

Abstract
The ubiquitin–proteasome system (UPS) is critical for specific degradation of cellular proteins and plays a pivotal role on protein breakdown in muscle atrophy. Here, we show that ZNF216 directly binds polyubiquitin chains through its N-terminal A20-type zinc-finger domain and associates with the 26S proteasome. ZNF216 was colocalized with the aggresome, which contains ubiquitinylated proteins and other UPS components. Expression of Znf216 was increased in both denervation- and fasting-induced muscle atrophy and upregulated by expression of constitutively active FOXO, a master regulator of muscle atrophy. Mice deficient in Znf216 exhibited resistance to denervation-induced atrophy, and ubiquitinylated proteins markedly accumulated in neurectomized muscle compared to wild-type mice. These data suggest that ZNF216 functions in protein degradation via the UPS and plays a crucial role in muscle atrophy.
Keywords: aggresome, muscular atrophy, proteasome, ubiquitin, zinc-finger protein
Introduction
The ubiquitin–proteasome system (UPS) is one of the major protein degradation pathways in eukaryotic cells. The UPS plays key regulatory roles in many cellular processes, including cell cycle control, the regulation of transcription and protein quality control (Hershko and Ciechanover, 1998; Pickart and Cohen, 2004). Aberrations of this system lead to many forms of pathogenesis, such as malignancies, neurodegenerative disease and inflammatory response (Glickman and Ciechanover, 2002). The UPS includes sequential, multistep reactions: ubiquitin-conjugation of target proteins by E1, E2 and E3 enzymes, recognition of ubiquitinylated proteins by ubiquitin-binding proteins or 19S subunits of proteasome and proteolysis in the proteasome.
Many catabolic conditions, such as low-insulin state, hyperthyroidism, sepsis and cancer cachexia lead to enhancement of protein breakdown in skeletal muscle known as muscle atrophy (Mitch and Goldberg, 1996; Lecker et al, 1999). In muscle atrophy, the UPS plays a pivotal role in protein breakdown (Price et al, 1996; Tawa et al, 1997). Several studies indicate that mRNAs encoding UPS components are increased in atrophying muscle (Medina et al, 1991; Wing and Goldberg, 1993; Bailey et al, 1996; Price et al, 1996; Jagoe et al, 2002). In particular, the E3 ubiquitin ligases MAFbx/Atrogin-1 and MuRF-1 (muscle RING finger 1) are known to be markers of muscle atrophy (Bodine et al, 2001; Gomes et al, 2001). Both are induced in multiple models of muscle atrophy including immobilization, denervation and hindlimb suspension, and mice deficient in either gene are resistant to denervation-induced muscle atrophy (Bodine et al, 2001). Goldberg and co-workers proposed that atrophy-related genes, whose expression is induced in multiple types of muscle atrophy, are called ‘atrogenes' (Sandri et al, 2004). Recently, it was demonstrated that the IGF-I/PI3K/Akt pathway is an important regulator of muscle mass in muscle hypertrophy and atrophy (Sacheck et al, 2004; Sandri et al, 2004; Stitt et al, 2004). In that case, the transcription factor FOXO plays a pivotal role in activating atrogenes such as MAFbx/Atrogin-1 (Gomes et al, 2001).
Although many UPS players such as E3 ligases have been characterized, the mechanism of how ubiquitinylated proteins are delivered to the proteasome have not been fully elucidated. A component of 19S proteasome, Rpn10/S5a, recognizes the ubiquitinylated proteins (Young et al, 1998; Wilkinson et al, 2000). It has been shown that yeast proteins, Rad23p and Dsk2p, bind to ubiquitinylated substrates and to the 26S proteasome through their UBA and Ubl domains, respectively, thereby functioning as shuttle proteins that present polyubiquitinylated proteins to the proteasome (Chen et al, 2001; Funakoshi et al, 2002; Elsasser and Finley, 2005). Loss-of-function of shuttle proteins results in abnormal accumulation of polyubiquitinylated proteins (Lambertson et al, 1999; Saeki et al, 2002). However, yeast can survive when both RAD23 and DSK2 genes are mutated, suggesting that other mechanisms or molecule(s) possessing a shuttle function exist (Saeki et al, 2002). Here, we show that ZNF216, a novel ubiquitin-binding protein containing an A20-type zinc-finger, is such a factor. Znf216 expression is upregulated in skeletal muscle in experimental models of muscle atrophy, and Znf216-deficient mice exhibit resistance to muscle atrophy accompanied by abnormal accumulation of polyubiquitinylated proteins in skeletal muscle. Our findings suggest that ZNF216, with its potential function of anchoring ubiquitinylated proteins to the proteasome, plays a critical role in degrading muscle proteins.
Results
ZNF216 directly binds to polyubiquitin
We have identified a gene, Znf216 (Za20d2, Mouse Genome Informatics), encoding an A20 zinc-finger (Znf-A20) motif-containing protein, as a RANKL-induced gene upregulated upon osteoclast formation using a microarray technique (Hishiya et al, 2005). Znf216 was originally identified as a candidate gene for hearing loss and is expressed in cochlear and skeletal muscle (Scott et al, 1998; Huang et al, 2004). To determine the function of ZNF216, we searched for molecules that associate with ZNF216 using yeast two-hybrid screening and isolated several clones encoding a gene for polyubiquitin C. To determine whether ZNF216 interacts with ubiquitin in mammalian cells, we transfected HEK293 cells with an expression vector for FLAG-tagged ZNF216 and HA-tagged ubiquitin and performed co-immunoprecipitation experiments. ZNF216 possesses A20-type (amino acids 11–35) and AN1-type (amino acids 154–191) zinc-finger domains at its N- and C-termini, respectively (Figure 1A). Endogenous ubiquitinylated proteins, which appear as smears, were co-immunoprecipitated with FLAG-tagged ZNF216 (Figure 1B). Notably, N-terminal deletion (ΔN; amino acids 36–213) or point mutants (M1 and M3) of the A20-type zinc-finger (ZnF-A20) domain abolished ubiquitin-binding ability of ZNF216, indicating that the ZnF-A20 domain is indispensable for binding to ubiquitin (Figures 1A and B). Whereas in nondenaturing conditions, ubiquitinylated molecules were present with FLAG-tagged ZNF216, these molecules completely disappear from immunoprecipitates following heat denaturation, which abolishes noncovalent protein–protein interactions (Figure 1C), suggesting that ZNF216 associates with ubiquitinylated proteins rather than being ubiquitinylated itself. Next, to determine whether ZNF216 binds to ubiquitin directly, we performed GST pull-down assays using GST-ZNF216 fusion proteins (Figure 1D) and purified polyubiquitin. As shown in Figure 1E, GST-ZNF216 but not GST bound to polyubiquitin chains. As expected, binding of ZNF216 to polyubiquitin chains was completely abolished by a point mutation in the ZnF-A20 domain (M1, Figure 1E). Furthermore, a GST fusion protein containing only the ZnF-A20 domain (amino acids 2–60) could bind to polyubiquitin chains, suggesting that ZNF216 directly binds to polyubiquitin chains, and that the ZnF-A20 domain is required for binding to polyubiquitin. As for other ZnF-A20 containing proteins, AWP1 (ZA20D3) also possessed polyubiquitin-binding activity but the ZnF-A20 domain(s) of Rabex-5 (Horiuchi et al, 1997) and A20/TNFAIP3 (Opipari et al, 1990) proteins did not (Supplementary Figure S1).
Figure 1.
