity upon addition of exogenous plasminogen was also higher in seladin-1-overexpressing cells than in control cells, indicating an increased ability of seladin-1-overexpressing cell-derived membranes to bind plasminogen (Figure 3H). This was further confirmed by Western blot analysis showing that while there were no significant differences on the endogenous levels of total plasminogen in control and seladin-1-overexpressing cells (Figure 3J and L), the amount of plasminogen bound to the membrane was clearly increased in the latter (247±14.7%, P=0.004; Figure 3I and K).
Figure 3.
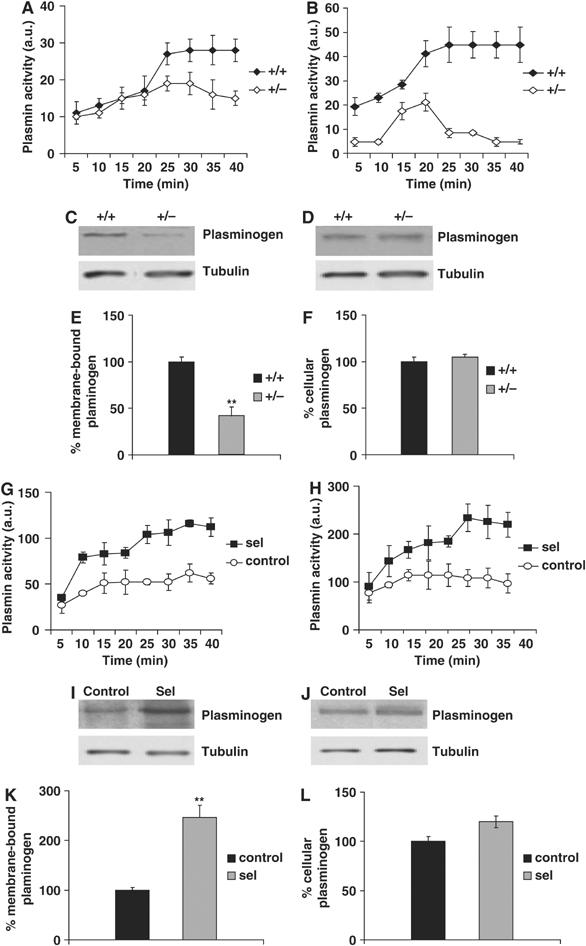
Plasminogen binding and plasmin activation depend on seladin-1 levels. Plasmin activity (expressed in arbitrary units) was measured at the indicated times in membranes isolated from wild-type (+/+) and seladin-1 heterozygous (+/−) mouse brains. The ability to bind plasminogen was monitored by measuring plasmin activity after the addition of exogenous plasminogen to the membrane fractions. Both endogenous plasmin activity (A) and the ability to bind plasminogen (B) were clearly reduced in the heterozygous mouse brains. Membrane-bound plasminogen was significantly decreased in heterozygous (+/−) brains (C, E). Endogenous levels of cellular plasminogen, however, were not altered in wild-type (+/+) and heterozygous mouse brains (D, F). The amount of tubulin is shown as a loading control in all Western blots (C, D, I, J). In contrast, seladin-1 overexpression in vitro resulted in increased endogenous plasmin activity (G). Moreover, when exogenous plasminogen was added to isolated membranes, plasmin activity was higher in membrane fractions isolated from seladin-1-overexpressing cells (H). Membrane-bound plasminogen was significantly elevated in these cells (I, K). Cellular plasminogen levels did not differ between overexpressing and control cells (J, L). Data in all the graphs correspond to mean value and standard error from three different mouse brains for each condition and from three independent seladin-1-overexpressing and control SH-SY5Y cultures. **P<0.006.
Seladin-1 alters BACE1–APP membrane compartmentalization, APP β-cleavage and Aβ generation in vitro and in vivo
DRMs segregate a major pool of the APP-β-secretase BACE1 from its non-DRM substrate APP, restricting APP β-cleavage and Aβ production in cultured primary neurons and CHO cells, respectively (Abad-Rodriguez et al, 2004). Hence, we determined whether low levels of seladin-1, leading to decreased cholesterol concentrations and DRM alterations, affect BACE1–APP membrane segregation and their functional interaction in vivo. The flotation profiles revealed that BACE1 was displaced from light fractions 4–6 to APP-containing heavy fractions in seladin-1 heterozygous mouse brains compared to wild-type brains. The percentages of total BACE1 in DRM fractions 4–6 in wild-type and heterozygous seladin-1 mice were 36.4±3.58 and 9.7±1.97%, respectively (P=0.003; Figure 4A and B). In contrast, no changes in the flotation profile of APP, as well as in its concentration, were detected between the genotypes (Figure 4C and D). To test if the changes in membrane compartmentalization had a functional consequence, we measured the levels of APP β-cleavage. The amount of the APP β-C-terminal fragment (β-CTF) was significantly increased (201±16%, P=0.04) in seladin-1 heterozygous mouse brains (Figure 4E and F and Supplementary Figure 4A).
