

Abstract
The unexpected encounter, 10 years ago, between human immunodeficiency virus (HIV) and the chemokine system has dramatically advanced our understanding of the pathogenesis of AIDS, opening new perspectives for the development of effective prophylactic and therapeutic measures. To initiate infection, the HIV-1 external envelope glycoprotein, gp120, sequentially interacts with two cellular receptors, CD4 and a chemokine receptor (or coreceptor) like CCR5 or CXCR4. This peculiar two-stage receptor-interaction strategy allows gp120 to maintain the highly conserved coreceptor-binding site in a cryptic conformation, protected from neutralizing antibodies. The differential use of CCR5 and CXCR4 defines three HIV-1 biological variants (R5, R5X4, X4), which vary in their prevalence during the disease course. The evolutionary choice of HIV-1 to exploit chemokine receptors as cellular entry gateways has turned their chemokine ligands into endogenous antiviral factors that variably modulate viral transmission, disease progression and vaccine responses. Likewise, the natural history of HIV-1 infection is influenced by specific polymorphisms of chemokine and chemokine-receptor genes. The imminent clinical availability of coreceptor-targeted viral entry inhibitors raises new hope for bridging the gap towards a definitive cure of HIV infection.
Keywords: chemokines, HIV, pathogenesis, therapy, viral receptors
The first close encounters
For more than 12 years after the discovery of human immunodeficiency virus (HIV) and its identification as the causative agent of AIDS (Gallo and Montagnier, 2003), the intimate relation that this virus entertains with the chemokine system has remained elusory. The first connecting threads were independently established, with a remarkable temporal coincidence, by two research teams between the end of 1995 and the spring of 1996, culminating a long quest for two critical ‘missing links' in HIV biology: on one side, the nature of the HIV-suppressive factor(s) released by CD8+ T cells, whose major components were identified as three inflammatory CC chemokines, RANTES (CC-chemokine ligand 5 or CCL5), MIP-1α (CCL3) and MIP-1β (CCL4) (Cocchi et al, 1995); on the other, the identity of a second HIV cellular receptor (or coreceptor), besides CD4, which was shown to be a chemokine receptor-like molecule (Feng et al, 1996), later named CXCR4. The extraordinary impact that these discoveries had on the HIV field is attested by the immediate chain reaction of breakthroughs that they triggered: within less than 1 year, five groups simultaneously reported the identity of a second major HIV coreceptor, CCR5; the discovery of a 32-bp deletion within the coding sequence of the CCR5 gene (CCR5-Δ32) provided the first conclusive evidence of genetically based resistance to HIV infection; and the CXC chemokine SDF-1 (CXCL12) was identified as a specific CXCR4 ligand (reviewed by Berger et al, 1999). This fortunate season was followed by a series of additional findings that, altogether, have greatly advanced our knowledge of the biology and pathogenesis of HIV infection, opening new perspectives for the development of effective measures for the control of AIDS.
New insights into the HIV entry mechanism
A two-stage receptor-interaction strategy
HIV has evolved a unique strategy of interaction with its cellular receptors, which provides an effective mechanism for concealing highly conserved neutralization epitopes from the attack of host antibodies. On the virion surface, the viral envelope is arranged in spike-like structures formed by trimeric complexes of gp120, the external subunit that mediates virion attachment, and gp41, the transmembrane subunit that mediates the fusion process. To trigger HIV entry, gp120 must sequentially engage two cellular receptor molecules: the CD4 glycoprotein and a coreceptor such as CCR5 or CXCR4. This two-stage receptor-interaction strategy allows HIV-1 to maintain the highly conserved coreceptor-binding surface in a cryptic conformation, unraveling it only upon binding of gp120 to CD4 (Figure 1). However, this occurs in a sterically and temporally constrained setting in close proximity to the cellular membrane, beyond the reach of complete antibody molecules (Labrijn et al, 2003). The detection, albeit infrequent, of primary strains of HIV-1 (Zerhouni et al, 2004; Decker et al, 2005) and HIV-2 (Reeves et al, 1999) capable of infecting coreceptor-expressing cells in a CD4-independent fashion has led to postulate the existence of an ancestral HIV that could directly bind to coreceptors without the facilitating effect of CD4. The Achilles' heel of this putative ancestor and its present-day descendants is their marked sensitivity to antibody-mediated neutralization due to a constitutive exposure of the coreceptor-binding region (Kolchinsky et al, 2001). Consistent with this concept, infected individuals possess high titers of antibodies specific for such region (Decker et al, 2005), most likely elicited by shed monomeric gp120 complexed with cell-surface CD4, which continuously patrol against the in vivo emergence of CD4-independent variants.
Figure 1.
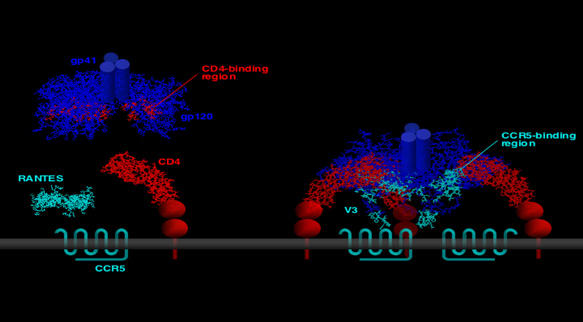
Two-stage interaction of the HIV envelope with its cellular receptors. The native, unbound envelope homotrimer (left panel; structure derived from Chen et al, 2005) exposes the CD4-binding surface but maintains the coreceptor-binding surface in a cryptic conformation. After binding to CD4, gp120 undergoes dramatic conformational changes (right panel; structure derived from Huang et al, 2005) that lead to de novo formation and/or unshielding of the high-affinity coreceptor-binding site.
Molecular anatomy of the coreceptor-binding surface of gp120
Mutagenesis and structural studies are unraveling with increasing definition the structural determinants of receptor and coreceptor recognition within the gp120 glycoprotein. In early domain-swapping experiments, the major determinants of HIV-1 coreceptor specificity were identified within the third variable (V3) domain of gp120 (Choe et al, 1996; Cocchi et al, 1996), which makes direct contact with the coreceptor during the viral entry process (Figure 1). However, the largest coreceptor-binding surface is provided by another gp120 region, the so-called ‘bridging sheet' and adjacent structures (Rizzuto et al, 1998). Being highly conserved, the ‘bridging sheet' is believed to provide the common determinants of coreceptor recognition, whereas the structure and charge of the variable V3 loop dictate the specificity for CCR5 or CXCR4.