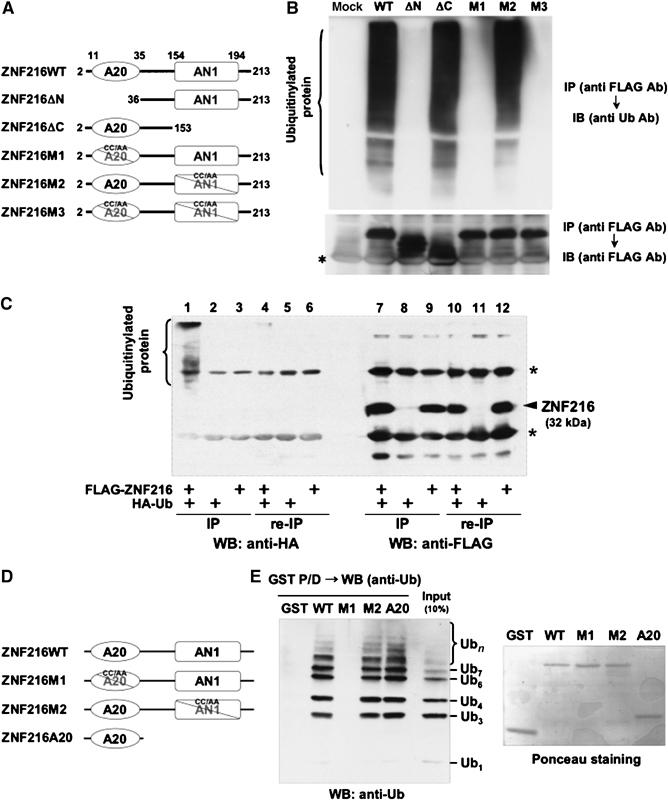
ZNF216 binds polyubiquitin directly through the ZnF-A20 domain. (A) Schematic representation of the primary structure of wild-type ZNF216 and its mutants. ZNF216ΔN (aa 36–213) and ZNF216ΔC (aa 2–153) constructs lack the ZnF-A20 (aa 11–35) and ZnF-AN1 (aa 154–194) domains, respectively. Cysteine residues at positions 30 and 33 within the ZnF-A20 were substituted with alanines (C30A/C33A) in ZNF216M1, and both cysteines 170 and 175 within the ZnF-AN1 were substituted with alanines (C170A/C175A) in ZNF216M2. Both ZnF-A20 and ZnF-AN1 domains were mutated in ZNF216M3. (B) Co-precipitation of ubiquitinylated proteins and ZNF216. FLAG-tagged ZNF216 or mutants were expressed in HEK293 cells, and cell extracts were immunoprecipitated with anti-FLAG antibody. Ubiquitinylated proteins detected with anti-ubiquitin antibody were precipitated with FLAG-tagged ZNF216 but not with ZnF-A20 mutants. Expression levels of FLAG-tagged ZNF216 constructs are shown at the bottom. Bands corresponding to immunoglobulin chains are marked by an asterisk. (C) ZNF216 is minimally ubiquitinylated. HEK293 cells expressing FLAG-tagged ZNF216 or HA-tagged ubiquitin were lysed and immunoprecipitation was performed using anti-FLAG antibody. Aliquots of precipitated beads were boiled and immunoprecipitated again (re-IP). Each sample was separated on gels and probed with anti-HA (left) or anti-FLAG antibody (right). Bands for immunoglobulin chains are marked by asterisks. (D) Constructs used for in vitro binding assay. ZNF216WT, ZNF216M1 and ZNF216 M2 were as indicated in (A). ZNF216A20 possesses only the A20 domain (aa 2–60). All constructs were produced as GST fusion proteins. (E) In vitro ubiquitin binding assay. Left panel: GST protein fused to the constructs indicated in (D) was incubated with purified K48-linked polyubiquitin chains, followed by precipitation with GSH beads. In all, 10% of purified polyubiquitin chains was separated without pull-down to evaluate protein amount (10% input). Right panel: the membrane was stained with ponceau to evaluate levels of GST fusion protein.
ZNF216 associates with the 26S proteasome
We also identified molecules associating with ZNF216 by proteomic analysis of complexes formed with FLAG-tagged ZNF216. Molecules expressed in HEK293 cells and that co-immunoprecipitated with FLAG-tagged ZNF216 were analyzed by tandem mass spectrometry. By this analysis, every subunit of the 26S proteasome complex was identified as associating with FLAG-tagged ZNF216 (data not shown). To identify the region of ZNF216 required for association with the 26S proteasome, lysates of cells expressing either FLAG-tagged ZNF216 or its mutants were immunoprecipitated with anti-FLAG antibody. Co-precipitation of proteasomal components was monitored by immunoblotting using an antibody against Rpn7p (S10a), a non-ATPase subunit of the 19S regulatory subunit. As shown in Figure 2A, this protein efficiently co-precipitated with FLAG-tagged ZNF216. The interaction was also observed with truncated or point mutants of ZnF-A20 (ΔN or M1), indicating that ubiquitin-binding ability is dispensable for association with the 26S proteasome. To determine whether endogenous ZNF216 proteins are also associated with the 26S proteasome, we performed a GST pull-down assay using the ubiquitin-like (Ubl) domain of hHR23B, a human homologue of Rad23, which is known to bind to the 26S proteasome. As shown in Figure 2B, GST-Ubl but not GST was pulled down with the endogenous 26S proteasome. Endogenous ZNF216 was also detected in the GST-Ubl/26S proteasome complex (upper panels, Figure 2B). Furthermore, purified recombinant ZNF216 did not bind to GST-Ubl (lower panel, Figure 2B), suggesting that endogenous ZNF216 is not directly bound to the Ubl domain but associates with the 26S proteasome.
Figure 2.
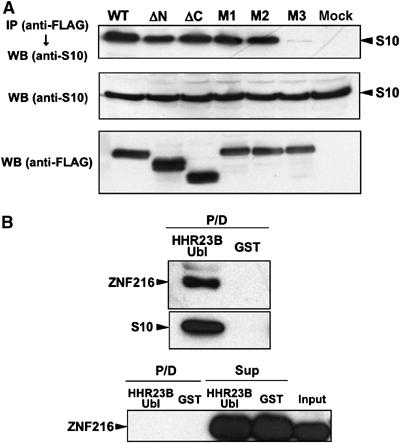
Interaction of ZNF216 with the 26S proteasome in mammalian cells. (A) Co-precipitation of the 26S proteasome and ZNF216. Co-precipitated proteins with FLAG-ZNF216 were resolved by SDS–PAGE and detected by immunoblotting using anti-S10a/Rpn7p antibody (anti-S10) or anti-FLAG antibody. Aliquots of cellular extracts were immunoblotted without immunoprecipitation to evaluate protein expression in the bottom panels. (B) ZNF216 was detected in the 26S proteasome fraction. Upper panel, cell lysates were incubated with a GST fusion of HHR23B Ubl (HHR23B Ubl) to isolate the 26S proteasome. Precipitated proteins (P/D) were separated and probed with anti-S10 or anti-ZNF216 antibody. Lower panel: purified recombinant ZNF216 was incubated with a GST fusion of HHR23B Ubl or GST protein. Precipitated (P/D) or not precipitated (Sup) proteins were probed with anti-ZNF216 antibody. No direct binding of ZNF216 to the Ubl domain of HHR23B was detected.