Figure 4.
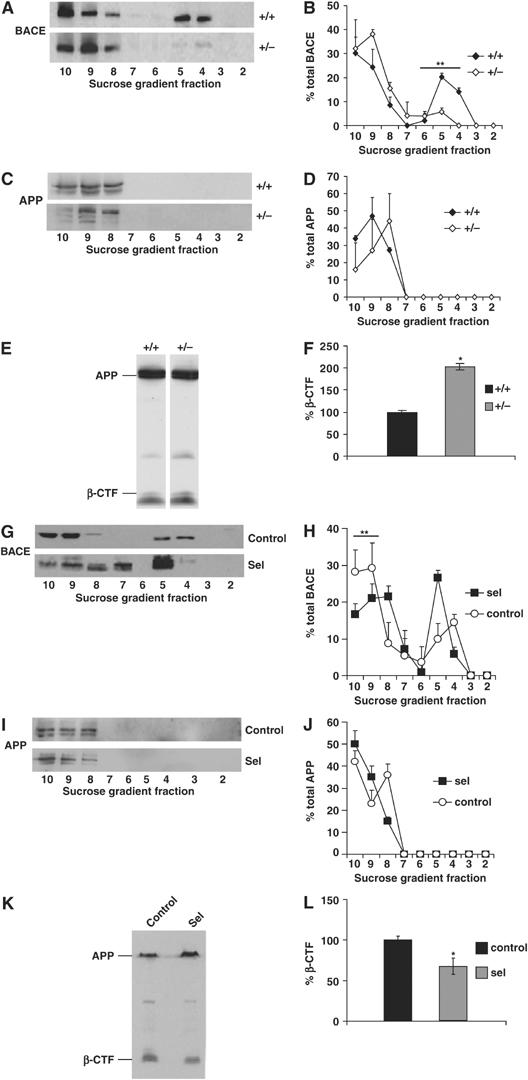
Seladin-1 alters BACE1–APP membrane segregation and APP β-cleavage. APP and BACE1 distribution along the sucrose gradient fractions of seladin-1 wild-type (+/+) and heterozygous (+/−) mouse brains (A–D) and from control cells and seladin-1-overexpressing SH-SY5Y (G–J) was analyzed by Western blot using specific antibodies. Left pictures show representative examples. Graphs on the right indicate the amount of BACE1 (B, H) and APP (D, J) in each fraction as a percentage of total BACE1 and APP, respectively. In seladin-1 heterozygous (+/−) mouse brains, BACE1 was displaced from fractions 4–6 (DRM fractions) to APP-containing heavy fractions 8–10 (A, B), whereas the APP flotation profile did not change between the groups and APP remained in the heavy fractions (C, D). In contrast, seladin-1 overexpression resulted in a significant decrease of BACE1 in the APP-containing fractions (G, H). There was no change in APP distribution in seladin-1-overexpressing compared to control cells (I, J). APP β-cleavage was analyzed by Western blot of cellular extracts containing an equal amount of protein prepared from wild-type (+/+) and heterozygous (+/−) mouse brains (E) and from control and seladin-1-overexpressing SH-SY5Y cells (K). Levels of APP β-CTF (see also Supplementary Figure 4A) were normalized to the amount of full-length APP (F, L). Seladin-1 deficiency revealed a significant two-fold increase in the amount of β-CTF in heterozygous (+/−) mouse brains (F). In concert to the in vivo data, overexpression of seladin-1 in SH-SY5Y cells led to a significant decrease (34%) of APP β-CTF (L). The ratio of β-CTF to APP in wild-type (+/+) mouse brain (F) and in control SH-SY5Y cells (L) was considered as 100%. Data in all the graphs correspond to mean value and standard error from three different mouse brains for each condition and from three independent seladin-1-overexpressing and control SH-SY5Y cultures. *P<0.05, **P<0.004.
To assess whether high levels of seladin-1 would reverse the effects observed upon its deficiency, the flotation profiles of BACE1 and APP were also analyzed in seladin-1-overexpressing and control cells. While APP always remained in the heavy fractions 9–10 (Figure 4I and J), more BACE1 was recruited in the light fractions upon seladin-1 overexpression (Figure 4G and H). Quantitative analysis revealed that 54.4±9.8% of BACE1 is in the APP-containing fractions 9–10 in the control cells, whereas the percentage significantly diminishes to 35.6±4.3% in seladin-1-overexpressing cells (P=0.04; Figure 4H and J). Consistent with the enhanced membrane segregation of BACE1 from APP, the generation of APP β-CTF was significantly reduced (34±11%, P=0.04) in seladin-1-overexpressing cells (Figure 4K and L). Taken together, the above observations reveal an important role of seladin-1 in the regulation of BACE1–APP interaction and thus in APP β-cleavage.