Gp120 has been crystallized in two alternative conformations. Early studies were performed with CD4-complexed HIV-1 gp120, that is the competent form for high-affinity coreceptor binding (Kwong et al, 1998): in this conformation, the ‘bridging sheet' appears as a four-stranded antiparallel β-sheet (β2–β3 from the V1/V2 stem and β20–β21 from the C4 region) connecting the two major gp120 domains (inner and outer). More recently, crystals were obtained with CD4-unbound simian immunodeficiency virus (SIV) gp120 (Chen et al, 2005), permitting one to visualize the native conformation before the dramatic structural reorganization induced by CD4 binding. Strikingly, the ‘bridging sheet' in this structure is split into two β-ribbons, separated by a 20–25 Å space occupied by the α1 helix, providing a structural basis for the lack of coreceptor-binding competence of CD4-unbound gp120. One of the major limitations of the available structural information is that it was derived using monomeric gp120, which is immunologically and functionally different from the native trimer presented on the surface of infectious virions. Moreover, despite the recent crystallization of a CD4-bound HIV-1 gp120 containing the V3 region, which appears as an extended domain protruding toward the target cell membrane (Huang et al, 2005), the structural basis of the coreceptor specificity of gp120 remains undefined.
Functional domains of the coreceptors
All the efforts aimed at defining the three-dimensional structure of the HIV coreceptors have so far been hampered by the inherent difficulties in crystallizing molecules belonging to the seven-transmembrane-domain G-protein-coupled receptor superfamily. Although studies with mutated or chimeric molecules have provided important information on the domains involved in the interaction with gp120, the emerging picture is complex. There is consensus that the N-terminal domain plays a critical role in the HIV-coreceptor function of both CCR5 (Doranz et al, 1997) and CXCR4 (Brelot et al, 1997). This domain is post-translationally modified by the addition of sulfate moieties on tyrosine residues, which should facilitate electrostatic interactions with positively charged amino acids in the ‘bridging sheet' and the V3 base (Farzan et al, 1999). Another domain that is certainly involved in the HIV entry process is the second extracellular loop, which is believed to interact with the tip of V3 (Figure 1). However, the available data are not univocal, particularly for CXCR4, and other domains may also play a role. The prevalent opinion is that the gp120-binding surface of the coreceptors is complex and varies, to some extent, according to the viral envelope examined, with critical residues dispersed throughout the extracellular domains. Of note, the signaling function of the coreceptors is not required for HIV entry (Cocchi et al, 1996; Doranz et al, 1997).
New insights into pathogenesis
A novel classification system
Long before the discovery of the viral coreceptors, different biological phenotypes of HIV-1 were recognized, and their prevalence was correlated with the clinical stage (Schuitemaker et al, 1992). The merging with the chemokine field has permitted to unveil the physiological basis of the biological variability of HIV-1, that is the differential usage of the two major coreceptors, CCR5 and CXCR4. As a result, a novel classification system has been devised, with three main viral variants distinguished on the basis of their ability to use selectively CCR5 (R5) or CXCR4 (X4), or either coreceptor interchangeably (R5X4). This classification has replaced the old and obsolete nomenclature based on cellular tropism (T-cell- versus macrophage-tropic) or giant multinucleated cell formation (syncytia versus non-syncytia inducer). It is now increasingly recognized that such distinctions were essentially laboratory artifacts: all primary HIV-1 isolates can indeed grow in primary CD4+ T cells and readily induce the formation of syncytia in cells that express the appropriate coreceptors; furthermore, despite a persisting prejudice, most primary HIV-1 isolates have the ability to replicate to some extent in primary macrophage cultures, regardless of their coreceptor preference (Scarlatti et al, 1997; Simmons et al, 1998; Verani et al, 1998). A restriction in cellular tropism is seen mainly with neoplastic CD4+ T-cell lines that, with rare exceptions, selectively express CXCR4 and therefore fail to support the growth of R5 isolates.
A virus with a ‘switch': in vivo evolution of HIV-1
Similar to other RNA viruses, HIV-1 is characterized by a high degree of genetic heterogeneity, classified into three hierarchical strata: genetic subtypes, isolates and quasispecies. Coreceptor specificity represents an additional, transversal stratum of heterogeneity, which often shows a characteristic evolution pattern during the natural course of HIV infection (Connor et al, 1997; Scarlatti et al, 1997). The HIV-1 strains that are most commonly responsible for transmission and predominate during the long asymptomatic phase are almost invariably restricted to CCR5 usage (R5). They replicate efficiently in activated/memory cells, which express high levels of CCR5 and are particularly abundant in the gut-associated lymphoid tissue, which is now recognized as a primary site of HIV and SIV replication (Veazey et al, 1998; Brenchley et al, 2004). Later in the infection course, concomitant with immunologic and clinical signs heralding disease progression, HIV-1 strains may emerge that use CXCR4, a homeostatic chemokine receptor expressed on a broader range of cells, including naïve/resting T cells and thymic precursors. This viral ‘phenotypic switch' is likely to be fostered by the selective pressure of endogenous CCR5-binding chemokines, to which CXCR4-using strains are resistant. Such strains often display a promiscuous coreceptor usage, not only maintaining their ability to enter cells via CCR5, but also variably utilizing a series of minor coreceptors, like CCR2b, CCR3, CCR8, CX3CR1, CXCR6, D6 and RDC1; the latter was recently identified as an alternative SDF-1 receptor, besides CXCR4, and provisionally named CXCR7 (Balabanian et al, 2005). Of note, patients may harbor mixed viral populations encompassing pure R5 strains along with CXCR4-using ones (Scarlatti et al, 1997). Whether a highly promiscuous coreceptor usage constitutes a bona fide virulence factor for HIV remains uncertain. Unlike CCR5 and CXCR4, which are expressed and functional in the two major HIV-1 target cells, CD4+ T lymphocytes and mononuclear phagocytes, most of the minor coreceptors show a low and/or tissue-specific expression pattern. Moreover, during the terminal stages of the disease, promiscuous viruses tend to be lost and replaced by pure X4 variants. However, it is possible that HIV-1 exploits minor coreceptors to colonize specific cellular or anatomical niches where the major coreceptors are limited or absent, as illustrated by D6-mediated infection of astrocytes (Neil et al, 2005).
What is the biological significance of the HIV-1 ‘phenotypic switch'? Based on its temporal relation with the disease onset, the emergence of CXCR4-using viral variants has traditionally been viewed as a ‘causal' event that triggers a rapid acceleration of the disease. Additional ground to this concept is provided by the higher virulence or pathogenicity displayed by CXCR4-using strains in some experimental models, including their ability to induce an early in vivo loss (via depletion and/or activation) of naïve/resting T cells in macaques (Ho et al, 2005). However, the simplistic model whereby HIV-1 evolves from a benign R5 form to a highly pathogenic CXCR4-using one that is the true cause of immunodeficiency is increasingly under challenge. One of the most evident inconsistencies is the fact that a large fraction of patients progress to full-blown AIDS without ever experiencing a ‘switch' to CXCR4 usage. Although late isolates from these patients seem to be inherently more pathogenic and RANTES-resistant than early isolates (Karlsson et al, 2003; Koning et al, 2003), they apparently remain ‘monogamous' CCR5 users throughout the disease course. This pattern is similar to that observed in non-human primates infected with SIV, which never evolves to use CXCR4 even though its pathogenicity may increase during the late disease stages (Kimata et al, 1999). Furthermore, studies by molecular amplification have proven that the emergence of CXCR4-using variants is neither a dominant nor an irreversible phenomenon: DNA sequences predictive of CXCR4 usage may be present in blood cells without any detectable replication of such variants, and in many patients CXCR4-using strains appear only transiently on a background of sustained R5 persistence (Ida et al, 1997; Shankarappa et al, 1999; Jensen et al, 2003). Likewise, CXCR4-using viruses are preferentially cleared after the implementation of effective antiretroviral therapy (Philpott et al, 2001). Altogether, these observations prove that CXCR4 usage is not an essential condition for the development of AIDS. If we revert the cause–effect relation, CXCR4-using HIV-1 might be regarded as a unique type of ‘consanguine' opportunistic agent, which can only emerge after the development of the immunodeficiency patiently prepared by its R5 counterpart. As is the case with opportunistic agents, the early in vivo emergence of CXCR4-using strains seems to be hindered by negative selective forces, which are then weakened or lost with the progressive deterioration of immune functions.