Colocalization with the aggresome
Next, we determined the subcellular localization of ZNF216. Indirect immunofluorescence of ZNF216 expressed in COS-7 cells showed that the protein was largely cytoplasmic but was seen to a lesser extent in the nucleus (Figure 3A). Aggresomes, which are insoluble aggregates of ubiquitinylated proteins complexed with the proteasome and induced by treatment with proteasome inhibitors, are known to mimic inclusions seen in pathogenic UPS disorders (Johnston et al, 1998; Kopito, 2000; Lelouard et al, 2002). As shown in Figures 3D–H, ZNF216 proteins were colocalized with aggresomes induced by treatment with the proteasome inhibitor MG132. ZNF216 itself was not ubiquitinylated as shown in Figure 1C.
Figure 3.
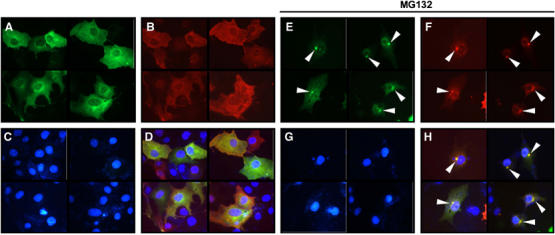
ZNF216 is localized in ‘aggresomes' with ubiquitinylated proteins. (A–H) COS cells were transfected with expression vectors for FLAG-tagged ZNF216 and HA-tagged ubiquitin. Fixed cells were subjected to indirect immunofluorescence using (A, E) anti-FLAG (with AlexaFluor 488 anti-mouse IgG, green) and (B, F) anti-HA (with AlexaFluor 546 anti-rat IgG antibodies, red) antibodies. (C, G) Nuclei were stained with DAPI in the same fields of each panel. (E–H) Transfected COS cells were treated with the proteasome inhibitor, MG132 (0.5 μM). Aggresomes formed are indicated by arrowheads. The merged images were shown in (D and H).
Induction of ZNF216 expression upon muscle atrophy
Biochemical and cell biological evidence presented here strongly suggests that ZNF216 functions in the UPS. In skeletal muscle, it is generally accepted that the UPS plays a critical role in muscular atrophy, and expression of atrophy-related genes including those encoding UPS components is induced in atrophying muscle (Jagoe et al, 2002; Lecker et al, 2004). As Znf216 was predominantly expressed in brain and skeletal muscle (Scott et al, 1998), we investigated the relationship between ZNF216 and muscle atrophy. To determine whether Znf216 expression is induced during muscle atrophy, an in vitro model of muscle atrophy was utilized. It has been reported that addition of dexamethasone to cultures of differentiated C2C12 myotubes causes formation of myotubes exhibiting signs of atrophy, including a reduction in myotube diameter (Stitt et al, 2004). Such treatment dramatically induced expression of Znf216 (Figure 4A).
Figure 4.
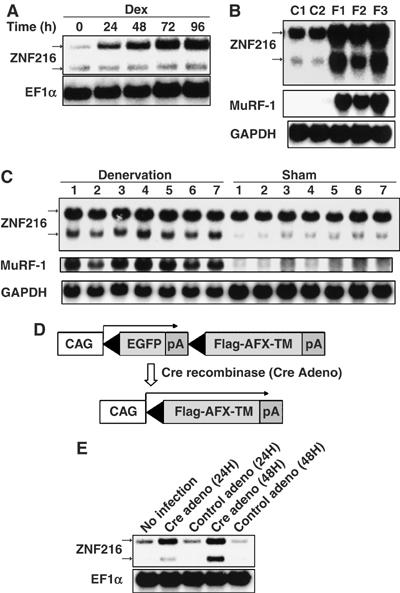
Expression of ZNF216 is induced by muscle atrophy. (A) C2C12 myoblast cells were differentiated into myotubes, and treated with 100 μM Dex for the indicated times. Northern blotting was performed to reveal the effect of Dex on ZNF216 expression. The entire coding region of ZNF216 was used as a probe, which recognized 2.4 and 1.5 kb mRNA species arising from alternative splicing and polyadenylation. The loading control was elongation factor α (EF1α). (B) Fasting-induced muscle atrophy. Three mice were fasted (F1∼F3), and two mice (C1, C2) were fed freely. After 2 days, RNA was purified from gastrocnemius muscle, and Northern blotting was performed to determine ZNF216 expression. The membrane was re-probed with MuRF-1 and GAPDH. (C) Denervation-induced muscle atrophy was induced by cutting the sciatic nerve of the hindlimb of seven mice (1∼7). The opposite limb was sham operated as the control. At 7 days after surgery, total RNA was purified from gastrocnemius muscles, and Northern blotting was performed to detect ZNF216 expression. The membrane was re-probed with MuRF-1 and GAPDH. (D) Cre-loxP-mediated, constitutively active FOXO expression system. cDNA encoding FLAG-tagged constitutively active FOXO4 (AFX-TM) is separated from the CAG promoter of an expression vector by a loxP-flanked EGFP-poly(A) cassette. Infection with adenovirus expressing Cre recombinase (Cre) results in excision of the DNA fragment located between the two loxP sequences and expression of FLAG-tagged AFX-TM. (E) ZNF216 is downstream of FOXO. Total RNAs were prepared from C2C12-AFX-TM cells at the indicated times after infection with adenovirus expressing Cre (Cre) or lacZ (control) and probed by Znf216 or EF1α. A marked increase in expression of Znf216 was observed only in Cre-infected cells.
Next, expression of Znf216 was determined in in vivo experimental models of muscle atrophy. Mice that undergo fasting for 2 days show significant decreases in body weight, as well as in the mass of the gastrocnemius muscles (data not shown). In this model, fasting for 2 days results in dramatic increases in Znf216 mRNA (Figure 4B) and protein (Supplementary Figure S3) in muscle. Although there were differences in induction patterns of two differently sized transcripts of Znf216 by atrophy-inducting stimuli, both transcripts encode the same protein (Supplementary Figures S2 and S3). Expression of MuRF-1 (Figure 4B) and MAFbx (Gomes et al, 2001) was also induced in fasting. Upregulation of Znf216 was also observed in a model of denervation-induced muscle atrophy. Neurectomy promotes significant reduction (∼20%) in the weight of gastrocnemius muscles within the first 7 days postsurgery. As expected, expression of Znf216 and MuRF-1 was induced in gastrocnemius muscles by denervation-induced muscle atrophy (Figure 4C). These results suggest that Znf216 expression is associated with atrophy in skeletal muscles.
The transcription factor FOXO has been reported to play a critical role in muscular atrophy by inducing atrophy-related genes, including MAFbx/Atrogin-1 (Sandri et al, 2004; Stitt et al, 2004). Therefore, we asked whether FOXO activation upregulated Znf216 expression. To do so, we employed a Cre-loxP system (Furukawa-Hibi et al, 2002) in which constitutively active FOXO4 (AFX-TM) created by mutation of the three Akt phosphorylation sites, T32A, S253A and S315A (Brunet et al, 1999), was expressed in C2C12-AFX-TM cells following infection by Cre recombinase-expressing adenovirus (Cre) (Figure 4D). Both AFX-TM mRNA and protein were induced 24 h after infection with Cre but not with control adenovirus (Furukawa-Hibi et al, 2002). ZNF216 mRNA was markedly increased in C2C12-AFX-TM cells as a result of infection with Cre but not following infection with control virus (Figure 4E). These results suggest that ZNF216 may function as a downstream effector of FOXO in muscle atrophy.
