

Abstract
The generally accepted role of iron-regulatory protein 1 (IRP1) in orchestrating the fate of iron-regulated mRNAs depends on the interconversion of its cytosolic aconitase and RNA-binding forms through assembly/disassembly of its Fe–S cluster, without altering protein abundance. Here, we show that IRP1 protein abundance can be iron-regulated. Modulation of IRP1 abundance by iron did not require assembly of the Fe–S cluster, since a mutant with all cluster-ligating cysteines mutated to serine underwent iron-induced protein degradation. Phosphorylation of IRP1 at S138 favored the RNA-binding form and promoted iron-dependent degradation. However, phosphorylation at S138 was not required for degradation. Further, degradation of an S138 phosphomimetic mutant was not blocked by mutation of cluster-ligating cysteines. These findings were confirmed in mouse models with genetic defects in cytosolic Fe–S cluster assembly/disassembly. IRP1 RNA-binding activity was primarily regulated by IRP1 degradation in these animals. Our results reveal a mechanism for regulating IRP1 action relevant to the control of iron homeostasis during cell proliferation, inflammation, and in response to diseases altering cytosolic Fe–S cluster assembly or disassembly.
Keywords: Fe–S protein, iron homeostasis, iron-regulatory protein 1, phosphorylation, protein degradation
Introduction
Iron-regulatory protein 1 (IRP1) is an iron-regulated RNA-binding protein that controls cellular iron metabolism. IRPs bind to iron-responsive elements (IRE) in mRNAs, thereby controlling mRNA fate (Eisenstein, 2000; Hentze et al, 2004). Mutation of IRP1, its functional ortholog IRP2, or the IRE in specific mRNAs disrupts iron metabolism and can lead to iron-related abnormalities in humans and in mice (Beaumont et al, 1995; LaVaute et al, 2001; Fleming, 2002; Mok et al, 2004). Modulation of IRP1 RNA-binding activity is conventionally believed to occur as a result of insertion of a [4Fe–4S] cluster into the protein converting it to cytosolic aconitase (c-acon). Consequently, recent work has focused on identifying cytosolic proteins that directly modulate IRP1 Fe–S cluster assembly (Roy et al, 2003). However, it has been suggested that IRP1 RNA-binding activity is also controlled through regulated changes in protein degradation (Mascotti et al, 1995; Neonaki et al, 2001; Fillebeen et al, 2003). The role of these Fe–S cluster assembly/disassembly and protein degradation mechanisms in controlling IRP1 function is the focus of this investigation.
Multiple lines of evidence suggest that there is a physiological need for an Fe–S cluster-independent mechanism to control accumulation of IRP1 RNA-binding activity. First, the c-acon form can be as much as 20–100-fold more abundant than the RNA-binding form of IRP1 (Chen et al, 1997; Meyron-Holtz et al, 2004a). This observation predicts that conditions that perturb the synthesis or enhance the destruction of Fe–S clusters might lead to maladaptive responses to changes in iron status due to IRP1 dysregulation. Furthermore, the activity of proteins required for cytosolic Fe–S cluster assembly and the level of reactive species (e.g. superoxide, O2•−) capable of disrupting Fe–S clusters differ between tissues (Tong and Rouault, 2000; Balk and Lill, 2004; Meyron-Holtz et al, 2004b). Consequently, a cluster-independent mechanism for iron regulation of IRP1 could provide an alternative pathway to normalize IRP1-binding activity in tissues or situations where the capacity for cytosolic Fe–S cluster assembly or disassembly differs.
Despite the clear physiologic role of Fe–S cluster assembly and disassembly in controlling IRP1 RNA-binding activity, little is known of the effect of alterations in these pathways of Fe–S cluster metabolism on IRP1 function. In this regard, the Fe–S clusters of aconitases are susceptible to reactive oxygen and nitrogen species (i.e. O2•− and NO); yet, the extent to which such cluster perturbants dictate the mechanism of IRP1 regulation remains largely unexplored. For example, the pathogenesis of inherited amyotrophic lateral sclerosis due to mutations in superoxide dismutase 1 (SOD1) might, in part, involve disturbance of Fe–S cluster metabolism (Maier and Chan, 2002). Additionally, primary Fe–S biosynthetic defects contribute to the inherited neurodegenerative and hematologic disorders associated with abnormal cellular iron metabolism, including Friedreich's ataxia (Rotig et al, 1997; Stehling et al, 2004; Seznec et al, 2005) and X-linked sideroblastic anemia with ataxia (XLSA/A) (Pagon et al, 1985; Raskind et al, 1991; Hellier et al, 2001; Fleming, 2002). In the present study, we have specifically asked how assembly and disassembly of the Fe–S cluster in IRP1 contributes to the regulation of iron metabolism in normal and pathological states, and have uncovered a novel iron-dependent mechanism for controlling IRP1 protein stability that can occur without participation of the Fe–S cluster.
Results
IRP1 undergoes iron-dependent protein turnover in the absence of cluster assembly
Formation and loss of the Fe–S cluster in IRP1 has been widely considered to be the major mechanism through which iron influences IRP1 RNA-binding activity. Iron-dependent protein turnover may also control IRP1 function (Mascotti et al, 1995; Neonaki et al, 2001; Fillebeen et al, 2003). However, the circumstances under which a protein turnover mechanism is invoked, and whether or not it involves the Fe–S cluster, have not been determined. We reasoned that enhanced protein turnover of IRP1 could control accumulation of IRP1 RNA-binding activity, particularly under conditions when iron is elevated, but conversion of IRP1 to c-acon is not efficient. Therefore, we determined the effect of iron on wild-type IRP1 (IRP1WT) and an IRP1 mutant in which all cluster-ligating cysteines (residues 437, 503, and 506) were mutated to Ser (IRP13C>3S). To avoid potential toxicity from overexpression of IRP1, a tetracycline (tet) inducible system for expression of myc-tagged WT and mutant forms of IRP1 was used.