Finally, we determined whether these alterations affect the levels of Aβ, utilizing ELISA systems that measure specifically murine and human Aβ peptides. In concert with higher amyloidogenic APP processing, Aβ40 was significantly increased (152±16%, P=0.027; Figure 5A) in total brain extracts of heterozygous mice. Because the endogenous mouse Aβ42 levels are extremely low in young wild-type mice, as previously reported (Refolo et al, 2001), the Aβ42 measurements were below the detection level. We therefore analyzed age-matched wild-type and seladin-1 heterozygous mice cross bred to the Tg2576 SwAPP mouse line (SwAPP/seladin-1). The SwAPP mice overexpress human APP carrying the Swedish mutations under the regulatory sequence of the neuronal Prp promoter. These mutations located at the N-terminus of the Aβ peptide lead to increased β-cleavage and therefore elevated levels of Aβ40 and Aβ42 (Hsiao et al, 1995). In accordance with the murine ELISA data, Aβ40 was significantly increased in heterozygous SwAPP/seladin-1 mouse brains (270±31%, P=0.024; Figure 5B). In these mice, Aβ42 levels also revealed a significant increase compared to the SwAPP littermates (159±0.3%, P=0.015; Figure 5C), whereas the ratio of Aβ40 to Aβ42 did not significantly differ between the genotype groups (136±31%; Figure 5D), suggesting that the changes in Aβ levels were not due to a change in the γ-secretase activity. These data demonstrate that the elevation of Aβ concentrations in the seladin-1-deficient mice is caused by enhanced β-cleavage of APP in vivo.
Figure 5.
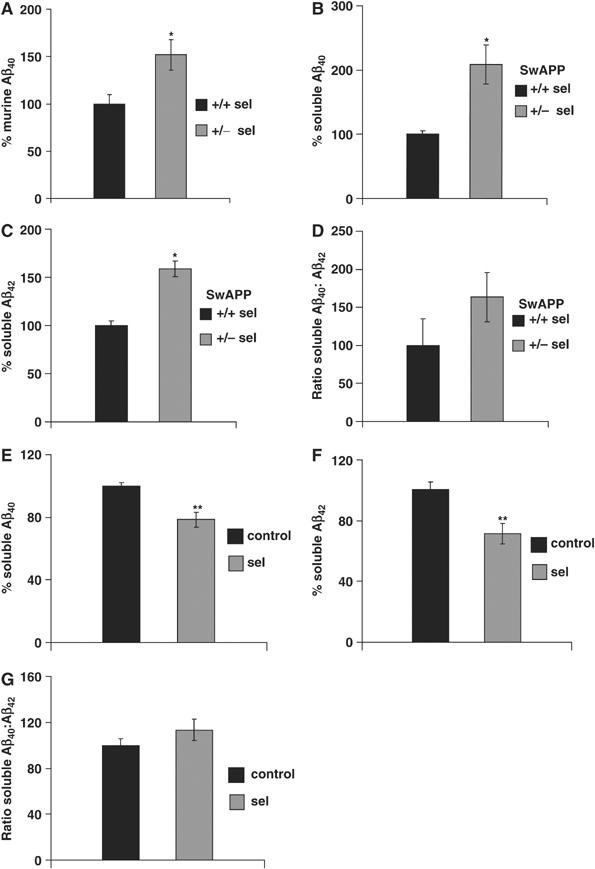
Seladin-1 affects Aβ generation in vivo and in vitro. Measurement of murine Aβ40 levels in wild-type (+/+ sel) and seladin-1 heterozygous (+/− sel) mouse brains without overexpression of the APP transgene revealed a significant increase of the murine Aβ40 peptide in the latter (n=4) (A). Aβ40 levels of wild-type mouse brains were considered as 100%. *P=0.027. Quantification of Aβ40 and Aβ42 in the brains of seladin-1 wild-type and heterozygous mice with the simultaneous overexpression of SwAPP confirmed that seladin-1 deficiency leads to higher Aβ steady-state levels. Brain Aβ40 levels showed a significant 2.7-fold increase (*P=0.024) in heterozygous (SwAPP/+/− sel) mouse brains compared to seladin-1 wild-type littermates expressing solely the SwAPP transgene (SwAPP/+/+ sel) (B). Levels of soluble Aβ42 were significantly increased by 1.6-fold (*P=0.015) in heterozygous (SwAPP/+/− sel) mouse brains (C). The ratio of Aβ40 to Aβ42 levels revealed no significant differences between the two genotypes (D). In seladin-1-overexpressing SH-SY5Y cultures, Aβ40 (E) and Aβ42 (F) were significantly decreased when compared to control cultures (n=10 for each, **P=0.001 and 0.003, for Aβ40 and Aβ42, respectively), whereas the ratio of Aβ40 to Aβ42 levels did not show significant differences between the two groups (G).