Generation of mice lacking ZNF216
To investigate the in vivo function of ZNF216, mice deficient for ZNF216 (Znf216lex/lex) were generated by gene trapping at Omnibank of Lexicon Genetics (Zambrowicz et al, 1998). The structure of the predicted trapped gene is shown in Figure 5A. The trapping vector, VICTR48, was inserted 3.3 kbp upstream of exon 3, which encodes the first methionine of mouse Znf216 (Figure 5A). Znf216lex/lex mice were born from interbred heterozygous Znf216+/lex mice in Mendelian ratios, indicating that ZNF216 is dispensable for embryogenesis or fetal development. No ZNF216 mRNA or protein was detected in Znf216lex/lex mice by Northern or immunoblot analyses, respectively (Figures 5B and C), indicating that the mice are ZNF216 nulls. Expression levels of ZNF216 in Znf216+/lex heterozygotes were nearly one-half those of wild-type mice. Znf216lex/lex mice were viable and fertile, without gross abnormalities or apparent pathological alteration, but they weighed less than sex- and age-matched controls (Figure 5D). At 45 weeks, the average weights of Znf216+/+ and Znf216lex/lex male mice were 42.66±7.06 g (n=14) and 33.16±4.44 g (n=9), respectively. The average weights of female Znf216+/+ and Znf216lex/lex mice were 34.46±4.21 g (n=14) and 26.85±5.38 g (n=11), respectively. After 30 weeks, both female and male Znf216lex/lex mice showed no or subtle increases in weight, whereas Znf216+/+ or Znf216+/lex mice gained weight as they aged (Figure 5D). The size of most organs in Znf216lex/lex mice was reduced in proportion with body weight. However, the fat volume of aged (>30 weeks of age) Znf216lex/lex mice was significantly decreased, suggesting that the marked difference in body weight between wild-type and aged Znf216lex/lex mice is mainly caused by decreased fat mass seen in Znf216lex/lex mice (not shown). Detailed phenotypic characterization of aged mutant mice will be provided elsewhere.
Figure 5.
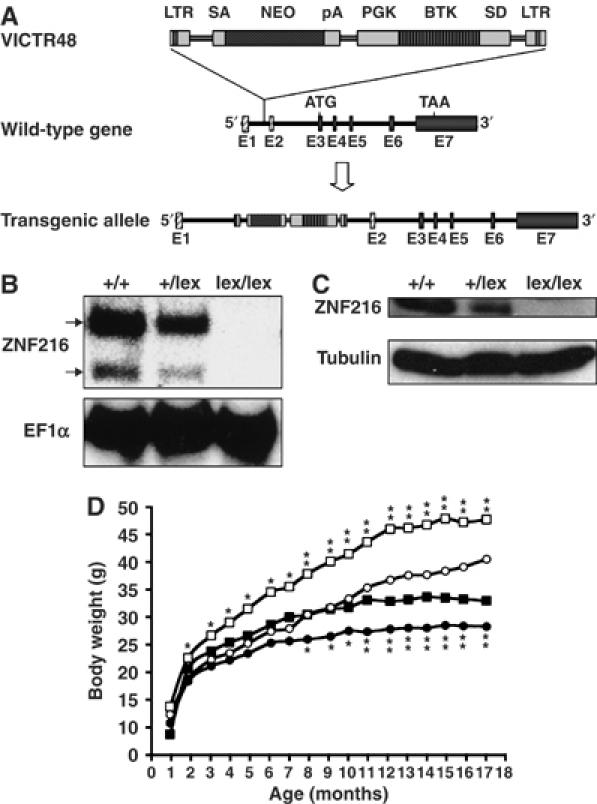
Disruption of Znf216 gene in mice. (A) Gene trap strategy of Znf216 gene. The structure of the trapping vector, VICTR48, is shown in the upper line. The wild-type allele and the trapped, transgenic allele follow the vector. The retroviral vector, VICTR48, was integrated between exons 1 and 2 of the Znf216 gene and transcription of downstream exons encoding ZNF216 was diminished. Exons are depicted by striped (noncoding exons) or shadowed boxes (protein-coding exons) and numbered (E1 and E2). LTR, long terminal repeat; SA, splice acceptor site; SD, splice donor site; pA, polyadenylation signal; PGK, PGK promoter. (B) Northern blot analysis. Total RNA was prepared from brains of Znf216+/+, ZnfF216+/lex or Znf216lex/lex mice. Full-length mouse ZNF216 cDNA was used as a probe. The membrane was re-probed using an EF1α probe. (C) Immunoblot analysis. Extracts from brain of Znf216+/+, Znf216+/lex or Znf216lex/lex mice were immunoblotted with antibody against ZNF216. The membrane was re-probed using anti-tubulin antibody. (D) Growth curve of Znf216lex/lex mice. Body weights at each time point of Znf216+/+ and Znf216lex/lex mice were indicated as open square boxes (males) or circles (females) and closed square boxes (males) or circles (females), respectively. *P<0.05; **P<0.005.
Znf216lex/lex mice exhibit partial resistance to denervation-induced muscle atrophy
To further explore the involvement of ZNF216 in muscle atrophy, neurectomy of sciatic nerve was undertaken in wild-type and Znf216lex/lex mice. As shown in Figure 6A, 7 days after denervation, significant muscle weight loss and reduction in fiber sizes of the gastrocnemius muscle were observed in wild-type mice. By contrast, such decreases in muscle weight were significantly attenuated in Znf216lex/lex mice (Figure 6A). Sections of gastrocnemius muscle also showed larger fibers in muscle from neurectomized Znf216lex/lex mice than in control muscle (Figure 6B). However, there was no significant difference in fiber area between sham-operated wild-type and Znf216lex/lex mice (wild type+sham operated, 1988±530 μm2; wild type+denervation, 1379±345 μm2; lex/lex+sham operated, 1776±484 μm2; lex/lex+denervation, 1393±344 μm2). As shown in Figure 6C, the reduction in fiber area was also less apparent in Znf216lex/lex mice compared to wild-type mice. These results suggest that ZNF216 plays a crucial role in reduction of muscle mass on denervation-induced muscle atrophy.
Figure 6.
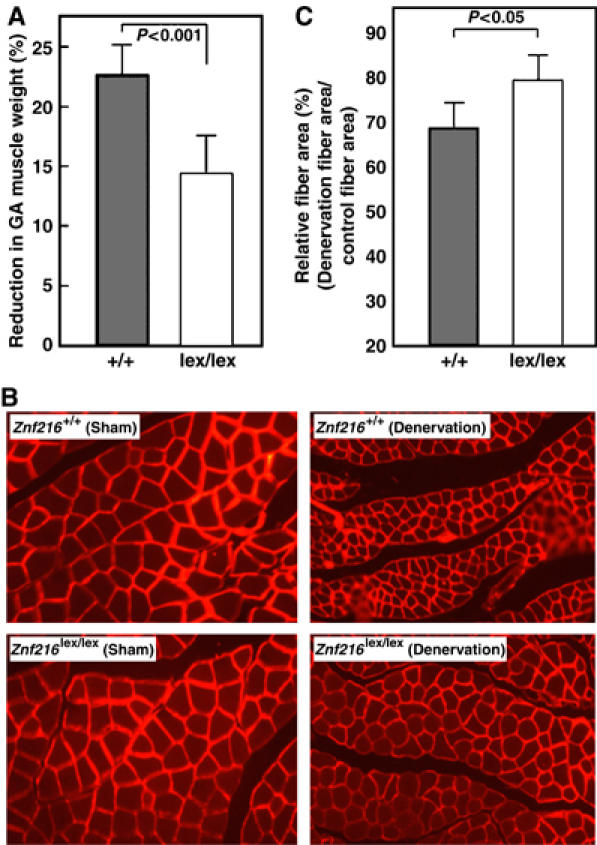
Denervation induced muscular atrophy was attenuated in ZNF216lex/lex mice. (A) Reduction of GA muscle weight upon neurectomy. Percent decreases in muscle weights are shown as a percent of control, calculated as the left/right muscle weights. (B) Cross-sections from gastrocnemius muscle were stained by indirect immunofluorescence with anti-laminin. The reduction in size was also significant in muscle fibers of control mice but less in Znf216lex/lex. (C) Muscle fiber cross-sectional areas were measured in transverse tissue section (B). Percent relative fiber area of denervated muscle to control fiber area (sham-operated) are shown.