Cells expressing IRP1WT or IRP13C>3S were treated for 24 h with hemin, an iron source, or desferal, an iron chelator. Hemin had little or no effect on the RNA-binding activity of IRP1WT at 24 h (Figure 1A and B), but with shorter exposure (8 h), RNA-binding activity of IRP1WT was reduced by 20–30% (results not shown). This is similar to the time-dependent effect of iron treatment on IRP1 in other cell types (Leibold and Munro, 1988). On the other hand, desferal-induced iron deficiency stimulated RNA binding by IRP1WT three-fold, consistent with recruitment of the RNA-binding form from the c-acon form (Figure 1A and B). As expected, in iron-sufficient cells the IRP13C>3S had RNA-binding activity similar to that observed for IRP1WT in iron-deficient conditions (Figure 1A, lanes 4 versus 3). In contrast, comparison of the activity of both proteins in iron-sufficient cells showed that IRP13C>3S had higher RNA-binding activity than IRP1WT (Figure 1A, lanes 4 versus 1). Furthermore, the RNA-binding activity of IRP13C>3S responded to iron even though this protein cannot form an Fe–S cluster; hemin decreased the RNA-binding activity of IRP13C>3S by 30%, while desferal increased binding by 45% (Figure 1A and B). Of note, iron excess and iron deficiency produced a similar fold change in the RNA-binding activity of IRP1WT and IRP13C>3S. However, for IRP13C>3S, the point at which this activity is maintained in untreated cells is more reflective of a response seen in iron deficiency or in proliferating cells. These results demonstrate that IRP1 RNA-binding activity responds to iron even when a Fe–S cluster cannot be inserted, and suggests that IRP1 can be iron-regulated by cluster-dependent and -independent means.
Figure 1.
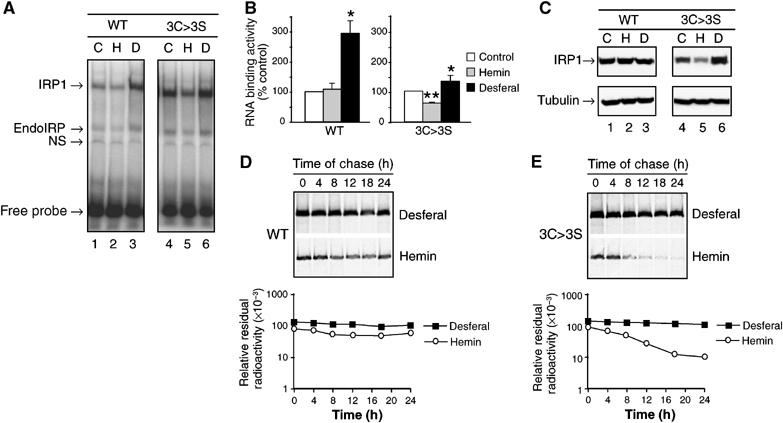
Iron-dependent protein turnover of an IRP1 cluster mutant: Cells expressing IRP1WT or IRP13C>3S were grown without or with 100 μM hemin or 100 μM desferal for 24 h. (A) RNA-binding activity of IRP1WT or IRP13C>3S determined by EMSA. Control cells are indicated by (C), hemin-treated cells by (H) and desferal-treated cells by (D). A representative gel is shown. EndoIRP is endogenous IRP and NS refers to nonspecific band. (B) Quantified RNA-binding results for IRP1WT or IRP13C>3S incubated with no addition (Control), 100 μM hemin or 100 μM desferal. Results are expressed as percent of control value for each clone and are mean±s.e.m. (n=3 experiments). An asterisk indicates that desferal value is significantly different from control (P<0.05), while two asterisks indicate a hemin effect relative to control or desferal. (C) Representative immunoblot of IRP1WT and IRP13C>3S in lysates from cells incubated as in panels A and B. (D) Pulse-chase analysis of the half-life of IRP1WT; half-life of IRP1WT was 18 h (n=3) irrespective of iron status (details in Materials and methods). (E) The half-life of IRP13C>3S was 3.9±0.7 h in the presence of hemin, and was greater than 18 h in the presence of desferal (n=3) (details in Materials and methods). Representative decay curves are shown.
We next determined if protein level contributed to the responses of IRP1WT and IRP13C>3S to hemin (Figure 1C). As expected, IRP1WT protein was not altered by hemin or desferal. In contrast, IRP13C>3S protein level was inversely related to iron status, and was closely related to changes in its RNA-binding activity. When treated with hemin, the abundance of IRP13C>3S decreased by 30%, while after desferal treatment it increased 80%. To determine if this change in protein level was due to post-translational regulation, we determined IRP1 protein half-life (Figure 1D and E). We found that IRP13C>3S had a half-life of 4 h in hemin-treated cells that increased to greater than 18 h in the presence of desferal. In contrast, IRP1WT did not exhibit this response. Hence, in the absence of Fe–S cluster assembly, IRP1 RNA-binding activity is substantially regulated through protein degradation.
S138 phosphomimetic mutants of IRP1 exhibit cluster-independent regulation of IRP1 protein stability
Previous studies have shown that S138 phosphomimetic mutants of IRP1 (e.g. IRP1S138E) can be converted to c-acon, but have markedly unstable Fe–S clusters (Brown et al, 1998). Since this would predict preferential accumulation of IRP1S138E in the RNA-binding form, we determined if this phosphomimetic mutant was also subject to cluster-independent regulation. We found that in transfected cells the fraction of IRP1S138E in the RNA-binding form was five-fold more than IRP1WT or nonphosphorylatable IRP1S138A (Supplementary Figure 1). In fact, IRP1S138E displayed RNA-binding characteristics similar to the IRP13C>3S cluster mutant. Hemin treatment reduced the RNA-binding activity of IRP1S138E by 60%, while desferal increased RNA binding by 30% (Figure 2A). In contrast, IRP1S138A behaved like its WT counterpart, showing no or little response to hemin at 24 h, but a two-fold increase in RNA-binding activity after desferal treatment (compare Figures 2A and 1B). Since IRP1S138E can be an aconitase (Brown et al, 1998) (Supplementary Figure 1), we determined if the Fe–S cluster was required for iron regulation of RNA binding by creating the IRP1S138E/3C>3S quadruple mutant. We found that IRP1S138E/3C>3S displayed changes in RNA-binding activity similar to IRP1S138E after hemin or desferal treatment, indicating that the Fe–S cluster is not required for iron regulation of IRP1S138E (Figure 2A).