In contrast, the generation of both Aβ40 and Aβ42 was reduced to 78.5±4.8 and 71.48±6.7% (P=0.001 and 0.003, respectively; Figure 5E and F) in seladin-1-overexpressing compared to control cells, whereas the ratio of Aβ40 to Aβ42 revealed no significant difference (Figure 5G). LDH and MTT assays confirmed that there was no interference with the viability of cells in both cultures, ruling out the potential effect of the transgene expression on cell survival. In the LDH and MTT assays, 102±3.2 and 98±1.8% viable cells were found in seladin-1-overexpressing cultures, respectively, when the corresponding levels of control cells were set as 100%. Altogether, these data confirm the crucial role of seladin-1 protein and cholesterol levels in modulating Aβ generation.
Discussion
The results of this study show that cholesterol levels are modulated by modifying the expression of the cholesterol-synthesizing enzyme, seladin-1. Our approach is different from previous work that relied on pharmacological inhibition of early steps of cholesterol synthesis or on using lipid-extracting drugs in cultured cells and in vivo that has led to controversial results (Simons et al, 1998; Sparks et al, 2000, 2002; Fassbender et al, 2001; Kirsch et al, 2003; Park et al, 2003; Abad-Rodriguez et al, 2004). We show that seladin-1 deficiency results in lower membrane cholesterol levels in mouse brains and, as a consequence, in altered DRMs. This in turn contributes to reduced membrane binding of plasminogen and plasmin activation and to increased APP β-cleavage and Aβ production in vivo. Importantly, the overexpression of seladin-1 caused the opposite biochemical changes as observed for seladin-1 deficiency.
In our colony, seladin-1 homozygous mice were born to published frequency (Wechsler et al, 2003). These mice, however, were extremely weak, half of the size of littermates and most of them (90%) died before reaching the age of 3 weeks (Supplementary Figure 4). Therefore, to study the effects of a moderate cholesterol reduction, we analyzed the seladin-1 heterozygous mice that develop normally and have no problem of health, fertility or longevity. Neuropathological and immunohistochemical examinations of these mice revealed no detectable differences between wild-type and heterozygous mice concerning morphology, cellular distribution and viability of the brain cells (unpublished data). Considering that cholesterol is an essential component of vertebrate cell membranes and exhibits essential biological roles, it is striking that seladin-1 knockout mice survive beyond birth. The question arises as to which mechanism/s enable the seladin-1 knockout mice to reach embryonic maturation and postnatal development, even if short. With mice, in contrast to humans, cholesterol passes the placenta. Therefore, one reasonable answer is that circulating cholesterol, contributed from the heterozygous mother, suffices for early development. In this scenario, soon after birth seladin-1 knockout mice are compromised due to the deficiency to synthesize cholesterol. Alternatively, embryonic development and the short postnatal survival in the seladin-1 knockout mice may depend on the substitution of cholesterol by desmosterol. In agreement with this possibility, one study indicated that desmosterol could replace cholesterol without deleterious effects in fibroblasts (Rothblat et al, 1970). Although this last scenario would imply a non-essential role of cholesterol in early developmental processes, it also indicates an essential requirement of cholesterol for full maturation. Indeed, seladin-1 heterozygous mice, which show a 29% reduction of brain membrane cholesterol, exhibit alterations in DRM composition and DRM-dependent functions. Moreover, although changes in seladin-1 expression also affect the amount of desmosterol in mouse brains, the distribution of this lipid in DRMs was not altered. In addition, experimental increase of desmosterol levels in cultured cells did not affect DRM-related functions analyzed in this work, whereas moderate reduction of cholesterol changed these parameters (Supplementary Figures 2 and 3). These results strongly support our conclusion that the reduction of cholesterol but not the increase of desmosterol levels mediates the seladin-1-induced alterations in DRMs observed in vivo. Altogether, our data reveal that proper brain maturation requires the maintenance of a certain steady-state level of brain cholesterol and that a modest reduction of cholesterol in brain could have deleterious consequences.
Seladin-1 can exert antiapoptotic function, and apoptosis may lead to elevated Aβ levels in the brains of AD patients and mouse models (Gervais et al, 1999; Mohajeri et al, 2002). We found, however, no apoptotic cell death in the heterozygous and wild-type mouse brains. Moreover, the viability of seladin-1-overexpressing and control cultures did not differ at any time points. Taken together, these data provide the evidence that the effects described in this work are not a consequence of the antiapoptotic function of seladin-1.