What is the nature of the selective forces that restrict the use of CXCR4 during the initial phases of HIV infection? A putative transmission bias at the mucosal level, mediated at least in part by constitutive SDF-1 secretion (Agace et al, 2000), is contradicted by transmission studies in non-human primates, as well as by the observation of an early R5 predominance also in hemophiliacs and intravenous drug users who acquired HIV-1 by the parenteral route. A biased in vivo expansion of R5 strains has been postulated based on the preferential tropism of such strains for CCR5hi activated/memory CD4+ T cells, which would result in a larger viral burst size (Eckstein et al, 2001); however, this model is contradicted by the fact that most activated/memory CD4+ T cells also express CXCR4, and that many primary CXCR4-using HIV-1 isolates are in fact dual tropic (R5X4) and therefore can also utilize CCR5 for entry. Thus, immunologic mechanisms (cellular or humoral) remain the most likely factors that prevent the early in vivo emergence of CXCR4-using HIV-1. Several lines of evidence corroborate this concept, including the selective purging of CXCR4-using variants observed at the time of antibody seroconversion in patients (Cornelissen et al, 1995; Lathey et al, 1997) and monkeys (Harouse et al, 2003) initially infected with a mixed (R5+X4) viral population, the re-emergence of X4 strains in dually infected monkeys after in vivo depletion of CD8+ T cells (Harouse et al, 2003) and the recent paradigm shift indicating that the sensitivity of primary HIV-1 strains to antibody-mediated neutralization may correlate with coreceptor usage (Lusso et al, 2005).
Manipulation of the chemokine system by opportunistic agents
The chemokine system is a primary target for microbial manipulation, further attesting to its pivotal role in the orchestration of protective immune responses. Two families of large DNA viruses, the Herpesviridae and Poxviridae, have perfected the art of ‘molecular piracy' during their evolution, hijacking and often reprogramming to their advantage multiple host genes involved in cell survival and antimicrobial immunity, including genes of the chemokine system (reviewed by Lusso, 2000). Specifically, human cytomegalovirus encodes a functional HIV coreceptor homologous to human CCR1, whereas human herpesvirus (HHV)-8 encodes MIP-1-related viral chemokines that block HIV infection. Although these two herpesviruses cause severe opportunistic diseases in AIDS, it is still unclear whether their chemokine-system homologues effectively modulate the cellular tropism and replication capacity of HIV-1 in vivo.
Another intriguing observation is that several agents that actively replicate in HIV-infected patients are potent inducers of HIV-suppressive chemokines (Margolis, 2003). For example, the favorable effect of coinfection with GB virus on the progression of HIV disease was associated with the ability of this flavivirus to induce the secretion of SDF-1 and CCR5-binding chemokines (Xiang et al, 2004). Likewise, HHV-6, a CD4+ T-lymphotropic virus frequently reactivated during the early symptomatic phase of HIV infection (Lusso and Gallo, 1995), was shown to induce high levels of RANTES in lymphoid tissue ex vivo, conferring a selective replication advantage to CXCR4-using strains (Grivel et al, 2001). This model suggests a novel mechanism, triggered by opportunistic infections, for the ‘phenotypic switch' of HIV-1.
Death by gp120: T-cell and neuronal apoptosis mediated by coreceptor engagement
Binding of virion-associated or soluble gp120 to coreceptors is not merely a mechanical event; quite to the contrary, it may induce physiological effects that are relevant to pathogenesis (Weissman et al, 1997). In particular, engagement of CCR5 (Algeciras-Schimnich et al, 2002) or CXCR4 (Berndt et al, 1998) was shown to trigger apoptosis of both infected and uninfected T cells, which may represent a fundamental mechanism of immunologic damage in AIDS. Considerable interest is also focused on neuronal damage secondary to CXCR4 engagement by gp120 as a potential mechanism in the development of AIDS–dementia complex (reviewed by Gonzalez-Scarano and Martin-Garcia, 2005).
Implications for natural and acquired immunity to HIV
Chemokines and other endogenous non-cytolytic mechanisms of virus control
The paradigm that soluble factors contribute to the control of viral infections without causing destruction of infected cells was established half a century ago with the discovery of type-I interferon (Isaacs and Lindenmann, 1957). Non-cytolytic mechanisms of virus control have been documented in different models, including HIV (Walker et al, 1986) and hepatitis B virus (Guidotti et al, 1999). Although interferon remains the antiviral cytokine par excellence, there is increasing awareness that viral replication can be controlled through the cooperative action of multiple factors secreted by different cells taking part in inflammatory reactions. In conflict with this prevalent view, however, the quest for the identification of the HIV-suppressive factors produced by CD8+ T cells, often referred to as CD8 antiviral factor (CAF), has long been dominated by the ‘single-factor' hypothesis, which has attempted to reconcile into a unifying theory a variety of results obtained in diverse experimental systems (Levy et al, 1996). When RANTES, MIP-1α and MIP-1β were identified as major components of the HIV-suppressive activity of CD8+ T cells (Cocchi et al, 1995), it was immediately evident that CAF could no longer be ascribed to a single factor. Although the dominant role of CCR5-binding chemokines as CD8-derived HIV-suppressive factors has been widely confirmed, it is indisputable that CD8+ T cells, and other immune cells alike, produce a complex array of substances with antiviral activity, some of which still await discovery. For example, both CD8+ T cells (Yang et al, 1997) and macrophages (Verani et al, 2002) release soluble factors that block CXCR4-using HIV-1 variants; albeit still unrecognized, such factors are by definition distinct from CCR5-binding chemokines. In addition, the non-lytic HIV-suppressive activity of CD8+ T cells is consistently stronger when a direct contact with infected target cells is allowed (Levy et al, 1996; Chun et al, 2001; Furci et al, 2002), indicating that non-soluble (i.e. cell-associated) mechanisms are also operational. Altogether, these considerations reinforce the concept that a unifying theory of CAF is insufficient to account for the variety and complexity of experimental results thus far accumulated.