Figure 2.
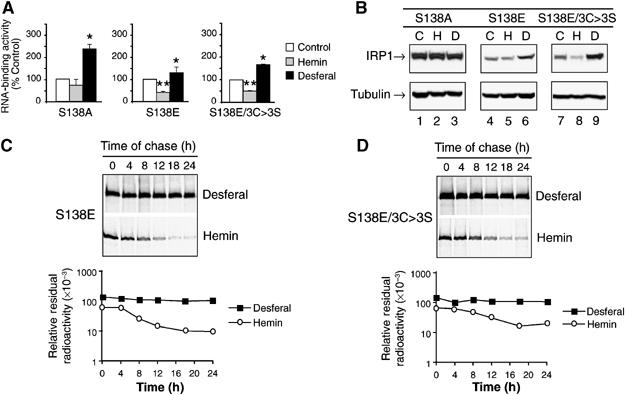
Altered iron regulation and protein degradation of the S138E phosphomimetic mutant of IRP1. Cells expressing IRP1S138A, IRP1S138E or IRP1S138E/3C>3S were grown without or with 100 μM hemin or 100 μM desferal for 24 h. (A) RNA-binding activity of IRP1S138A, IRP1S138E or IRP1S138E/3C>3S determined by quantitative EMSA. Cells were incubated with no addition (Control), 100 μM hemin or 100 μM desferal for 24 h. Results are expressed as percent of control value for each clone and are mean±s.e.m. (n=3). An asterisk indicates that desferal value is significantly different from control (P<0.05), while two asterisks indicate a hemin effect relative to control or desferal. (B) Representative immunoblot of IRP1S138A, IRP1S138E, and IRP1S138E/3C>3S in lysates from cells incubated as in panel A. Treatments are indicated by Control (C); hemin (H); and desferal (D). (C) The half-life of IRP1S138E was 4.1±0.3 h in hemin, but greater than 18 h in desferal (n=3) (details in Materials and methods). (D) The half-life of IRP1S138E/3C>3S was 5.1±1.1 h in the presence of hemin, and was greater than 18 h in the presence of desferal (n=3) (details in Materials and methods). Representative decay curves are shown.
To extend the parallelism with IRP13C>3S, we examined the protein level of the IRP1S138E and IRP1S138E/3C>3S mutants, and found that their protein levels were reduced by 40 and 30%, respectively, after hemin treatment (Figure 2B). Furthermore, desferal increased the protein level of both mutants by 60%. Like IRP1WT, the protein level of nonphosphorylatable IRP1S138A was not affected by hemin or desferal treatment. Similar to IRP13C>3S, IRP1S138E had a half-life of 4 h in hemin-treated cells, and desferal treatment stabilized the protein, resulting in a half-life of >18 h (Figure 2C); similar results have been observed by others (Fillebeen et al, 2003). Likewise, IRP1S138E/3C>3S had a half-life of 5 h in hemin-treated cells and >18 h when cells were exposed to desferal (Figure 2D). Hence, iron-mediated degradation independent of the Fe–S cluster accounts for the alterations in protein abundance and RNA-binding activity of IRP13C>3S as well as the IRP1S138E and IRP1S138E/3C>3S mutants.
S138 phosphorylation targets the RNA-binding form of IRP1 for iron-dependent degradation
Phosphorylation state-specific antibodies were developed and used to demonstrate that IRP1 is phosphorylated at S138 in control HEK cells and the level of S138 phosphorylation increased after treatment of the cells with the protein kinase C (PKC) activator phorbol 12-myristate 13-acetate (PMA) (Supplementary Figures 2 and 3). The S138 site was also recognized by these antibodies after incubation of purified IRP1WT, but not IRP1S138A, with PKC (Supplementary Figure 2). These antibodies were used to determine the properties of S138-phosphorylated IRP1 in HEK cells. In order to determine the fraction of IRP1 that is phosphorylated in cells, two-dimensional gel electrophoresis was employed. Cells expressing IRP1WT were labeled with [35S]Met/Cys or [32P]orthophosphate in the presence or absence of PMA, and IRP1 was collected by immunoprecipitation. In control cells, two species of [35S]IRP1 were observed, with the more acidic form (form 2) present in lower abundance (Figure 3A). After PMA treatment, this acidic isoform (form 2) increased in relative abundance by about 20% and two additional more acidic isoforms (forms 3 and 4) of [35S]IRP1 were observed (compare Figure 3A with B). When labeled with [32P], form 3 and possibly form 4 were detected in control cells (Figure 3C). After PMA treatment, the amount of [32P]IRP1 increased substantially and [32P] was detected in all acidic isoforms (forms 2–4) (Figure 3D). Hence, forms 2–4 correspond to phosphorylated IRP1. The S138 phosphospecific antibody recognized forms 2–4 in PMA-treated cells (Figure 3E); S138 phosphorylated IRP1 was present at a lower level in control cells (Supplementary Figure 3). Since IRP1 can also be phosphorylated at S711 (Pitula et al, 2004), the phosphorylated forms 2 and 3 likely represent mono- and diphosphorylated IRP1. Form 4 may represent IRP1 phosphorylated at sites in addition to S138 and S711 as additional phosphorylation sites have been observed in cultured cells and with purified IRP1 protein (S Anderson and R Eisenstein, data not shown). On the basis of the intensity of the 35S-labeled protein species, approximately 2% of IRP1 is phosphorylated in control cells, while in PMA-treated cells phosphorylation increased six-fold such that 12% of the protein was phosphorylated. Our previous work using purified IRP1 demonstrated that the free apoprotein form is the preferred substrate for phosphorylation; the c-acon and the RNA-bound forms of IRP1 are poor substrates for phosphorylation (Schalinske et al, 1997). The level of IRP1 phosphorylation observed in HEK cells suggests that this is the case in vivo. In agreement with this, desferal treatment of cells stimulated phosphorylation of IRP1 at S138 several fold, and IRP13C>3S is more highly phosphorylated than IRP1WT in untreated cells (A Vasanthakumar and R Eisenstein, data not shown).
Figure 3.