The finding that low expression of seladin-1 paralleled a reduction in brain membrane cholesterol offers a new perspective on the role that this protein might play in AD pathology. Because seladin-1 is downregulated in vulnerable areas of brains in AD patients (Greeve et al, 2000), it is reasonable to think that low levels of seladin-1, promoted by yet to be identified causes, are responsible for the membrane cholesterol reduction found in such brains (Ledesma et al, 2003a). This would consequently lead to Aβ accumulation via a combination of inefficient Aβ degradation (due to low plasmin activity) and increased APP amyloidogenic cleavage. Iivonen et al (2002), in contrast, could not find an association between reduced seladin-1 transcription levels in AD brains and Aβ content. The fact that cholesterol levels were not measured in this study together with the low number of samples analyzed precludes drawing a definitive conclusion. Indeed, cholesterol loss has been observed in a significant number but not in all AD brains (Ledesma et al, 2003a). Altogether, these data suggest that compensatory mechanisms to the effects of seladin-1 deficiency on cholesterol levels might exist that would differ among AD patients. Moreover, alternative mechanisms (i.e. mutations in APP or presenilins) would be responsible for Aβ accumulation in certain AD cases. Further research, including large-scale studies, is required to clarify these matters.
Seladin-1 also participates in the Ras/p53 pathway by binding to p53 leading to p53 accumulation (Wu et al, 2004). Although the domain of seladin-1 involved in cholesterol synthesis is apparently not required for binding to p53, Ras signaling is modulated in a cholesterol- and DRM-dependent manner (Prior et al, 2003; Parton and Hancock, 2004). These examples suggest that altered seladin-1 levels could contribute to disease conditions through both cholesterol-dependent and -independent modulation of cell survival/death pathways.
In conclusion, our data provide the evidence for a role of seladin-1 in providing enough cholesterol to generate and maintain proper DRM composition and function (Simons and Toomre, 2000). In this regard, the alterations produced by seladin-1 deficiency, such as increased APP amyloidogenic cleavage, Aβ peptide accumulation and decreased plasmin levels, could explain both amyloid build-up and cytopathology in AD in vivo. Our study demonstrates the consequences of cholesterol loss in brain cells in vivo and provides groundwork to better understanding of the roles of cholesterol in health and disease.
Materials and methods
Cell culture
The open reading frame of human seladin-1 (KIAA0018;DHCR24) (Swiss-Prot: Q15392) was cloned in pcDNA3 expression plasmid (Invitrogen). A hemagglutinin tag was added in-frame to the C-terminus of the protein. The expression was controlled by the CMV promoter. SH-SY5Y cells were cultured in DMEM NUT-F12 medium (Invitrogen) containing 10% fetal calf serum (FCS), 5% horse serum (HS) and 5000 U/ml penicillin and 5000 μg/ml streptomycin. Transfections were performed using Lipofectamine 2000 (Invitrogen). Stably transfected cultures were established by addition of 125 μM G418 (Invitrogen) to the medium. SH-SY5Y cells stably expressing the EGFP protein under the control of the same promoter were used as controls.
Cell viability assays
Before harvesting the cells for quantification of Aβ levels, the viability of the seladin-1-overexpressing and SH-SY5Y control cultures was determined using the MTT (Sigma) and the LDH assay (Sigma) according to the manufacturers' instructions.
Mice
Heterozygous breeding pairs with target depletion of one seladin-1 allele were received from Dr E Feinstein (Quark Biotech Inc.). Seladin-1-deficient mice were bred and genotyped as previously described (Wechsler et al, 2003). In addition, seladin-1 heterozygous mice were bred to SwAPP transgenic mice overexpressing human APP harboring the Swedish double mutation (Hsiao et al, 1995) (SwAPP/seladin-1). All animal experiments and husbandry were performed compliant with national guidelines. All mice were analyzed at 3 weeks of age.