What is the in vivo relevance of chemokines and other soluble factors as virus control mechanisms? Although several studies have attempted to establish a correlation between the clinical stage of HIV infection and the ability of CD8+ or CD4+ T cells to produce HIV-suppressive chemokines (or other suppressive factors), the results are still inconclusive. To further confuse the puzzle, there is growing awareness that such ability is determined, at least in part, by constitutive factors, as shown by the elevated chemokine levels measured in selected individuals or monkeys without previous retroviral exposure (Lehner et al, 1996; Rosok et al, 1997; Furci et al, 2002) as well as, more compellingly, in subjects carrying specific genetic traits (Paxton et al, 1998; Liu et al, 1999; Gonzalez et al, 2005). Despite the lack of clearcut correlation with the clinical stage, it is unquestionable that the endogenous production of soluble antiviral factors within the genital mucosa, lymphoid tissue or other anatomical sites may significantly affect the transmission, tissue tropism and replication of HIV-1, as well as, ultimately, the pace of disease progression. For example, the sustained production of RANTES, MIP-1α and MIP-1β detectable in HIV-infected lymph nodes since the early stages of infection (Trumpfheller et al, 1998) is likely to provide an effective barrier against the spread of R5 strains, thereby contributing to the slow disease pace commonly observed in HIV-infected individuals. With the progression of the disease, even though chemokine levels may remain sustained under the drive of chronic inflammation, viral strains with reduced or lost sensitivity to such chemokines start to emerge, progressively demoting this endogenous mechanism of virus control.
Unlike CCR5-binding chemokines, the CXCR4 ligand, SDF-1, is a homeostatic chemokine that is constitutively expressed irrespective of inflammation. High levels are produced by epithelial cells of the genital and intestinal mucosae (Agace et al, 2000), suggesting a possible ‘gatekeeper' effect against the transmission and early expansion of CXCR4-using HIV-1 strains. However, its rapid inactivation by serum exopeptidases (Delgado et al, 2001) and its limited activity against primary HIV-1 strains (Scarlatti et al, 1997) argue against a major role of this chemokine in the long-term control of HIV-1 in vivo.
Lucky defects: genetic basis of HIV resistance
Insightful clinicians have long focused their attention on two peculiar groups of individuals, the so-called exposed-uninfected (EU; subjects who remain persistently HIV-seronegative despite repeated high-risk sexual contacts) and the long-term non-progressors (HIV-infected subjects who maintain normal levels of CD4+ T cells and low levels of viremia for prolonged periods in the absence of therapy), hoping that they might hold the key to elucidating mechanisms of natural resistance to HIV infection and disease. Soon after the connection between HIV and the chemokine system was established, genetic studies in EU subjects provided the first conclusive evidence that HIV resistance may be genetically determined, with the identification of a 32-bp deletion within the coding region of the CCR5 gene (CCR5-Δ32) that introduces a premature stop codon, resulting in a defective molecule that is not transported to the cellular surface (Liu et al, 1996; Samson et al, 1996). The CCR5-Δ32 allele shows the highest frequency in Europe with a distinct north-to-south gradient, whereas it is extremely rare among east Asians, native Americans and Africans. In homozygotes, this ‘lucky defect' confers a high degree of protection against HIV infection. Only a handful of cases of infected homozygotes has been reported, all carrying exclusively CXCR4-tropic HIV-1 strains; interestingly, the viral load in these subjects tends to be low (Sheppard et al, 2002), reinforcing the concept that CXCR4-using viral strains face inherent obstacles in their replication in immunocompetent hosts. Although CCR5-Δ32 heterozygotes are not resistant to HIV infection, their disease progression is significantly delayed, most likely as a consequence of reduced CCR5 expression levels. Strikingly, the complete absence of CCR5 in homozygotes is not associated with any obvious clinical phenotype, even though the intensity of certain immune responses may be reduced (Fischereder et al, 2001). A second crippling polymorphism, m303, that introduces a premature stop codon in the CCR5 gene has been identified in EU subjects (Quillent et al, 1998). These extraordinary gene knockout ‘experiments of nature' reiterate the central role played by CCR5 in the physiology of HIV infection and provide an important proof-of-principle of the potential safety of CCR5-targeted therapeutic strategies.
Studies of large epidemiologic cohorts have permitted to identify a series of other polymorphisms or duplications of chemokine and chemokine-receptor genes that exert variable effects on the progression of HIV disease (for a summary and references, see Supplementary Table 1). An important concept that is emerging from these studies is that the disease pace in each individual is not determined by a single genetic polymorphism, but rather by the combined effects, often divergent, of multiple genes, and is further modulated by the genetic background of the racial group.
Implications for an HIV vaccine
The connection between HIV and the chemokine system also has implications for the development of an effective HIV vaccine. First, it has fostered our understanding of the early interactions between the viral envelope and its cellular receptors, which involve most of the conserved epitopes targeted by broadly neutralizing antibodies, the primary ingredient of a protective vaccine (Zinkernagel, 2003). Unfortunately, HIV-1 has evolved an extraordinary array of shielding mechanisms that prevent the elicitation of and recognition by such antibodies (Wyatt et al, 1998). One of the most effective, as discussed above, is the cryptic conformation adopted by the coreceptor-binding site of gp120. Thus, despite its critical role in the viral entry process and its documented immunogenicity in humans (Decker et al, 2005), this region is generally discounted as a vaccine target. Nevertheless, some epitopes overlapping or neighboring the coreceptor-binding surface are at least partially accessible in the native, CD4-unbound envelope oligomer (Moulard et al, 2002; Labrijn et al, 2003), providing a basis for the use of the CD4-triggered envelope or rationally designed synthetic immunogens mimicking this region as a means to induce broadly protective antibodies.
A second implication is the potential value of chemokines as correlate markers of vaccine-elicited protection, as suggested by studies in non-human primate models (Lehner et al, 1996). The mechanisms of chemokine induction by vaccination are still incompletely understood. Chemokine secretion is physiologically associated not only with innate but also with adaptive immune responses, as seen with nominal antigen-stimulated CD8+ and CD4+ T cells (Yang et al, 1997). However, it is evident that the magnitude of chemokine responses is also significantly modulated by genetic factors. Will chemokines effectively play a role in the protective immunity elicited by an HIV vaccine? In any kind of vaccine, locally released chemokines contribute to the generation of protective responses through the recruitment and costimulation of specific immune cells, providing a rationale for the use of chemokines as ‘intelligent' vaccine adjuvants. In the case of HIV vaccines, however, the production of CCR5-binding chemokines will have the bonus effect of creating a refractory milieu against the R5 HIV-1 strains commonly implicated in HIV-1 transmission.
A new therapeutic principle
The past decade has witnessed extraordinary advancements in the therapy of HIV infection. The introduction of viral protease inhibitors in 1995 has permitted to formulate multidrug combination protocols, commonly referred to as highly active anti-retroviral therapy (HAART), which have dramatically changed the natural history of HIV disease, leading to long-term disease-free survival associated with durable containment of HIV replication. Despite their undeniable efficacy, HAART protocols nonetheless suffer from important limitations, including the rapid virus-replication rebound after withdrawal, the increasing emergence and transmission of multiresistant viral strains, difficulties in schedule compliance and severe side effects. Most strikingly, in the face of long-term virus suppression, not a single case of HIV eradication has been convincingly documented to date. Altogether, these considerations underscore the necessity to develop novel and more effective therapeutic tools.