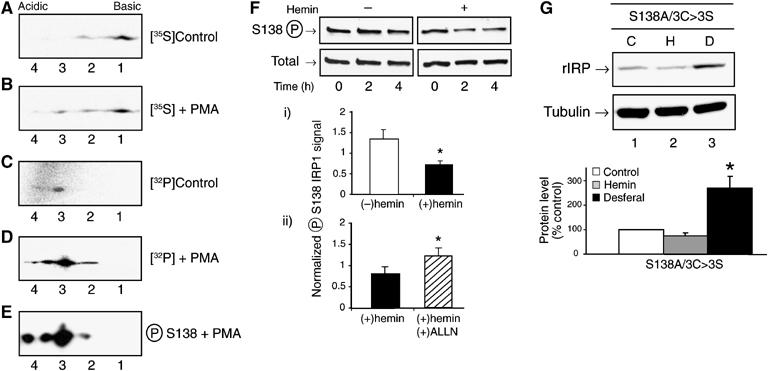
Abundance and iron-dependent loss of S138 phosphorylated IRP1. Panels (A) and (B): HEK cells were incubated with 100 μCi/ml of [35S]Met/Cys for 2 h before the addition of vehicle (DMSO, control) or PMA (1 μM) for an additional 2 h. Myc-tagged IRP1 was immunoprecipitated and analyzed by 2-D gel electrophoresis. (C) HEK cells were incubated with 0.75 mCi/ml of [32P]orthophosphate for 4 h. (D) HEK cells were incubated with 0.75 mCi/ml of [32P]orthophosphate for 2 h before addition of PMA and incubation for an additional 2 h. Immunoprecipitated myc-tagged IRP1 was analyzed in panels C and D. (E) Myc-tagged IRP1 was immunoprecipitated from unlabeled HEK cells treated with 1 μM PMA for 2 h. After 2D gel separation, S138 phosphorylated IRP1 was detected by phosphoblotting. (F) HEK cells were treated with 1 μM PMA for 2 h. The media was removed and replaced with normal media with or without 100 μM hemin. After 2 or 4 h, myc-tagged IRP1 was immunoprecipitated from cell lysates and analyzed for S138-phosphorylated or total IRP1 by immunoblotting. Graph (i) represents the 4-h time point. Graph (ii) represents the 4-h time point from an experiment where cells were treated with PMA for 2 h and then hemin for 4 h. In one set of plates, the protease inhibitor ALLN (10 μM) (N-acetyl-leucyl-leucyl-norleucinal, Calbiochem) was added with the PMA and hemin. Results in graphs i and ii are mean±s.e.m. for three separate experiments. (G) Cells expressing IRP1S138A/3C>3S were treated without or with 100 μM hemin or 100 μM desferal for 24 h. Representative immunoblot of myc-tagged IRP1. The graph shows quantified results for n=3 independent pools of clones (mean±s.e.m.). Panels F and G: an asterisk indicates significant difference (P<0.05).
To further establish the physiological relevance of phosphorylation, we determined the effect of hemin treatment on the abundance of S138-phosphorylated IRP1. To do so, cells were treated with PMA for 2 h and then incubated in the presence or absence of hemin for another 2 or 4 h. Addition of hemin reduced the level of S138-phosphorylated IRP1 by 45% at 4 h (Figure 3F, graph i). In contrast, the level of S138-phosphorylated and total IRP1 did not change in cells that were not hemin-treated. In the presence of the protease inhibitor ALLN (plus hemin), the level of S138-phosphorylated IRP1 protein was 52% greater than in the presence of hemin alone (Figure 3F, graph ii). This shows that S138-phosphorylated IRP1 undergoes iron-dependent degradation, consistent with the notion that IRP1 undergoes cluster-independent regulation when phosphorylated at S138.
Phosphorylation at S138 is not required for iron regulation of protein stability of IRP1 cluster mutants
We next determined whether phosphorylation is required for, or merely induces, cluster-independent regulation of IRP1. The requirement for S138 phosphorylation for turnover of the IRP1 cluster mutants was evaluated by creating the nonphosphorylatable IRP1S138A/3C>3S mutant. Hemin treatment reduced the level of IRP1S138A/3C>3S protein by 25%, whereas desferal promoted a 150% increase in its abundance (Figure 3G). Hence, S138 phosphorylation is not required for iron regulation of the turnover of IRP1 cluster mutants. Nonetheless, these findings do suggest that IRP1 S138 phosphorylation is one of the physiological mechanisms that impairs the accumulation of the c-acon form, thereby rendering IRP1 susceptible to iron-dependent turnover. Overall, these results indicate that S138 phosphorylation promotes, but is not required for, iron-dependent control of IRP1 protein turnover, and that other scenarios where cluster accumulation is impaired are likely to promote a similar mode of regulation.
Sod1−/− mice exhibit iron-dependent regulation of IRP1 protein levels
To establish the relevance of our findings in vivo, we used two animal disease models with perturbed cytosolic Fe–S cluster metabolism. As the Fe–S cluster in aconitases is known to be sensitive to superoxide, we first examined the effect of loss of the Fe–S cluster in vivo in mice lacking the copper-zinc superoxide dismutase, SOD1 (Huang et al, 1997; Elchuri et al, 2005). SOD1 deficiency is well established to promote cytosolic oxidative stress, which can damage Fe–S clusters, including that of c-acon (Strain et al, 1998; Missirilis et al, 2003; Starzynski et al, 2005). Iron-replete Sod1−/− mice had 50% more IRP1 RNA-binding activity in liver compared to their WT littermates (Figure 4A). Treatment with 2-mercaptoethanol (2-ME) activates RNA binding by c-acon and other inactive forms of IRP1, allowing an indirect assessment of total IRP1 protein. In WT mouse liver, IRP1 RNA-binding activity was increased 260-fold after 2-ME treatment, whereas in Sod1−/− mice a 15-fold increase was observed (Figure 4B). In addition, the total level of 2-ME inducible RNA-binding activity was lower in Sod1−/− liver. In agreement with these observations, IRP1 protein abundance was reduced by 80% (Figure 4C) and c-acon activity was not detectable in Sod1−/− liver (Figure 4D); others have obtained similar results (Starzynski et al, 2005). Mitochondrial aconitase activity was not reduced in liver of Sod1−/− mice (Figure 4D). To determine if this effect of loss of SOD1 on IRP1 protein accumulation was a phenomenon applicable to other cytosolic Fe–S proteins, xanthine oxidase (XO) was examined. In contrast to IRP1, XO protein levels were not altered in Sod1−/− mouse liver (Figure 4C), even though XO activity was reduced by 74% (Figure 4D). Hence, the reduction in IRP1 protein in Sod1−/− liver reflects regulation of the stability of only certain cytosolic Fe–S proteins.