Abnormal accumulation of ubiquitinylated proteins in muscle from Znf216lex/lex mice
To investigate what abnormalities occur during denervation-induced muscle atrophy in Znf216lex/lex mice, we examined expression levels of factors involved in muscle atrophy. As expected, expression of MAFbx/Atrogin-1 and MuRF-1 was dramatically induced by denervation-induced muscle atrophy in gastrocnemius muscle from wild-type mice (Figure 6A). In Znf216lex/lex mice, expression of MAFbx/Atrogin-1 and MuRF-1 was also induced at levels comparable to those seen in wild-type mice. Induction of Pmsa1 and Pmsd11, genes encoding the 26S proteasome subunits α6 and Rpn6, respectively, was also indistinguishable between Znf216lex/lex and wild-type mice (Figure 7A). Furthermore, proteasome activities in gastrocnemius muscles were comparable between wild-type and Znf216lex/lex mice (Figure 7B). Thus, induction of relevant ubiquitin ligases or proteasome components was not affected in Znf216lex/lex mice. It is known that ubiquitinylated proteins accumulate during muscle atrophy (Medina et al, 1991; Wing et al, 1995). As shown in Figure 7C, following denervation, ubiquitinylated proteins accumulated in the gastrocnemius muscle of wild-type mice, but higher levels of ubiquitinylated proteins accumulated in muscle derived from Znf216lex/lex mice (∼2-fold: P<0.001 in neurectomized Znf216lex/lex versus wild-type muscle). Similar results were obtained by fasting-induced muscle atrophy, although no difference in the levels of ubiquitinylated proteins from controls (sham-operated or fed) was observed between genotypes (Figure 7C). These results indicate that ZNF216 is a critical regulator of muscle atrophy, most likely functioning to regulate degradation of muscle proteins without altering expression of proteasomal components or known E3 ligases.
Figure 7.
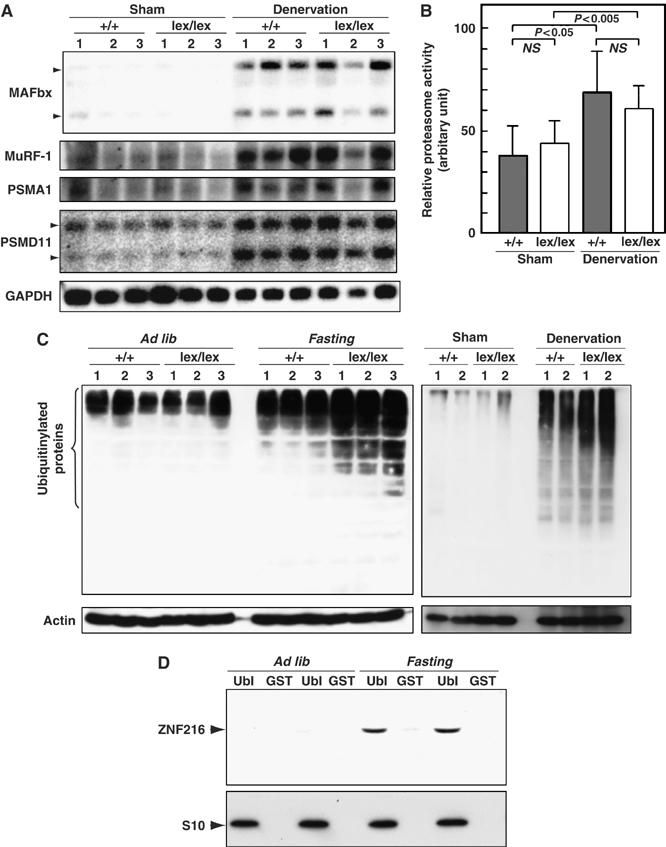
Changes in UPS upon muscular atrophy. (A) Expression of UPS components in denervation-induced muscular atrophy. Total RNAs were purified from gastrocnemius muscle, and Northern blotting was performed using indicated probes. Expression of genes for ubiquitin-ligases, such as MAFbx or MuRF-1, and proteasome subunits PSMA1 and PSMD11 was induced by muscle atrophy at comparable levels between wild-type and ZNF216lex/lex mice. (B) Proteasome activity. Proteasome activities in muscle extracts from wild-type or ZNF216lex/lex mice were measured and are shown as arbitrary units. No significant difference in proteasome activity between wild-type and ZNF216lex/lex was observed. (C) High levels of ubiquitinylated proteins accumulated in muscles from ZNF216lex/lex mice than in muscles from wild-type mice. Muscle extracts from wild-type or ZNF216lex/lex mice were subjected to immunoblotting using anti-ubiquitin antibody to analyze levels of ubiquitinylated proteins. Left and right panels show fasting-induced and denervation-induced muscle atrophy, respectively. Each membrane was re-probed with anti-actin antibody. (D) Association of ZNF216 with the proteasome was increased upon atrophy. The proteasome fractions in muscle extracts from fed (ad lib) or fasted (fasting) mice were precipitated with GST-Ubl or GST only as a negative control. Endogenous ZNF216 protein was co-precipitated with the proteasome, which is probed by the anti-S10 antibody.
Effect of ZNF216 on UPS-mediated protein degradation
Accumulation of ubiquitinylated protein under any circumstance might be because of loss of inhibition of ubiquitinylation and/or deubiquitinylation (DUB). However, no inhibition or DUB activity was observed (Supplementary Figures S4 and S5). As shown in Figure 7D, association of ZNF216 protein to the proteasome was significantly increased when atrophy was induced, suggesting that ZNF216 may be involved in association of ubiquitinylated proteins and the proteasome. The biochemical activity of ZNF216 is similar to that of the UPS proteins, hHR23 and hPLIC, both of which have a shuttle function and are known to bind to both polyubiquitinylated proteins and the 26S proteasome (Hartmann-Petersen and Gordon, 2004; Elsasser and Finley, 2005). Interestingly, overexpression of hHR23 and hPLIC results in stabilization of unstable proteins such as p53 (Kleijnen et al, 2000; Glockzin et al, 2003). To determine if ZNF216 functioned similarly, we employed a degradation system using unstable GFP (Bence et al, 2001). In this system, the CL1 peptide, which functions as a degron, is fused to EGFP (EGFP-CL1). Degradation by conjugation with the degron is mediated by the UPS (Bence et al, 2001). EGFP-CL1, constitutively expressed in HEK293 cells, is unstable and the estimated half-life (t1/2) of EGFP-CL1 in this system is about 11 min. Ubiquitinylated EGFP-CL1 protein stabilized by treatment with a proteasome inhibitor was associated with ZNF216 but EGFP itself was not (not shown). As shown in Figure 8A, protein degradation was markedly retarded in the presence of ectopic ZNF216 (t1/2>30 min) compared to cells transfected with the loss of function mutant ZNF216M3 or mock-transfected cells. Rapid turnover of EGFP-CL1 protein was inhibited by treatment with the proteasome inhibitor MG132 (MG132, Figure 8B). The levels of the proteins stabilized by MG132 were comparable among cells transfected with ZNF216 constructs, indicating that protein synthesis of EGFP-CL1 was not significantly affected by ectopic expression of ZNF216 (MG132, Figure 8B). ZNF216WT, and to a lesser extent the mutants M1 and M2 but not M3, attenuated degradation (NT, Figure 8B). Thus, as is the case with other shuttle proteins, overexpression of ZNF216 inhibits degradation of unstable proteins via the UPS.