By interfering with the function of the HIV coreceptors, chemokines provide a new therapeutic principle. At variance with the viral-enzyme inhibitors used in HAART, coreceptor inhibitors lock HIV outside the target cell and thereby should limit the ability of the virus to replenish its latent cellular reservoirs, which represent the primary hurdle on the way toward HIV eradication. In this regard, encouraging results have recently been reported using the prototype HIV-entry inhibitor, the gp41-targeted peptide T20, in combination with valproic acid (Lehrman et al, 2005). Coreceptor inhibitors are also receiving increasing attention as topical ‘microbicides' for the prophylaxis of HIV transmission. Proof-of-principle of the efficacy of CCR5 blockade in preventing transmission was recently obtained in female macaques using a potent chemokine analogue, PSC-RANTES (Lederman et al, 2004).
Why are chemokine receptors attractive targets for antiviral therapy? A major advantage is that they are highly conserved cellular structures and, therefore, not liable to escape mutation. Moreover, no evidence of coreceptor-independent infection has so far been reported, suggesting that coreceptors are inescapable gateways for HIV. Finally, chemokine receptors belong to the superfamily of seven-transmembrane-domain receptors, which includes several successful drug targets.
Of targets and caveats
Efforts aimed at developing HIV-1 coreceptor inhibitors have focused on CCR5 and CXCR4, the only two coreceptors of recognized biological relevance. CCR5 is undoubtedly a primary target because of its pivotal role in the transmission and spread of HIV-1 during the entire course of the infection; by contrast, CXCR4 is used almost exclusively by strains that emerge during the advanced stages of the disease. Thus, a rational strategy would be to hit CCR5 early and CXCR4 only later in the infection course. However, a popular concern related to the early use of CCR5-targeted inhibitors is the putative risk of exerting a selective pressure on the viral population, favoring the premature emergence of CXCR4-using strains. Is this a real issue? A first important consideration is that CCR5-targeted inhibitors will never be employed in monotherapy, but rather as components of effective drug combination protocols, which should minimize viral replication and, thereby, the risk of emergence of escape mutants. Moreover, as discussed above, individuals that are still immunocompetent possess inherent restrictive mechanisms that impede the emergence of CXCR4-using variants. The potency of such forces is illustrated by the lack of switch to CXCR4 usage throughout the long asymptomatic phase of HIV-1 infection despite the sustained selective pressure of endogenous CCR5-binding chemokines produced at high levels in lymphoid tissue (Trumpfheller et al, 1998). Moreover, evidence obtained both in vitro with HIV-1 (Kuhmann et al, 2004) and in vivo with SIV (Biancotto et al, unpublished) indicates that resistance to CCR5-targeted inhibitors may develop without a switch in coreceptor usage, implying that the viral envelope can devise alternative modes of interaction with CCR5. Regardless, the addition of an effective CXCR4-targeted inhibitor to the drug cocktail for asymptomatic patients might be a reasonable safety measure. The therapeutic efficacy of blocking CCR5 was recently proven in a short-term phase IIa monotherapy trial with the most advanced CCR5 antagonist, Maraviroc, which induced a >1.6 log10 reduction in plasma viral load (Fatkenheuer et al, 2005).
The use of coreceptor inhibitors late in the course of HIV infection poses a different set of caveats. Targeting CXCR4 during this phase is rational, but it has to be recalled that many CXCR4-using isolates exhibit a promiscuous coreceptor usage and therefore can bypass CXCR4 for entry. In fact, the clinical development of a small-molecule CXCR4 antagonist, AMD3100, was halted because of limited efficacy (Hendrix et al, 2004), emphasizing that blockade of CXCR4 alone is insufficient for an effective HIV control. Thus, the addition of CCR5 inhibitors is warranted even in the advanced disease stages, also in consideration of the frequent back-switch to pure CCR5 usage documented in patients responding to HAART treatment (Philpott et al, 2001).
An important concern with the use of coreceptor inhibitors is the risk of interfering with the physiology of either the inflammatory (CCR5) or homeostatic (CXCR4) chemokine systems. Blocking CCR5 is generally considered safe because its congenital deficiency in CCR5-Δ32 homozygotes and knockout mice is not associated with any obvious pathology; conversely, the use of CCR5 agonists could in principle induce inflammatory side effects. Regarding CXCR4, despite the lethal phenotype documented in knockout mice (Zou et al, 1998), its role in the adult life—that is, the physiological recirculation of leukocytes and hematopoietic precursors—is apparently less vital. Although there is no doubt that an ideal coreceptor inhibitor should be devoid of both agonistic and antagonistic activities on the chemokine-receptor function, most of the inhibitors identified to date are not. Finally, another critical issue that needs to be considered is selectivity: coreceptor inhibitors may crossreact with other receptors of the same superfamily, which are widespread in higher organisms, causing side effects unrelated to interference with the chemokine system.
Uncoupling HIV blockade from receptor activation
Two main strategies have been undertaken for the generation of coreceptor-targeted inhibitors: the high-throughput screening of large chemical libraries and rational design based on the structure of HIV-suppressive chemokines. A series of inhibitors targeting CCR5 and/or CXCR4 have been obtained, including small-molecule inhibitors, modified full-length chemokines, shorter derivative peptides, antibodies and gene therapy approaches (for a summary and references, see Supplementary Table 2). Some of these molecules have already progressed through the initial stages of clinical experimentation. Of note, the development of two small-molecule allosteric CCR5 antagonists, SCH-C and Aplaviroc, was recently halted because of cardiac and hepatic toxicity, respectively, most likely due to crossreactivity with structurally related receptors in these organs. These problems emphasize the potential limitations of high-throughput screening strategies for the discovery of inhibitors with high target selectivity.
A variety of approaches have been devised for the rational design of coreceptor inhibitors. One of the most widely explored has been the introduction of targeted alterations into native chemokines with the aim of reducing their receptor-agonistic activity and/or enhancing their antiviral potency. The addition of hydrophobic groups to the amino-terminus of RANTES has yielded potent anti-HIV molecules, such as AOP-, NNY- and PSC-RANTES, which induce sustained CCR5 downmodulation (Hartley and Offord, 2005). However, these analogues maintain a strong agonistic activity, which is undesirable in the perspective of a clinical use; moreover, the need for chemical modifications makes them unsuitable for in vivo expression strategies. A partial dissociation of the antiviral and receptor-triggering functions was obtained with another analogue, C1C5-RANTES, in which the N-terminal domain is profoundly rearranged owing to the introduction of a new disulfide bond (Polo et al, 2000). As this analogue contains only natural amino acids, it can be expressed in vivo using viral or bacterial vectors.