Figure 4.
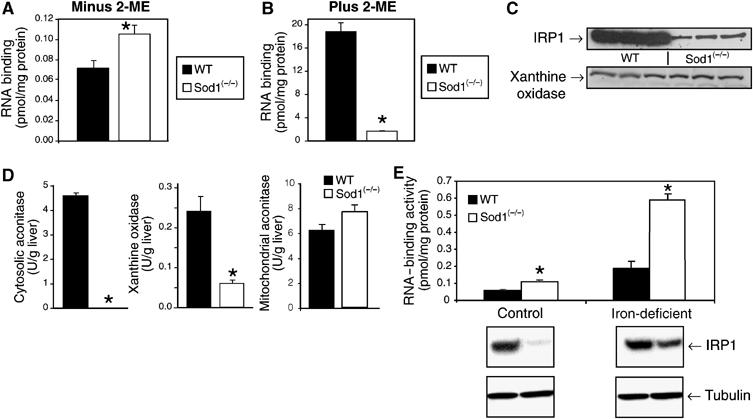
Iron regulates IRP1 protein abundance in the liver of Sod1−/− mice. Liver IRP1 RNA-binding activity, enzyme activities and protein levels were determined in the liver of wild-type (WT) or Sod1−/− mice. (A) IRP1 RNA-binding activity was determined by EMSA of liver cytosol from 8-week-old mice (n=4). IRP1 RNA-binding activity was 0.072 pmol/mg protein (WT) and 0.11 pmol/mg protein (Sod1−/−). (B) RNA binding after treatment of cytosol with 4% 2-ME. IRP1 RNA-binding activity was 19 pmol/mg protein in WT liver and 1.6 pmol/mg protein in Sod1−/− liver. (C) Immunoblot for IRP1 and XO in the liver of WT or Sod1−/− mice fed a diet containing 50 ppm iron. (D) c-Acon, XO, and mitochondrial aconitase activity in WT and Sod1−/− liver. Unit activity is μmol cis-aconitate produced/min for aconitases and is μmol Amplex-Red consumed per min for XO. Results for n=3 animals/group. (E) IRP1 RNA-binding activity and protein level (immunoblot) in WT or Sod1−/− mice fed an iron-replete (50 ppm Fe) or iron-deficient (<2 ppm Fe) diet for 3 weeks. Immunoblot of IRP1 or α-tubulin in the liver cytosol of WT or Sod1−/− mice fed either the iron-sufficient or -deficient diet for 3 weeks. For all panels an asterisk indicates that Sod1−/− is significantly different from WT (P<0.05). In panel E graph, iron-deficient values are different from iron-sufficient values for both genotypes (P<0.05).
The iron-dependent regulation of IRP1 protein abundance observed with the cluster mutants (IRP13C>3S, IRP1S138/3C>3S) in cultured cells was recapitulated in Sod1−/− mice. Iron-deficient WT mice had three-fold more IRP1-binding activity in liver than did iron-replete WT animals (Figure 4E) and, as expected, there was no change in IRP1 protein level. However, there was five-fold more IRP1-binding activity in iron-deficient versus iron-replete Sod1−/− mice. Furthermore, and unexpectedly, iron deficiency resulted in a 2.5-fold increase in IRP1 protein levels in mutant animals, compared to iron-sufficient mutant mice. No such effect was seen in wild-type mice. Thus, one-half of the increase in IRP1 RNA-binding activity in the liver of iron-deficient Sod1−/− mice can be attributed to an increase in IRP1 protein.
Impaired assembly of cytosolic Fe–S clusters in Abcb7lv/Y mice leads to iron regulation of IRP1 protein abundance
To confirm and extend the findings in Sod1−/− mice, an alternative method predicted to impair accumulation of c-acon from IRP1 was used. Here, the expression of Abcb7, a protein implicated in cytosolic Fe–S cluster assembly, that is defective in the human disorder X-linked sideroblastic anemia with ataxia (XLSA/A), was specifically ablated in liver using a tissue-specific deletion strategy (Pondarré et al, submitted). Male mice containing the Abcb7 conditional allele and a liver-specific Cre recombinase transgene are hereafter referred to as Abcb7lv/Y mice. Consistent with the prediction that cytosolic Fe–S cluster assembly would be impaired, a significant loss of c-acon and XO activity was observed in Abcb7lv/Y liver (Pondarré et al, submitted). In order to specifically evaluate if iron-dependent regulation of IRP1 protein could be detected in this model, Abcb7lv/Y mice were fed a control diet (200 ppm Fe), or a diet that was deficient (2 ppm Fe), or high (4000 ppm Fe) in iron. Compared to WT mice, IRP1 RNA-binding activity was elevated by six-fold in livers of Abcb7lv/Y mice (Figure 5A). As expected, 2-ME increased IRP1-binding activity in WT liver, demonstrating the predominance of the c-acon form (compare Figure 5A and B). In contrast, 2-ME had little effect on IRP1-binding activity in Abcb7lv/Y samples, indicating a predominance of the RNA-binding form in mutant liver. Furthermore, the level of 2-ME inducible RNA-binding activity was markedly reduced in Abcb7lv/Y liver, suggesting a substantial reduction in IRP1 protein. In fact, IRP1 protein was reduced in the liver of Abcb7lv/Y mice fed any of the three diets (compare Figure 5C and D). Furthermore, when Abcb7lv/Y mice were fed the iron-deficient diet, a 2.2-fold increase in IRP1 protein level was seen relative to mutant mice fed the high-iron diet (Figure 5D), while no effect of diet on IRP1 protein level was seen in WT animals (Figure 5C). Similar to Sod1−/− liver, the level of XO protein was not altered in Abcb7lv/Y mice (Pondarré et al, submitted).