Figure 8.
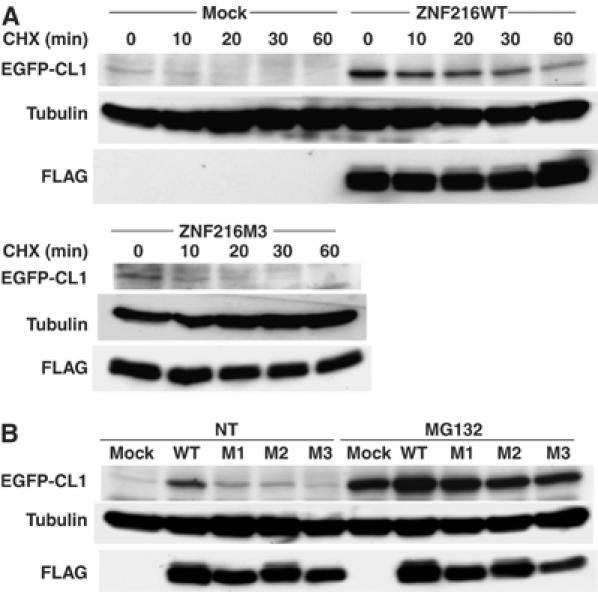
Ectopic expression of ZNF216-affected protein degradation. (A) Degradation of EGFP-CL1 protein was delayed by overexpression of ZNF216. 293 cells stably expressing EGFP-CL1 were transfected with plasmid of ZNF216WT, ZNF216M3 or pcDNA3 (mock). Estimated half-lives of the EGFP-CL1 are 35, 11 and 11 min in ZNF216WT-, ZNF216M3- and mock-transfected cells, respectively. De novo protein synthesis was arrested by cycloheximide (CHX). The membrane was re-probed with tubulin antibody to control for protein loading (tubulin) and FLAG antibody to detect ZNF216 expression (FLAG). (B) Degradation of EGFP-CL1 protein in the presence of various ZNF216 constructs. HEK293 cells stably expressing EGFP-CL1 were transfected with plasmids expressing the indicated mutants. Transfected cells were not treated (NT) or MG132-treated (MG132), and EGFP-CL1 protein was detected with an anti-GFP antibody (EGFP-CL1). The membrane was re-probed with tubulin antibody to control for protein loading (tubulin) and FLAG antibody to detect ZNF216 expression (FLAG).
Discussion
ZNF216 is an atrogene
In this report, we show that Znf216lex/lex mice exhibit resistance to denervation-induced muscle atrophy. It has been shown that TNFα induces catabolic conditions through UPS during cancer cachexia (Mitch and Price, 2001). Recently, it has been reported that mice deficient in molecules involved in the NF-κB pathway exhibit resistance to muscular atrophy (Cai et al, 2004; Hunter and Kandarian, 2004; McKinnell and Rudnicki, 2004). On the other hand, the IGF-FOXO axis has been suggested to regulate muscle mass through induction of ‘atrogenes' such as Murf1 and MAFbx/Atrogin-1 (Sandri et al, 2004; Stitt et al, 2004). Although we provide evidence that Znf216 is downstream of FOXO, the NF-κB pathway could represent an alternative signal inducing ZNF216. Indeed, we have identified Znf216 as a gene induced by RANKL, a TNF family ligand (Hishiya et al, 2005) which activates the NF-κB pathway through RANK (Anderson et al, 1997; Lacey et al, 1998). Moreover, TNFα and IL-1β upregulate expression of ZNF216 in fibroblasts and macrophages (Hishiya et al, 2005). These results suggest that Znf216 may be activated by NF-κB. Huang et al (2004) recently reported that ZNF216 inhibits the NF-κB pathway. Whereas treatment with TNFα or overexpression of TRAF6 dramatically activated a reporter driven by NF-κB response elements, ectopic expression of A20/TNFAIP3 but not ZNF216 inhibited NF-κB activation (not shown). Using mouse embryonic fibroblasts, splenocytes or bone marrow cells from Znf216lex/lex or wild-type mice, no significant differences were observed in TNFα-dependent NF-κB activation, LPS-induced cytokine expression or proliferation (unpublished data). Therefore, ZNF216 seems to function as a downstream effector (i.e., a component of the UPS) rather than a specific negative regulator of NF-κB signaling, although ZNF216 function in that pathway is still under investigation. Whereas expression of ZNF216 is not restricted to muscle, such expression was induced upon muscular atrophy and loss of function of Znf216 promotes resistance to denervation-induced atrophy, thereby suggesting that it fulfills the definition of an ‘atrogene'.
As it is in skeletal muscle, ZNF216 is highly expressed in the brain (Scott et al, 1998). Aberrations in the UPS have been documented in the pathogenesis of neurodegenerative diseases such as Parkinson's and Huntington's diseases (Ross and Poirier, 2004). Massive accumulation of ubiquitinylated proteins, which are often aggregated and impair the UPS leading to neuronal degeneration, has been observed in these pathogenic conditions (Ciechanover and Brundin, 2003; Korhonen and Lindholm, 2004). In cultured cells, blocking the UPS by proteasome inhibitors leads to accumulation of ubiquitinylated proteins. These ubiquitinylated proteins are then transferred to perinuclear locations and form aggresomes (Johnston et al, 1998). As shown here, ZNF216 is localized in aggresomes together with ubiquitinylated proteins. Interestingly, proteomic analysis of a protein complex containing HDAC6, a protein often associated with aggresomes (Kawaguchi et al, 2003), showed that the complex included AWP1, a structural homologue of ZNF216 (Seigneurin-Berny et al, 2001). Although it is unclear whether ZNF216 is involved in aggresome formation, there is great interest in the role of ZNF216 in the pathogenesis of neurodegenerative diseases.
Molecular function of an A20-containing protein, ZNF216
In muscle atrophy, more ubiquitinylated proteins accumulate in muscle from Znf216lex/lex mice than in muscle from wild-type mice, suggesting an abnormal UPS function. Inhibition of neither polyubiquitinylation nor DUB activity was observed in ZNF216. Although our in vivo data showed significant accumulation of polyubiquitinylated proteins in muscle from Znf216lex/lex mice, there is a possibility that ZNF216 is a ubiquitin-ligase. It has been recently reported that A20/TNFAIP3 protein possesses ubiquitin ligase activity against RIP through its ZnF-A20 repeats (Wertz et al, 2004). We asked whether the ZnF-A20 of ZNF216 exhibited activity similar to A20/TNFAIP3, but in vitro ubiquitinylation assays were negative (Supplementary Figure S6). In fact, the ZnF-A20 of A20/TNFAIP3 protein does not bind polyubiquitin chains as does the ZnF-A20 of ZNF216 (Supplementary Figure S1). Furthermore, there are seven ZnF-A20 motifs in A20/TNFAIP3 and only the fourth is responsible for E3 activity, suggesting that the ZnF-A20 motif is not inherently active enzymatically (Wertz et al, 2004). However, we cannot exclude the possibility that ZNF216 may possess DUB or E3 activity highly specific to an unknown substrate without nonspecific or self-ubiquitinylating activity.