Antibodies, Western blots and quantification
Monoclonal anti-flotillin 1 (clone 18; Transduction Laboratories), monoclonal anti-PrPc POM-1 (kindly provided by Dr A Aguzzi, University of Zurich), monoclonal anti-TfR (clone CD-71; Santa Cruz Biotechnology Inc.), polyclonal anti-human plasminogen (Biogenesis), monoclonal anti-N-terminal APP (clone 22C11; Roche), polyclonal chicken anti-BACE1 (raised against Fc-Asp 2-fusion protein, kindly provided by Dr C Dingwall, GlaxoSmithKline), polyclonal anti-C-terminal APP (Sigma) and monoclonal 6E10 (Signet) antibodies were used for Western blot analysis. Monoclonal anti-tubulin (Calbiochem) was used as an internal loading control and for normalization of densitometric analysis of the immunoreactive bands. All antibodies were diluted in 5% fat-free milk in 35 mM Tris–HCl (pH 7.4) and 140 mM NaCl (TBS buffer). For Western blotting, 15% polyacrylamide–SDS gels and Novex 10–20% Tricine gels (Invitrogen) were used. Proteins were transferred to a nitrocellulose membrane (0.45 μm pore; Bio-Rad). Species-specific peroxidase-conjugated secondary antibodies and the ECL detection method (Amersham) were subsequently used. Quantification of immunoreactive bands was carried out by densitometry of the scanned autoradiograms under conditions of non-saturated signal using the NIH-image software.
Aβ measurement
Hemi-brains from 3-week-old mice with the depletion of one seladin-1 allele (heterozygous) and wild-type littermates (n=4 for each) as well as their littermates carrying the SwAPP transgene were homogenized in a buffer containing 100 mM Tris and 150 mM NaCl (pH 7.4) and proteinase inhibitors (Roche). These total extracts were analyzed by a modified sandwich ELISA that detects specifically either Aβ40 or Aβ42 (Takeda, Japan) according to the provider's protocol. Aβ was captured with a specific anti-Aβ antibody (BNT77). Aβ species ending in residue 40 or 42 were measured using horse radish peroxidase (HRP)-coupled monoclonal antibodies specific for Aβ40 (HRP-conjugated BA27) and Aβ42 (HRP-conjugated BC05) sequence. Seladin-1-overexpressing SH-SY5Y and control cells were cultured in a minimum volume of serum-free OPTIMEM medium (Invitrogen) for 30 h and homogenized in their supernatant by 10 passages through a 22-gauge syringe on ice. Aβ40 and Aβ42 levels were analyzed by ELISA as described above.
Quantitative RT–PCR
Total RNA from cells and brain tissue was extracted using TRIzol reagent (Invitrogen) following the manufacturer's instruction. Primers specific to human seladin-1 transgene, 5′-CCGTCCGAAAACTCAG-3′ and 5′-GCGGTGGTAGTAGTGT-3′, and to murine seladin-1, 5′-CATCGTCCCACAAGTATG-3′ and 5′-CTCTACGTCGTCCGTCA-3′, were designed using the program LC probe design software (Roche). Seladin-1 mRNA was quantified in three independent cell cultures and mouse brains (n=5 for each group). The housekeeping genes phosphoglucokinase and porphobilinogen deaminase were used as reference genes. These two genes were selected based on our unpublished results showing a constant expression of these genes in various experimental conditions in vitro and in vivo, respectively. Light Cycler quantitative real-time PCR was performed with an RNA SYBR Green kit (Roche Diagnostics).
Total and membrane extract preparation
SH-SY5Y cells or mouse brains were homogenized in phosphate buffer saline containing 9% sucrose and protease inhibitors (CLAP: pepstatin, antipain, chymostatin, each at a final concentration of 25 μg/ml) using 10 strokes in a dounce homogenizer and 10 passages through a 22-gauge syringe on ice. The samples were centrifuged for 10 min at 4°C and 700 g and the supernatants considered as total extracts. A further centrifugation of the supernatant was performed at 100 000 g for 1 h at 4°C to pellet the membrane fraction. Protein concentration was quantified by the BCA method (Bio-Rad).
Detergent extraction and DRM isolation
Total SH-SY5Y cell or mouse brain extracts were incubated for 1 h at 4°C in 1% Triton X-100, 25 mM MES pH 7.0, 5 mM DTT, 2 mM EDTA and CLAP. The extracts were mixed with 90% sucrose prepared in MBS buffer (25 mM MES pH 7.0, 150 mM NaCl and CLAP) to reach a final concentration of 60% and over-layered in an SW40 centrifugation tube with a step gradient of 35 and 5% sucrose in MBS. After centrifugation at 100 000 g for 18 h at 4°C, 11 fractions were collected from the top of each tube. Fractions 4–6 were identified as the DRM fraction by the presence of the DRM markers flotillin 1, PrPc and GM1 in the control samples.
Lipid analysis
Lipids were extracted from membrane pellets as described (Bligh and Dyer, 1959). Cholesterol, sphingomyelin or desmosterol was subsequently analyzed by thin-layer chromatography (TLC) on silica gel 60 HPTLC plates using a two-solvent system (hydrophilic running solvent: chloroform/acetone/acetic acid/methanol/water (50:20:10:10:5); and hydrophobic solvent: hexane/ethyl acetate (5:2)). The ganglioside GM1 was analyzed by slot-blot using cholera toxin subunit B linked to a peroxidase (Sigma). Quantification was carried out by densitometry of the scanned TLCs or autoradiograms under conditions of non-saturated signal using the NIH-image software. Phospholipids were determined using the Phospholipid B kit (WAKO) according to the manufactures' protocol.