Figure 5.
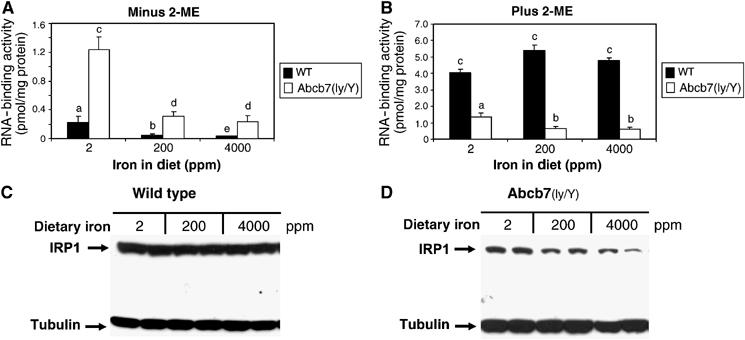
Liver-specific knockout of Abcb7 leads to iron-dependent regulation of IRP1 protein. WT or Abcb7lv/Y mice were fed a diet ranging in Fe level from 2 to 4000 ppm for 4 weeks. (A) IRP1 RNA-binding activity determined by EMSA for n=4 mice except the 4000 ppm Abcb7lv/Y group (n=3). Values for WT mice: 0.22±0.09, 0.051±0.011, and 0.035±0.0037 pmol/mg protein for the 2, 200, and 4000 ppm groups. (B) IRP1 RNA-binding activity after 2-ME induction. (C) Immunoblot of IRP1 and α-tubulin protein level in WT liver cytosol. (D) Same as panel C, except for Abcb7lv/Y mice. Lanes in panels C and D represent individual animals. For all panels, bars with different letters are significantly different (P<0.05).
Overall, in these animal models of impaired cytosolic Fe–S cluster assembly/disassembly IRP1 protein level dropped significantly, was inversely related to iron status, and directly related to RNA-binding activity. Taken together with our cell culture findings, the animal models support a role for an iron-dependent control of IRP1 protein stability through a mechanism not requiring the Fe–S cluster.
Discussion
In this paper, we have studied the responses of IRP1 to disruption of Fe–S cluster metabolism. In cultured cells, we have approached this issue by using IRP1 mutants that destabilize (Brown et al, 1998), or block assembly of, the Fe–S cluster. Unexpectedly, the protein levels and rate of degradation of these IRP1 mutants were iron-regulated. Importantly, a similar response was found for S138-phosphorylated IRP1, indicating that the WT protein can also be targeted for iron-induced degradation in a regulated manner. To model a potential role of each of these pathways in pathologic states in vivo, we examined IRP1 functional regulation in murine tissues deficient in: (1) Sod1, instrumental in neutralizing cytoplasmic oxidative stress, or (2) the mitochondrial ATP-binding cassette transporter B7 (Abcb7), implicated in the assembly of the holo-forms of cytosolic Fe–S proteins (Balk and Lill, 2004), such as the c-acon form of IRP1. We found that c-acon activity and IRP1 protein level fell dramatically in the livers of both Sod1−/− and liver-specific deletion of Abcb7 (Abcb7lv/Y) mice, and exhibited iron-dependent changes in abundance not seen in wild type (WT) mice. In total, these studies suggest that iron-dependent degradation of IRP1 serves as an alternative mechanism for controlling accumulation of IRP1 RNA-binding activity, which does not require participation of the Fe–S cluster.
These data show that IRP1 can be controlled via reversible and irreversible means, and establish several unique points concerning how IRP1 function is controlled (Figure 6): (1) it affirms that IRP1 can be regulated without participation of the Fe–S cluster; (2) this cluster-independent mechanism responds to changes in iron status; (3) iron-responsive degradation of IRP1 may have a role in cell proliferation, oxidative stress, or inflammation, where normal regulation of iron metabolism is disturbed and PKC can be activated (Eisenstein, 2000); (4) diseases leading to defects in Fe–S cluster assembly or disassembly may involve altered iron-sensing by IRP1, suggesting that IRP1 may be involved in the pathogenesis of these disorders.
Figure 6.
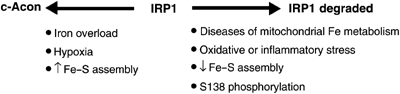
Mechanisms for inactivating IRP1 RNA-binding activity: IRP1 is iron-regulated by two mechanisms. Cluster-dependent regulation (left arrow) is reversible and allows for the maintenance of a large reserve of IRE-binding activity as c-acon. The cluster-independent mechanism (right arrow) is not reversible since IRP1 is degraded, but also provides a means for growth factors and other extracellular agents to uncouple IRP1 from regulation by the Fe–S cluster in order to alter the sensitivity and timing of the response of IRP1 to iron. S138 phosphorylation can activate RNA binding by destabilizing the Fe–S cluster, reducing the accumulation of IRP1 in the c-acon form. However, when iron levels rise to a sufficient level, S138 phosphorylated IRP1 is degraded.
The most salient point of our findings is that iron can participate in an IRP1 protein degradation response that is independent of the Fe–S cluster in that the cluster is not required for the action of iron on the IRP1 apoprotein. While no such protein level regulation was experimentally apparent in liver of WT animals, we note that only a small portion (∼1–5%) of IRP1 is present in the RNA-binding form in liver and many other tissues (Chen et al, 1997; Meyron-Holtz et al, 2004a) (see also Figures 4 and 5). Consequently, changes in the level of the RNA-binding form would be masked by the larger, more stable, c-acon pool. Previous studies of apo-metalloproteins, including apo-Fe–S proteins, have demonstrated that they are more rapidly degraded compared to their holo-protein counterparts (Grandoni et al, 1989; Li and Merchant, 1995; Mettert and Kiley, 2005). Our earlier work on IRP1 has demonstrated that the apoprotein form of IRP1 is more susceptible to proteolysis in vitro (Schalinske et al, 1997). However, our current findings indicate that the apoprotein form of IRP1 is uniquely regulated in that it can be stabilized either by insertion of its cofactor (i.e. Fe–S cluster) or by iron deficiency. This conclusion is supported by studies with an IRP1 mutant unable to form a cluster (IRP13C>3S), as well as one with an unstable cluster (IRP1S138E) and S138-phosphorylated IRP1. The components required for turnover of IRP1 protein have yet to be identified, but may involve the proteasome and/or other protein degradation systems (Fillebeen et al, 2003) (Figure 3). For example, it is not clear whether iron acts directly or indirectly to promote IRP1 protein degradation, or if activity of the protease(s) responsible is itself iron-regulated. It is well established that significant changes in IRP1 conformation occur on cluster insertion (Schalinske et al, 1997; Brazzolotto et al, 2002). Consequently, it is possible that stabilization of IRP1 by cluster insertion involves the accessibility of residues required for iron-dependent degradation of the apoprotein. We also note that identification of a cluster-independent protein degradation mechanism promoted by iron mimics the regulation of the functional orthologue IRP2, which is regulated by protein turnover in response to cellular iron status (Guo et al, 1995; Ishikawa et al, 2005). The extent of mechanistic overlap between these regulatory mechanisms awaits further study.