An alternative approach has stemmed from structure–function studies of RANTES (Nardese et al, 2001). The major determinants of anti-HIV activity have been mapped to two clusters of aromatic residues within the N-loop and β1-strand regions, respectively, which contribute to the formation of a large solvent-exposed hydrophobic surface postulated to serve as the major CCR5-binding interface (Figure 2). Dimeric peptides derived from such regions exhibit specific activity against R5 HIV-1 strains, but fail to induce receptor activation, which in RANTES is mediated by the N-terminal domain (Gong et al, 1996); to the contrary, they act as receptor antagonists and therefore might exert anti-inflammatory ‘side effects' of potential benefit in the treatment of AIDS. These results have provided the most conclusive evidence that the antiviral and signaling functions of CCR5-binding chemokines can be uncoupled, an essential principle for the development of safe and effective coreceptor inhibitors. The structure–function relations are different in SDF-1, where both the antiviral and signaling activities are mediated by the N-terminal domain (Heveker et al, 1998). Structure-guided modifications of the native N-loop/β1-strand sequence of RANTES have led to the production of second- and third-generation peptides with antiviral activity within the nanomolar range (P Lusso et al, unpublished). This approach illustrates how, in the absence of structural information on the target receptor, the structure of a naturally selected ligand can be exploited as a guide to the rational design of specific inhibitors.
Figure 2.
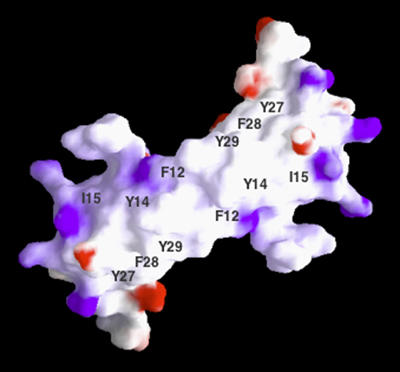
Structure of the putative CCR5-binding region of RANTES. Molecular surface representation (by GRASP) of the NMR solution structure of the RANTES dimer showing a large hydrophobic surface, lined by charged residues, where critical amino acids for CCR5 interaction and anti-HIV activity were mapped. Blue denotes positive electrical charge; red, negative charge; white, no charge (adapted from Nardese et al, 2001).
Conclusions and perspectives
The collision between the worlds of HIV and the chemokine system has had extraordinary inseminating effects, disclosing new horizons to our knowledge of the biology and pathogenesis of HIV infection. The advancements made in this field over the past 10 years have raised a cautious optimism on the possibility of developing novel therapeutic and prophylactic measures for the control of the AIDS epidemics. One of the most successful examples is the imminent availability of safe and effective CCR5- and CXCR4-targeted inhibitors. Their inclusion in multidrug combination protocols will permit to fully evaluate the impact of viral-entry blockade on the formation of latent HIV cellular reservoirs, which constitute the primary stumbling block on the way toward the final goal of HIV eradication.
Supplementary Material
Supplementary Material
References
- Agace WW, Amara A, Roberts AI, Pablos JL, Thelen S, Uguccioni M, Li XY, Marsal J, Arenzana-Seisdedos F, Delaunay T, Ebert EC, Moser B, Parker CM (2000) Constitutive expression of stromal derived factor-1 by mucosal epithelia and its role in HIV transmission and propagation. Curr Biol 10: 325–328 [DOI] [PubMed] [Google Scholar]
- Algeciras-Schimnich A, Vlahakis SR, Villasis-Keever A, Gomez T, Heppelmann CJ, Bou G, Paya CV (2002) CCR5 mediates Fas- and caspase-8 dependent apoptosis of both uninfected and HIV infected primary human CD4T cells. AIDS 16: 1467–1478 [DOI] [PubMed] [Google Scholar]
- Balabanian K, Lagane B, Infantino S, Chow KY, Harriague J, Moepps B, Arenzana-Seisdedos F, Thelen M, Bachelerie F (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J Biol Chem 280: 35760–35766 [DOI] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol 17: 657–700 [DOI] [PubMed] [Google Scholar]
- Berndt C, Mopps B, Angermuller S, Gierschik P, Krammer PH (1998) CXCR4 and CD4 mediate a rapid CD95-independent cell death in CD4+ T cells. Proc Natl Acad Sci USA 95: 12556–12561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brelot A, Heveker N, Pleskoff O, Sol N, Alizon M (1997) Role of the first and third extracellular domains of CXCR-4 in human immunodeficiency virus coreceptor activity. J Virol 71: 4744–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC (2004) CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200: 749–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC (2005) Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433: 834–841 [DOI] [PubMed] [Google Scholar]
- Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J (1996) The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85: 1135–1148 [DOI] [PubMed] [Google Scholar]
- Chun TW, Justement JS, Moir S, Hallahan CW, Ehler LA, Liu S, McLaughlin M, Dybul M, Mican JM, Fauci AS (2001) Suppression of HIV replication in the resting CD4+ T cell reservoir by autologous CD8+ T cells: implications for the development of therapeutic strategies. Proc Natl Acad Sci USA 98: 253–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P (1995) Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science 270: 1811–1815 [DOI] [PubMed] [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Cara A, Gallo RC, Lusso P (1996) The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat Med 2: 1244–1247 [DOI] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR (1997) Change in coreceptor use coreceptor use correlates with disease progression in HIV-1-infected individuals. J Exp Med 185: 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen M, Mulder-Kampinga G, Veenstra J, Zorgdrager F, Kuiken C, Hartman S, Dekker J, van der Hoek L, Sol C, Coutinho R, Goudsmit J (1995) Syncytium-inducing (SI) phenotype suppression at seroconversion after intramuscular inoculation of a non-syncytium-inducing/SI phenotypically mixed human immunodeficiency virus population. J Virol 69: 1810–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM (2005) Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med 201: 1407–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MB, Clark-Lewis I, Loetscher P, Langen H, Thelen M, Baggiolini M, Wolf M (2001) Rapid inactivation of stromal cell-derived factor-1 by cathepsin G associated with lymphocytes. Eur J Immunol 31: 699–707 [DOI] [PubMed] [Google Scholar]
- Doranz BJ, Lu ZH, Rucker J, Zhang TY, Sharron M, Cen YH, Wang ZX, Guo HH, Du JG, Accavitti MA, Doms RW, Peiper SC (1997) Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J Virol 71: 6305–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, Roederer M, Sherman MP, Chin PS, Goldsmith MA (2001) HIV-1 actively replicates in naive CD4+ T cells residing within human lymphoid tissues. Immunity 15: 671–682 [DOI] [PubMed] [Google Scholar]
- Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H (1999) Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96: 667–676 [DOI] [PubMed] [Google Scholar]
- Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, Zavolan M, van der Ryst E (2005) Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11: 1170–1172 [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE, Berger EA (1996) HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272: 872–877 [DOI] [PubMed] [Google Scholar]