Sterol analysis
Confluent cells (80%) were cultured in FCS/HS free DMEM NUT-F12 medium without phenol red during 24 h, washed once with ice-cold PBS containing 2 mg/ml BSA (Sigma) and twice with PBS. The sterols were extracted twice from confluent cells grown in a tissue culture dish (diameter 10 cm) using 4 ml hexan/isopropanol (3:2) containing 1 μg epicoprostanol as an internal standard. Similarly, sterols were extracted from fontal brain regions expanding from interaural regions 6 to 4 and from each fraction of the flotation gradient. Gas chromatography–mass spectrometry was performed to determine the total levels of the extracted sterols as described (Lutjohann et al, 2002).
As an alternative method, we also measured cholesterol in total and membrane extracts containing equal amounts of protein (40 μg) using the Ecoline 25 cholesterol kit (Merck). The optical density was measured at 500 nm. Pure cholesterol solutions (Sigma) were used as standards. The amounts of all sterols are normalized to the corresponding protein amounts (brains) or the number of cultured cells.
Desmosterol addition and cholesterol reduction in SH-SY5Y cells
Desmosterol and methyl-β-cyclodextrin (Sigma) were complexed as described previously for cholesterol by Klein et al (1995). These complexes containing 0.3 mM desmosterol were added to the medium of SH-S5Y5 cells at a final 1:10 dilution together with 2 μg/ml free desmosterol and incubated for 1 h at 37°C. In certain experiments, before the addition of desmosterol complexes, the cells were incubated for 48 h with 0.4 μM mevilonin (Sigma) and 1 mM methyl-β-cyclodextrin to extract 30% cholesterol as described by Abad-Rodriguez et al (2004). The incorporation of desmosterol was monitored by TLC using pure desmosterol (Sigma) as standard. Cholesterol reduction was monitored by the Ecoline 25 kit as described above. Cell viability after these treatments was not affected (Estus et al, 1997).
Plasminogen binding and plasmin activity
A 200 μg portion of freshly prepared membrane extracts from SH-SY5Y cells or mouse brains was resuspended in Hank's balanced saline solution and 0.1% ovalbumin (Sigma) and was placed in a 96-multiwell plate in the presence of 2 mM chromogenic peptide to measure endogenous plasmin activity. In parallel experiments, plasminogen binding to cellular membranes was determined by addition of 2 μM human plasminogen to the same extracts. Plasmin enzymatic activity was assayed using the chromogenic substrate S-2251 (Chromogenix) specific for this protease. Absorbance was measured at 37°C and 405 nm in an ultramicroplate reader Elx808iu (BioteK, Instruments Inc.) every 5 min.
Quantification of APP C-terminal fragments
Equal amounts of total extracts (40 μg) from either SH-SY5Y cells or mouse brains were submitted to 15% PAGE–SDS, transferred to nitrocellulose and blotted with either 6E10 or the anti-APP C-terminal antibody to detect full-length APP and its C-terminal fragments, respectively. Quantification was carried out by densitometry of the scanned autoradiograms using the NIH-image software. The amount of the C-terminal fragments was normalized to the amount of full-length APP in each lane.
Statistical analysis
Data were collected by investigators blinded to the experimental setup and were statistically analyzed by non-parametric Mann–Whitney U-test. In all graphs, mean±s.e. (standard error of the mean) are shown. P-values <0.05 were considered to be statistically significant.
Supplementary Material
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4
Acknowledgments
We thank Dr E Feinstein (Quark Biotech Inc.) for providing the seladin-1-deficient mice and Takeda Pharmaceutical Company Limited for providing antibodies against Aβ for their quantification by ELISA. This work was supported by grants from the Swiss National Science Foundation, the University of Zurich, NCCR, SAMW, Hermann Klaus, Hartmann Müller and the Novartis Foundations to MHM, Regione Piemonte to MDL and by EU contract LSHM-CT-2003-503330 (APOPIS) to RN and CGD.