Previous studies found that elimination of cluster ligands in IRP1WT impaired iron regulation of RNA binding (Hirling et al, 1994; DeRusso et al, 1995; Wang and Pantopoulos, 2002), and concluded that a dysregulation of iron metabolism occurred when an IRP1 cluster assembly mutant was expressed in mammalian cells (DeRusso et al, 1995; Wang and Pantopoulos, 2002). Here we clearly show that iron regulation of RNA binding is not entirely eliminated in such mutants. Additionally, an alternative view of the change in iron-regulatory characteristics of IRP1 on loss of cluster-dependent regulation, as illustrated by the extreme case of the IRP13C>3S cluster mutant, is that it reflects a physiologically appropriate response in cells with unique iron needs, or with a limited capacity to suppress accumulation of reactive oxygen or nitrogen species. Hence, tissue-specific regulation of IRP1 may occur through the classical Fe–S switch or through protein stability, or both, according to the iron needs and metabolic capacity of specific cell types. For example, cells with limited ability to suppress oxidative stress might depend more heavily on the protein degradation mechanism.
Regulation of IRE RNA-binding activity through S138 phosphorylation confers properties on IRP1 that allow cells to meet unique metabolic needs. First, S138 phosphorylation allows for IRP1 RNA-binding activity to be biased toward the iron-deficient state. For instance, in proliferating cells reduced inactivation of IRP1 in response to increased iron uptake would help target iron to specific metabolic fates (Teixeira and Kühn, 1991; Testa et al, 1991; Thomson et al, 2000). Activation of PKC has an important role in these and other models of cell proliferation (Eisenstein, 2000). A related situation concerns the activation of IRP1 activity and TfR1 mRNA accumulation in cells with specialized needs for iron such as developing red cells and differentiating monocytes (Testa et al, 1989; Chan et al, 1994). In these situations IRP1 is resistant to iron-induced downregulation; S138 phosphorylation may promote this by altering the set-point for iron regulation of IRP1. Second, by coupling altered iron regulation with irreversible inactivation of RNA-binding activity (i.e. protein degradation), S138 phosphorylation may facilitate transient activation of the protein during inflammation (Oliveira and Drapier, 2000). By altering the timing and sensitivity of the response to iron, S138 phosphorylation broadens the regulatory profiles under which IRP1 can operate, and provides a means through which extracellular agents can alter cellular iron metabolism.
IRP1-regulatory mechanisms may also contribute to the pathophysiology of human disorders as well as the adaptive response of animal cells in mitigating damage caused by the dysregulation of Fe–S cluster assembly or disassembly. For example, diseases of mitochondrial iron metabolism can cause disruption of cytosolic Fe–S cluster formation or disassembly (Pagon et al, 1985; Raskind et al, 1991; Rotig et al, 1997; Hellier et al, 2001; Fleming, 2002; Stehling et al, 2004; Seznec et al, 2005). It is possible that the extent of iron-dependent inactivation of the IRP1 apoprotein through the cluster-independent protein degradation mechanism may dictate the degree of cellular damage in these situations. Particular examples include Friedreich's ataxia and X-linked sideroblastic anemia with ataxia (XLSA/A), the latter of which is caused by defects in Abcb7. Studies described here and elsewhere (Pondarré et al, submitted) show that specific ablation of Abcb7 expression in liver results in nonlethal changes in hepatic IRP1 regulation and iron metabolism. In contrast, in other cell types, such as hematopoietic cells and brain, loss of Abcb7 function is lethal (C Pondarré and MD Fleming, data not shown). It is not clear whether it is the loss of an essential Fe–S protein or the dysregulation of the IRP1 system, or both, that causes this differential response. However, evidence supporting a critical role of IRP1 dysregulation induced by a defect in Fe–S metabolism is illustrated by the rescue from the consequences of a mutation in a mitochondrial glutaredoxin involved in Fe–S cluster biogenesis by ablation of IRP1 or the IRE in a heme-biosynthetic gene (Wingert et al, 2005). Altogether, it is apparent that multiple mechanisms are required to control accumulation of IRP1 RNA-binding activity, illustrating its central role in the adaptive changes in iron metabolism in diverse settings. Furthermore, these data suggest that IRP1 dysregulation has the potential to be substantially more damaging than complete loss of the protein (Galy et al, 2004; Meyron-Holtz et al, 2004a).
Materials and methods
Animals
Mice lacking SOD1 were generously provided by Huang et al (1997). To generate mice lacking Abcb7 in hepatocytes, a loxP site and a loxP-flanked neomycin resistance cassette (NeoR) were targeted into introns 8 and 10 of Abcb7 of embryonic stem (ES) cells to create a conditionally targeted allele flanked by loxP sites (Pondarré et al, submitted). Mice carrying the targeted Abcb7 allele were bred to a hepatocyte-specific Cre-transgenic line: B6.Cg-Tg(Alb-Cre)21Mgn/J [Alb-Cre] (Pondarré et al, submitted). Male animals carrying the Abcb7fl allele and Alb-Cre are referred to as Abcb7lv/Y to reflect the hepatocyte-specific deletion; deletion of exons 9 and 10 was confirmed by PCR (data not shown). Animal use met the requirements of Children's Hospital Boston or UW-Madison. Sod1−/− mice and their WT littermates were fed a diet with 2 or 50 ppm iron made as described (Chen et al, 1998). For the experiments with Abcb7lv/Y mice, a commercial iron-deficient diet (TD 80396, Harlan Teklad) contained 20% casein, 0.3% methionine, 54.99% sucrose, 15% cornstarch, 5% corn oil, 3.5% iron-deficient mineral mix (TD 81062), 1% vitamin mix (40077), 0.2% choline bitartrate, and 0.0001% ethoxyquin; dietary iron content was 2 ppm. For the 200 and 4000 ppm iron diets, ferric ammonium citrate was added.