- Fischereder M, Luckow B, Hocher B, Wuthrich RP, Rothenpieler U, Schneeberger H, Panzer U, Stahl RA, Hauser IA, Budde K, Neumayer H, Kramer BK, Land W, Schlondorff D (2001) CC chemokine receptor 5 and renal-transplant survival. Lancet 357: 1758–1761 [DOI] [PubMed] [Google Scholar]
- Furci L, Lopalco L, Loverro P, Sinnone M, Tambussi G, Lazzarin A, Lusso P (2002) Non-cytotoxic inhibition of HIV-1 infection by unstimulated CD8+ T lymphocytes from HIV-exposed-uninfected individuals. AIDS 16: 1003–1008 [DOI] [PubMed] [Google Scholar]
- Gallo RC, Montagnier L (2003) The discovery of HIV as the cause of AIDS. N Engl J Med 349: 2283–2285 [DOI] [PubMed] [Google Scholar]
- Gong JH, Uguccioni M, Dewald B, Baggiolini M, Clark-Lewis I (1996) RANTES and MCP-3 antagonists bind multiple chemokine receptors. J Biol Chem 271: 10521–10527 [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, Murthy KK, Rovin BH, Bradley W, Clark RA, Anderson SA, O'Connell RJ, Agan BK, Ahuja SS, Bologna R, Sen L, Dolan MJ, Ahuja SK (2005) The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307: 1434–1440 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J (2005) The neuropathogenesis of AIDS. Nat Rev Immunol 5: 69–81 [DOI] [PubMed] [Google Scholar]
- Grivel JC, Ito Y, Faga G, Santoro F, Shaheen F, Malnati MS, Fitzgerald W, Lusso P, Margolis L (2001) Suppression of CCR5- but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat Med 7: 1232–1235 [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV (1999) Viral clearance without destruction of infected cells during acute HBV infection. Science 284: 825–829 [DOI] [PubMed] [Google Scholar]
- Harouse JM, Buckner C, Gettie A, Fuller R, Bohm R, Blanchard J, Cheng-Mayer C (2003) CD8+ T cell-mediated CXC chemokine receptor 4-simian/human immunodeficiency virus suppression in dually infected rhesus macaques. Proc Natl Acad Sci USA 100: 10977–10982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley O, Offord RE (2005) Engineering chemokines to develop optimized HIV inhibitors. Curr Protein Pept Sci 6: 207–219 [DOI] [PubMed] [Google Scholar]
- Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, Jackson JB, Coombs RW, Glesby MJ, Flexner CW, Bridger GJ, Badel K, MacFarland RT, Henson GW, Calandra G (2004) Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr 37: 1253–1262 [DOI] [PubMed] [Google Scholar]
- Heveker N, Montes M, Germeroth L, Amara A, Trautmann A, Alizon M, Schneider-Mergener J (1998) Dissociation of the signalling and antiviral properties of SDF-1-derived small peptides. Curr Biol 8: 369–376 [DOI] [PubMed] [Google Scholar]
- Ho SH, Shek L, Gettie A, Blanchard J, Cheng-Mayer C (2005) V3 loop-determined coreceptor preference dictates the dynamics of CD4+ T-cell loss in simian–human immunodeficiency virus-infected macaques. J Virol 79: 12296–12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD (2005) Structure of a V3-containing HIV-1 gp120 core. Science 310: 1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ida S, Gatanaga H, Shioda T, Nagai Y, Kobayashi N, Shimada K, Kimura S, Iwamoto A, Oka S (1997) HIV type 1 V3 variation dynamics in vivo: long-term persistence of non-syncytium-inducing genotypes and transient presence of syncytium-inducing genotypes during the course of progressive AIDS. AIDS Res Hum Retroviruses 13: 1597–1609 [DOI] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J (1957) Virus interference. I. The interferon. Proc R Soc London B 147: 258–26713465720 [Google Scholar]
- Jensen MA, Li FS, van't Wout AB, Nickle DC, Shriner D, He HX, McLaughlin S, Shankarappa R, Margolick JB, Mullins JI (2003) Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J Virol 77: 13376–13388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson I, Antonsson L, Shi Y, Karlsson A, Albert J, Leitner T, Olde B, Owman C, Fenyo EM (2003) HIV biological variability unveiled: frequent isolations and chimeric receptors reveal unprecedented variation of coreceptor use. AIDS 17: 2561–2569 [DOI] [PubMed] [Google Scholar]
- Kimata JT, Kuller L, Anderson DB, Dailey P, Overbaugh J (1999) Emerging cytopathic and antigenic simian immunodeficiency virus variants influence AIDS progression. Nat Med 5: 535–541 [DOI] [PubMed] [Google Scholar]
- Kolchinsky P, Kiprilov E, Bartley P, Rubinstein R, Sodroski J (2001) Loss of a single N-linked glycan allows CD4-independent human immunodeficiency virus type 1 infection by altering the position of the gp120 V1/V2 variable loops. J Virol 75: 3435–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning FA, Kwa D, Boeser-Nunnink B, Dekker J, Vingerhoed J, Hiemstra H, Schuitemaker H (2003) Decreasing sensitivity to RANTES neutralization of CC chemokine receptor 5-using, non-syncytium-inducing virus variants in the course of human immunodeficiency virus type 1 infection. J Infect Dis 188: 864–872 [DOI] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, Strizki JM, Riley J, Baroudy BM, Wilson IA, Korber BT, Wolinsky SM, Moore JP (2004) Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol 78: 2790–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393: 648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, Venturi M, Petropoulos CJ, Wrin T, Dimitrov DS, Robinson J, Kwong PD, Wyatt RT, Sodroski J, Burton DR (2003) Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. J Virol 77: 10557–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathey JL, Pratt RD, Spector SA (1997) Appearance of autologous neutralizing antibody correlates with reduction in virus load and phenotype switch during primary infection with human immunodeficiency virus type 1. J Infect Dis 175: 231–232 [DOI] [PubMed] [Google Scholar]
- Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, Piatak M Jr, Lifson JD, Salkowitz JR, Rodriguez B, Blauvelt A, Hartley O (2004) Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306: 485–487 [DOI] [PubMed] [Google Scholar]
- Lehner T, Wang Y, Cranage M, Bergmeier LA, Mitchell E, Tao L, Hall G, Dennis M, Cook N, Brookes R, Klavinskis L, Jones I, Doyle C, Ward R (1996) Protective mucosal immunity elicited by targeted iliac lymph node immunization with a subunit SIV envelope and core vaccine in macaques. Nat Med 2: 767–775 [DOI] [PubMed] [Google Scholar]
- Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM (2005) Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366: 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JA, Mackewicz CE, Barker E (1996) Controlling HIV pathogenesis: the role of the noncytotoxic anti-HIV response of CD8+ T cells. Immunol Today 17: 217–224 [DOI] [PubMed] [Google Scholar]
- Liu H, Chao D, Nakayama EE, Taguchi H, Goto M, Xin X, Takamatsu JK, Saito H, Ishikawa Y, Akaza T, Juji T, Takebe Y, Ohishi T, Fukutake K, Maruyama Y, Yashiki S, Sonoda S, Nakamura T, Nagai Y, Iwamoto A, Shioda T (1999) Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proc Natl Acad Sci USA 96: 4581–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR (1996) Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86: 367–377 [DOI] [PubMed] [Google Scholar]