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG (2004) Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol 167: 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917 [DOI] [PubMed] [Google Scholar]
- Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C (1998) The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J Biol Chem 273: 3205–3211 [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165: 1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG (2000) A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature 408: 975–979 [DOI] [PubMed] [Google Scholar]
- Ciana A, Balduini C, Minetti G (2005) Detergent-resistant membranes in human erythrocytes and their connection to the membrane-skeleton. J Biosci 30: 317–328 [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923 [DOI] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE (1997) Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant ‘apoptotic' pattern of gene induction. J Neurosci 17: 7736–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T (2001) Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA 98: 5856–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97: 395–406 [DOI] [PubMed] [Google Scholar]
- Greeve I, Hermans-Borgmeyer I, Brellinger C, Kasper D, Gomez-Isla T, Behl C, Levkau B, Nitsch RM (2000) The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer's disease-associated neurodegeneration and oxidative stress. J Neurosci 20: 7345–7352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356 [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G (1995) Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15: 1203–1218 [DOI] [PubMed] [Google Scholar]
- Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI (1999) Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274: 37111–37116 [DOI] [PubMed] [Google Scholar]
- Iivonen S, Hiltunen M, Alafuzoff I, Mannermaa A, Kerokoski P, Puolivali J, Salminen A, Helisalmi S, Soininen H (2002) Seladin-1 transcription is linked to neuronal degeneration in Alzheimer's disease. Neuroscience 113: 301–310 [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D (2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408: 979–982 [DOI] [PubMed] [Google Scholar]
- Kirsch C, Eckert GP, Mueller WE (2003) Statin effects on cholesterol micro-domains in brain plasma membranes. Biochem Pharmacol 65: 843–856 [DOI] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F (1995) Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34: 13784–13793 [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Abad-Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A, Dingwal C, Dotti CG (2003a) Raft disorganization leads to reduced plasmin activity in Alzheimer's disease brains. EMBO Rep 4: 1190–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B (2000) Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep 1: 530–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Da Silva JS, Schevchenko A, Wilm M, Dotti CG (2003b) Proteomic characterisation of neuronal sphingolipid–cholesterol microdomains: role in plasminogen activation. Brain Res 987: 107–116 [DOI] [PubMed] [Google Scholar]
- Lutjohann D, Brzezinka A, Barth E, Abramowski D, Staufenbiel M, von Bergmann K, Beyreuther K, Multhaup G, Bayer TA (2002) Profile of cholesterol-related sterols in aged amyloid precursor protein transgenic mouse brain. J Lipid Res 43: 1078–1085 [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Saini K, Schultz JG, Wollmer MA, Hock C, Nitsch RM (2002) Passive immunization against beta-amyloid peptide protects central nervous system (CNS) neurons from increased vulnerability associated with an Alzheimer's disease-causing mutation. J Biol Chem 277: 33012–33017 [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982–985 [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I (2003) Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging 24: 637–643 [DOI] [PubMed] [Google Scholar]
- Parton RG, Hancock JF (2004) Lipid rafts and plasma membrane microorganization: insights from Ras. Trends Cell Biol 14: 141–147 [DOI] [PubMed] [Google Scholar]
- Prior IA, Muncke C, Parton RG, Hancock JF (2003) Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol 160: 165–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE (2001) A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 8: 890–899 [DOI] [PubMed] [Google Scholar]
- Rothblat GH, Burns CH, Conner RL, Landrey JR (1970) Desmosterol as the major sterol in L-cell mouse fibroblasts grown in sterol-free culture medium. Science 169: 880–882 [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rutten BP, Pielen A, Schafer S, Wirths O, Tremp G, Czech C, Blanchard V, Multhaup G, Rezaie P, Korr H, Steinbusch HW, Pradier L, Bayer TA (2004) Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am J Pathol 164: 1495–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Clearing the brain's amyloid cobwebs. Neuron 32: 177–180 [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31–39 [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K (1998) Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA 95: 6460–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small DH, Mok SS, Bornstein JC (2001) Alzheimer's disease and Abeta toxicity: from top to bottom. Nat Rev Neurosci 2: 595–598 [DOI] [PubMed] [Google Scholar]
- Sparks DL, Martin TA, Gross DR, Hunsaker JC III (2000) Link between heart disease, cholesterol, and Alzheimer's disease: a review. Microsc Res Tech 50: 287–290 [DOI] [PubMed] [Google Scholar]
- Sparks DL, Martins R, Martin T (2002) Cholesterol and cognition: rationale for the AD cholesterol-lowering treatment trial and sex-related differences in beta-amyloid accumulation in the brains of spontaneously hypercholesterolemic Watanabe rabbits. Ann NY Acad Sci 977: 356–366 [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, Hennekam RC, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI, Wanders RJ (2001) Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet 69: 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler A, Brafman A, Shafir M, Heverin M, Gottlieb H, Damari G, Gozlan-Kelner S, Spivak I, Moshkin O, Fridman E, Becker Y, Skaliter R, Einat P, Faerman A, Bjorkhem I, Feinstein E (2003) Generation of viable cholesterol-free mice. Science 302: 2087. [DOI] [PubMed] [Google Scholar]
- Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K (2004) Regulation of cellular response to oncogenic and oxidative stress by Seladin-1. Nature 432: 640–645 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Supplementary Information 4