ZNF216 likely acts as a bridging or a shuttle factor of ubiquitinylated proteins targeted to the proteasome. Shuttle proteins, such as Rad23p and Dsk2p, share interfaces for ubiquitinylated proteins and the proteasome (Hartmann-Petersen and Gordon, 2004; Elsasser and Finley, 2005). Although shuttle proteins are required for efficient protein degradation, ectopic expression of hHR23 or hPLIC, the human homologues of Rad23p or Dsk2p, respectively, lead to stabilization of p53 protein (Kleijnen et al, 2000; Glockzin et al, 2003). These outcomes may be caused by titration effects due to overexpression and are commonly observed following misexpression of shuttle proteins in yeast and mammals (Hartmann-Petersen and Gordon, 2004; Verma et al, 2004). Here, we show that ZNF216 has a ubiquitin binding domain and can associate with the 26S proteasome even in the absence of ubiquitin binding, and that overexpression of the zinc-finger protein attenuates protein degradation rate. There is no structural counterpart of ZNF216 in the yeast genome. We asked whether ZNF216 could rescue the bridging function of RAD23 or DSK2 mutants by introducing ZNF216 into Δrad23Δdsk2 yeast cells, but the phenotype could not be rescued (data not shown). This suggests that ZNF216 is not the functional orthologue of these proteins. Recently, the presence of an alternative pathway of Rad23p/Dsk2p in protein targeting to the proteasome has been suggested (Bazirgan and Hampton, 2005; Richly et al, 2005). It has been reported that tetra-ubiquitin constitutes the minimum proteasomal targeting signal and that the length of polyubiquitin chain may determine the targeting route (Thrower et al, 2000; Bazirgan and Hampton, 2005; Richly et al, 2005). Notably, ZNF216 preferentially binds polyubiquitin chains longer than di- or tri-ubiquitin (Figure 1D). Therefore, these data suggest that ZNF216 is a novel ubiquitin recognition factor, required for efficient protein degradation via a pathway different from the canonical Rad23p/Dsk2p pathway. Although it is now under investigation, the characterization of ZnF-AN1, an AN1-type zinc-finger domain located at the C-terminus of ZNF216, may reveal the precise molecular function of ZNF216.
Materials and methods
Antibodies
An anti-ZNF216 antibody was raised by immunizing rabbits against synthesized peptide corresponding to the C-terminal sequence of mouse ZNF216. Mouse monoclonal antibodies for FLAG (Sigma, St Louis, MO) and ubiquitin (Santa Cruz Biotechnology, CA), rabbit polyclonal antibodies for ubiquitin (Affiniti Research Products) and actin (Neo Markers, CA), a rat monoclonal antibody for HA (Roche Diagnostics, Mannheim, Germany), and a rabbit polyclonal antibody against S10a/Rpn7p (Affiniti Research Products) were purchased from the indicated manufacturers. For indirect immunofluorescence staining, AlexaFluor 488 goat anti-mouse IgG or AlexaFluor 546 goat anti-rat IgG antibody was obtained from Molecular Probes, OR.
Identification of interacting proteins
RNA was purified from RAW264.7 cells stimulated by RANKL, and used to construct the yeast library (MatchMaker Library Construction & Screening Kit, Clontech). Yeast two-hybrid screening with pGBKT7-ZNF216 was performed as described previously (Masuda et al, 2001). Identification of the co-immunoprecipitated proteins with N- or C-terminally FLAG-tagged ZNF216 (ZA20D2) or AWP1 (ZA20D3) was essentially done by a nano-LC/MS/MS system as previously described (Natsume et al, 2002; Komatsu et al, 2004).
Experimental models of muscle atrophy
For fasting-induced muscle atrophy, 8-week-old C57BL6 male mice were deprived of food but given free access to water. After 2 days, gastrocnemius muscles were harvested for each experiment. Denervation-induced muscle atrophy was performed by dissecting the sciatic nerve of one hindlimb, and the other hindlimb was sham operated as the control. After 7 days, the contralateral normal and denervated gastrocnemius muscles were harvested for each experiment. All animal experiments were approved in advance by the Ethics Review Committee for Animal Experimentation of the National Institute for Longevity Sciences and the National Center for Geriatrics and Gerontology. Student's t-tests were used to evaluate statistical differences between the two groups.
Znf216-deficient mice
Generation of heterozygous Znf216+/lex mice was essentially done by the gene trap method at Lexicon Genetics (Zambrowicz et al, 1998). Briefly, ES cells heterozygous for the trapped Znf216 gene were microinjected into eight-cell-stage ICR mouse embryos and transplanted into uteri. Chimeric mice were crossed to C57BL/6J mice. Northern and immunoblot analyses confirmed disruption of the gene (see text). For genotyping, primers were as follows: KO-A, ACCGACAGGATAGACAATGGCAGAG; KO-B, CGATTTTAAGAAAGGAGGCTCTGACC; LTR2, AAATGGCGTTACTTAAGCTAGCTTGC. The wild-type and inserted alleles were detected by PCR using KO-A and KO-B (0.5 kb), and LTR2 and KO-B (0.3 kb), respectively.
EGFP-CL1 degradation assay
The nucleotide sequence encoding the CL1 peptide (ACKNWFSSLSHFVIHL) (Gilon et al, 1998) was inserted into the XhoI/EcoRI site of pEGFP-C3, and the resulting plasmid was designated pEGFP-CL1. A cell line stably expressing EGFP-CL1 (293EGFP-CL1) was generated by transfection of pEGFP-CL1 into 293 cells. For the degradation assay, ZNF216 expression vectors were transfected into 293EGFP-CL1 cells and cells were harvested 48 h after transfection. MG132 (final 10 μM) or cycloheximide (final 100 μg/ml) was added to the culture at 12 or 1 h before harvest, respectively. Protein extraction was as described above.
For more details on supplementary Materials and methods, see Supplementary data
Supplementary Material
Supplementary Information
Acknowledgments
We are grateful to Drs Kazuhiro Iwai (Osaka City University) and Noboru Motoyama (NCGG) for reagents, helpful comments and suggestions throughout this study. We also thank Drs Akio Matsuda and Tatsuo Furuyama for experimental instruction and advice; Dr Aya Sasaki for pathological determinations; Ms Miho Kamiya and Ms Kumi Tsutsumi for technical assistance; and Dr Elise Lamar for proofreading the manuscript. This study is supported in part by the Program for Promotion of Fundamental Studies in Health Sciences of the Organization for Pharmaceutical Safety and Research of Japan, and by a Research Grant for Longevity Sciences from the Ministry of Health, Labor and Welfare.