- Lusso P (2000) Chemokines and viruses: the dearest enemies. Virology 273: 228–240 [DOI] [PubMed] [Google Scholar]
- Lusso P, Earl PL, Sironi F, Santoro F, Ripamonti C, Scarlatti G, Longhi R, Berger EA, Burastero SE (2005) Cryptic nature of a conserved, CD4-inducible V3 loop neutralization epitope in the native envelope glycoprotein oligomer of CCR5-restricted, but not CXCR4-using, primary human immunodeficiency virus type 1 strains. J Virol 79: 6957–6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusso P, Gallo RC (1995) Human herpesvirus 6 in AIDS. Immunol Today 16: 67–71 [DOI] [PubMed] [Google Scholar]
- Margolis L (2003) Cytokines—strategic weapons in germ warfare? Nat Biotechnol 21: 15–16 [DOI] [PubMed] [Google Scholar]
- Moulard M, Phogat SK, Shu Y, Labrijn AF, Xiao X, Binley JM, Zhang MY, Sidorov IA, Broder CC, Robinson J, Parren PW, Burton DR, Dimitrov DS (2002) Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120–CD4–CCR5 complexes. Proc Natl Acad Sci USA 99: 6913–6918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardese V, Longhi R, Polo S, Sironi F, Arcelloni C, Paroni R, DeSantis C, Sarmientos P, Rizzi M, Bolognesi M, Pavone V, Lusso P (2001) Structural determinants of CCR5 recognition and HIV-1 blockade in RANTES. Nat Struct Biol 8: 611–615 [DOI] [PubMed] [Google Scholar]
- Neil SJ, Aasa-Chapman MM, Clapham PR, Nibbs RJ, McKnight A, Weiss RA (2005) The promiscuous CC chemokine receptor D6 is a functional coreceptor for primary isolates of human immunodeficiency virus type 1 (HIV-1) and HIV-2 on astrocytes. J Virol 79: 9618–9624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxton WA, Liu R, Kang S, Wu L, Gingeras TR, Landau NR, Mackay CR, Koup RA (1998) Reduced HIV-1 infectability of CD4+ lymphocytes from exposed-uninfected individuals: association with low expression of CCR5 and high production of β-chemokines. Virology 244: 66–73 [DOI] [PubMed] [Google Scholar]
- Philpott S, Weiser B, Anastos K, Kitchen CM, Robison E, Meyer WA, Sacks HS, Mathur-Wagh U, Brunner C, Burger H (2001) Preferential suppression of CXCR4-specific strains of HIV-1 by antiviral therapy. J Clin Invest 107: 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S, Nardese V, De Santis C, Arcelloni C, Paroni R, Sironi F, Verani A, Rizzi M, Bolognesi M, Lusso P (2000) Enhancement of the HIV-1 inhibitory activity of RANTES by modification of the N-terminal region: dissociation from CCR5 activation. Eur J Immunol 30: 3190–3198 [DOI] [PubMed] [Google Scholar]
- Quillent C, Oberlin E, Braun J, Rousset D, Gonzalez-Canali G, Metais P, Montagnier L, Virelizier JL, Arenzana-Seisdedos F, Beretta A (1998) HIV-1-resistance phenotype conferred by combination of two separate inherited mutations of CCR5 gene. Lancet 351: 14–18 [DOI] [PubMed] [Google Scholar]
- Reeves JD, Hibbitts S, Simmons G, McKnight A, Azevedo-Pereira JM, Moniz-Pereira J, Clapham PR (1999) Primary human immunodeficiency virus type 2 (HIV-2) isolates infect CD4-negative cells via CCR5 and CXCR4: comparison with HIV-1 and simian immunodeficiency virus and relevance to cell tropism in vivo. J Virol 73: 7795–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J (1998) A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280: 1949–1953 [DOI] [PubMed] [Google Scholar]
- Rosok B, Voltersvik P, Larsson BM, Albert J, Brinchmann JE, Asjo B (1997) CD8+ T cells from HIV type 1-seronegative individuals suppress virus replication in acutely infected cells. AIDS Res Hum Retroviruses 13: 79–85 [DOI] [PubMed] [Google Scholar]
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M (1996) Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382: 722–725 [DOI] [PubMed] [Google Scholar]
- Scarlatti G, Tresoldi E, Bjorndal A, Fredriksson R, Colognesi C, Deng HK, Malnati MS, Plebani A, Siccardi AG, Littman DR, Fenyo EM, Lusso P (1997) In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med 3: 1259–1265 [DOI] [PubMed] [Google Scholar]
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, Tersmette M (1992) Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol 66: 1354–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI (1999) Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73: 10489–10502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard HW, Celum C, Michael NL, O'Brien S, Dean M, Carrington M, Dondero D, Buchbinder SP (2002) HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr 29: 307–313 [DOI] [PubMed] [Google Scholar]
- Simmons G, Reeves JD, McKnight A, Dejucq N, Hibbitts S, Power CA, Aarons E, Schols D, De Clercq E, Proudfoot AE, Clapham PR (1998) CXCR4 as a functional coreceptor for human immunodeficiency virus type 1 infection of primary macrophages. J Virol 72: 8453–8457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpfheller C, Tenner-Racz K, Racz P, Fleischer B, Frosch S (1998) Expression of macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES genes in lymph nodes from HIV+ individuals: correlation with a Th1-type cytokine response. Clin Exp Immunol 112: 92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA (1998) Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280: 427–431 [DOI] [PubMed] [Google Scholar]
- Verani A, Pesenti E, Polo S, Tresoldi E, Scarlatti G, Lusso P, Siccardi AG, Vercelli D (1998) CXCR4 is a functional coreceptor for infection of human macrophages by CXCR4-dependent primary HIV-1 isolates. J Immunol 161: 2084–2088 [PubMed] [Google Scholar]
- Verani A, Sironi F, Siccardi AG, Lusso P, Vercelli D (2002) Inhibition of CXCR4-tropic HIV-1 infection by lipopolysaccharide: evidence of different mechanisms in macrophages and T lymphocytes. J Immunol 168: 6388–6395 [DOI] [PubMed] [Google Scholar]
- Walker CM, Moody DJ, Stites DP, Levy JA (1986) CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science 234: 1563–1566 [DOI] [PubMed] [Google Scholar]
- Weissman D, Rabin RL, Arthos J, Rubbert A, Dybul M, Swofford R, Venkatesan S, Farber JM, Fauci AS (1997) Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature 389: 981–985 [DOI] [PubMed] [Google Scholar]
- Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393: 705–711 [DOI] [PubMed] [Google Scholar]
- Xiang J, George SL, Wunschmann S, Chang Q, Klinzman D, Stapleton JT (2004) Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1α, MIP-1β, and SDF-1. Lancet 363: 2040–2046 [DOI] [PubMed] [Google Scholar]
- Yang OO, Kalams SA, Trocha A, Cao H, Luster A, Johnson RP, Walker BD (1997) Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolytic mechanisms. J Virol 71: 3120–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerhouni B, Nelson JA, Saha K (2004) Isolation of CD4-independent primary human immunodeficiency virus type 1 isolates that are syncytium inducing and acutely cytopathic for CD8+ lymphocytes. J Virol 78: 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel RM (2003) On natural and artificial vaccinations. Annu Rev Immunol 21: 515–546 [DOI] [PubMed] [Google Scholar]
- Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR (1998) Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393: 595–599 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material