References
- Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390: 175–179 [DOI] [PubMed] [Google Scholar]
- Bailey JL, Wang X, England BK, Price SR, Ding X, Mitch WE (1996) The acidosis of chronic renal failure activates muscle proteolysis in rats by augmenting transcription of genes encoding proteins of the ATP-dependent ubiquitin-proteasome pathway. J Clin Invest 97: 1447–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazirgan OA, Hampton RY (2005) Cdc48-Ufd2-Rad23: the road less ubiquitinated? Nat Cell Biol 7: 207–209 [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE (2004) IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119: 285–298 [DOI] [PubMed] [Google Scholar]
- Chen L, Shinde U, Ortolan TG, Madura K (2001) Ubiquitin-associated (UBA) domains in Rad23 bind ubiquitin and promote inhibition of multi-ubiquitin chain assembly. EMBO Rep 2: 933–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427–446 [DOI] [PubMed] [Google Scholar]
- Elsasser S, Finley D (2005) Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 7: 742–749 [DOI] [PubMed] [Google Scholar]
- Funakoshi M, Sasaki T, Nishimoto T, Kobayashi H (2002) Budding yeast Dsk2p is a polyubiquitin-binding protein that can interact with the proteasome. Proc Natl Acad Sci USA 99: 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N (2002) FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem 277: 26729–26732 [DOI] [PubMed] [Google Scholar]
- Gilon T, Chomsky O, Kulka RG (1998) Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J 17: 2759–2766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Glockzin S, Ogi FX, Hengstermann A, Scheffner M, Blattner C (2003) Involvement of the DNA repair protein hHR23 in p53 degradation. Mol Cell Biol 23: 8960–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann-Petersen R, Gordon C (2004) Protein degradation: recognition of ubiquitinylated substrates. Curr Biol 14: R754–R756 [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425–479 [DOI] [PubMed] [Google Scholar]
- Hishiya A, Ikeda K, Watanabe K (2005) A RANKL-inducible gene Znf216 in osteoclast differentiation. J. Receptor Signal Transduct 25: 199–216 [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Lippe R, McBride HM, Rubino M, Woodman P, Stenmark H, Rybin V, Wilm M, Ashman K, Mann M, Zerial M (1997) A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell 90: 1149–1159 [DOI] [PubMed] [Google Scholar]
- Huang J, Teng L, Li L, Liu T, Li L, Chen D, Xu LG, Zhai Z, Shu HB (2004) ZNF216 is an A20-like and IkappaB kinase gamma-interacting inhibitor of NFkappaB activation. J Biol Chem 279: 16847–16853 [DOI] [PubMed] [Google Scholar]
- Hunter RB, Kandarian SC (2004) Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest 114: 1504–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagoe RT, Lecker SH, Gomes M, Goldberg AL (2002) Patterns of gene expression in atrophying skeletal muscles: response to food deprivation. FASEB J 16: 1697–1712 [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115: 727–738 [DOI] [PubMed] [Google Scholar]
- Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 6: 409–419 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K (2004) A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J 23: 1977–1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10: 524–530 [DOI] [PubMed] [Google Scholar]
- Korhonen L, Lindholm D (2004) The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. J Cell Biol 165: 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93: 165–176 [DOI] [PubMed] [Google Scholar]
- Lambertson D, Chen L, Madura K (1999) Pleiotropic defects caused by loss of the proteasome-interacting factors Rad23 and Rpn10 of Saccharomyces cerevisiae. Genetics 153: 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51 [DOI] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, Goldberg AL (1999) Muscle protein breakdown and the critical role of the ubiquitin–proteasome pathway in normal and disease states. J Nutr 129: 227S–237S [DOI] [PubMed] [Google Scholar]
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P (2002) Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417: 177–182 [DOI] [PubMed] [Google Scholar]
- Masuda Y, Sasaki A, Shibuya H, Ueno N, Ikeda K, Watanabe K (2001) Dlxin-1, a novel protein that binds Dlx5 and regulates its transcriptional function. J Biol Chem 276: 5331–5338 [DOI] [PubMed] [Google Scholar]
- McKinnell IW, Rudnicki MA (2004) Molecular mechanisms of muscle atrophy. Cell 119: 907–910 [DOI] [PubMed] [Google Scholar]
- Medina R, Wing SS, Haas A, Goldberg AL (1991) Activation of the ubiquitin-ATP-dependent proteolytic system in skeletal muscle during fasting and denervation atrophy. Biomed Biochim Acta 50: 347–356 [PubMed] [Google Scholar]
- Mitch WE, Goldberg AL (1996) Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N Engl J Med 335: 1897–1905 [DOI] [PubMed] [Google Scholar]
- Mitch WE, Price SR (2001) Transcription factors and muscle cachexia: is there a therapeutic target? Lancet 357: 734–735 [DOI] [PubMed] [Google Scholar]
- Natsume T, Yamauchi Y, Nakayama H, Shinkawa T, Yanagida M, Takahashi N, Isobe T (2002) A direct nanoflow liquid chromatography-tandem mass spectrometry system for interaction proteomics. Anal Chem 74: 4725–4733 [DOI] [PubMed] [Google Scholar]
- Opipari AW Jr, Boguski MS, Dixit VM (1990) The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 265: 14705–14708 [PubMed] [Google Scholar]
- Pickart CM, Cohen RE (2004) Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 5: 177–187 [DOI] [PubMed] [Google Scholar]
- Price SR, Bailey JL, Wang X, Jurkovitz C, England BK, Ding X, Phillips LS, Mitch WE (1996) Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J Clin Invest 98: 1703–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120: 73–84 [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA (2004) Protein aggregation and neurodegenerative disease. Nat Med 10 (Suppl): S10–S17 [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL (2004) IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab 287: E591–E601 [DOI] [PubMed] [Google Scholar]
- Saeki Y, Saitoh A, Toh-e A, Yokosawa H (2002) Ubiquitin-like proteins and Rpn10 play cooperative roles in ubiquitin-dependent proteolysis. Biochem Biophys Res Commun 293: 986–992 [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Greinwald JH Jr, Marietta JR, Drury S, Swiderski RE, Vinas A, DeAngelis MM, Carmi R, Ramesh A, Kraft ML, Elbedour K, Skworak AB, Friedman RA, Srikumari Srisailapathy CR, Verhoeven K, Van Gamp G, Lovett M, Deininger PL, Batzer MA, Morton CC, Keats BJ, Smith RJ, Sheffield VC (1998) Identification and mutation analysis of a cochlear-expressed, zinc finger protein gene at the DFNB7/11 and dn hearing-loss loci on human chromosome 9q and mouse chromosome 19. Gene 215: 461–469 [DOI] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S (2001) Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol 21: 8035–8044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403 [DOI] [PubMed] [Google Scholar]
- Tawa NE Jr, Odessey R, Goldberg AL (1997) Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest 100: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19: 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania R, Graumann J, Deshaies RJ (2004) Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin–proteasome system. Cell 118: 99–110 [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430: 694–699 [DOI] [PubMed] [Google Scholar]
- Wilkinson CR, Ferrell K, Penney M, Wallace M, Dubiel W, Gordon C (2000) Analysis of a gene encoding Rpn10 of the fission yeast proteasome reveals that the polyubiquitin-binding site of this subunit is essential when Rpn12/Mts3 activity is compromised. J Biol Chem 275: 15182–15192 [DOI] [PubMed] [Google Scholar]
- Wing SS, Goldberg AL (1993) Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am J Physiol 264: E668–E676 [DOI] [PubMed] [Google Scholar]
- Wing SS, Haas AL, Goldberg AL (1995) Increase in ubiquitin–protein conjugates concomitant with the increase in proteolysis in rat skeletal muscle during starvation and atrophy denervation. Biochem J 307 (Part 3): 639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Deveraux Q, Beal RE, Pickart CM, Rechsteiner M (1998) Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J Biol Chem 273: 5461–5467 [DOI] [PubMed] [Google Scholar]
- Zambrowicz BP, Friedrich GA, Buxton EC, Lilleberg SL, Person C, Sands AT (1998) Disruption and sequence identification of 2000 genes in mouse embryonic stem cells. Nature 392: 608–611 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information