Liver subcellular fractionation and enzyme assays
Cytosol and mitochondria were obtained as described (Chen et al, 1998). Aconitase assays were performed at 25°C for 10 min as described (Kennedy et al, 1983). For XO, a kit (A-22182) from Molecular Probes, Inc., (Eugene, OR) was used. The recovery of cytosol from liver homogenate was 88 and 95% for WT and Sod1−/− liver, respectively, using lactate dehydrogenase as marker enzyme; mitochondrial contamination of cytosol was 6 and 5% on the basis of glutamate dehydrogenase activity in cytosol.
Immunoblotting
Cell lysates were immunoblotted as described (Chen et al, 1998). Myc-tagged IRP1 protein level was determined using anti-c-myc antibody (9E10). To detect S138-phosphorylated IRP1 in cells, 0.5–1.0 mg of lysate protein was immunoprecipitated using 9E10 antibodies for 2 h at 4°C and Ultra-Link Protein G (Pierce). For determination of XO or IRP1 level, liver homogenate or cytosol, respectively, was used as described (Eisenstein et al, 1993). For XO, blots were blocked in 5% non-fat dry milk (NFDM) in Tris-buffered saline (TBS) with 0.02% Tween-20. Primary antibody was used at 1:2000 in 0.5% NFDM in TBS with 0.02% Tween-20. Secondary antibody (goat anti-rabbit, Southern Biotechnology) was used at 1:30 000 and developed with SuperSignal (Pierce).
Antibodies
IRP1 antibodies were described (Eisenstein et al, 1993). Rabbit phosphospecific antibodies against the S138 phosphopeptide FNRRAD(p)S138LQKNQDLC, where (p)S denotes phosphoserine, were made and affinity purified (Biosource). Anti-XO (ab6194) was from Abcam Ltd (Cambridgeshire, UK).
Cell culture and lysis
Reagents were from Invitrogen unless noted. The Flp-In T-Rex-293 line of HEK cells were grown in DMEM plus 10% FBS, 100 μg/ml Zeocin, 15 μg/ml Blasticidin (Invivogen), 100 U/ml penicillin, and 100 μg/ml streptomycin. The cell pellet was resuspended in lysis buffer plus 5 μg/ml butylated hydroxytoluene and phosphatase inhibitor cocktail set I (Calbiochem) (Eisenstein et al, 1993). Protein assays used the BCA system (Pierce).
Generation of cell lines stably expressing IRP1
The Flp-In TRex system (Invitrogen) for producing isogenic transfected cell lines was used. Cells were transfected with plasmids encoding IRP1 plus the Flp recombinase plasmid pOG44 using Lipofectamine 2000. At 24 h after transfection, cells were trypsinized and replated onto 10 cm plates in a nonselective growth medium that was replaced with media containing 250 μg/ml hygromycin (Calbiochem) and 15 μg/ml blasticidin. Hygromycin-resistant colonies were pooled from each plate and subsequently screened for expression of myc-tagged IRP1 after induction with 1 μg/ml tetracycline for 48 h.
Site-directed mutagenesis
Mutagenesis used Quik-Change (Stratagene). The S138 codon (AGC) in the rabbit IRP1 cDNA with an encoded C-terminal myc tag (Brown et al, 1998) was replaced with GCC and GAA to create IRP1S138A or IRP1S138E, respectively. Codons for Cys 437, 503, and 506 were mutated to AGC to create IRP13C>3S. Mutations were confirmed by DNA sequencing.
IRE RNA-binding activity
IRP1 RNA-binding activity in liver was determined by electrophoretic mobility shift assay (EMSA) (Chen et al, 1997). To separate myc-tagged IRP1 from endogenous IRP1, gel supershift assays were performed. Cell lysate (1–2 μg protein) was diluted into binding buffer (Eisenstein et al, 1993) containing protein G purified 9E10 IgG (0.05 mg/ml final concentration). After 90 min at 4°C, [32P]RNA was added at a final concentration of 1 nM and the incubation continued for 10 min. Heparin was added and bound and free RNA separated as described (Eisenstein et al, 1993), except that gels were run at 4°C.
IRP1 protein half-life by pulse-chase analysis
HEK cells were incubated with 10 μM desferal overnight, and were then labeled with 100 μCi/ml [35S]Met/Cys (MP Biomedicals) for 2 h in 5% dialyzed FBS/DMEM lacking Met/Cys. Cells were washed with 10% FBS/DMEM and chased with complete media plus 10 mM each of unlabeled Met/Cys in the presence of 100 μM hemin or 100 μM desferal. Cells were lysed and IRP1 was immunoprecipitated using the 9E10 antibody (Eisenstein et al, 1993). Results were quantified by phosphorimaging.
Two-dimensional (2D) gel electrophoresis
2D electrophoresis was performed as described (O'Farrell, 1975) by Kendrick Labs, Inc. (Madison, WI).
In vitro phosphorylation
His-tagged IRP1 protein expressed in yeast was purified as described (Brown et al, 1998). IRP1 was incubated with rat brain PKC as described (Eisenstein et al, 1993).
Supplementary Material
Supplementary Material
Acknowledgments
We thank P Bertics for PKC and D Dean for NifS, and E Craig, D Eide, J Goforth, G Groblewski, J Kaplan, P Kiley, and W Walden for helpful comments and DR Campagna and BB Antiochos for assistance with the Abcb7 experiments. This work was supported in part by NIH grants DK47219 (RSE), DK62474 (MDF), AG16998 (CJE) and the USDA grant #01-35200-10683 (RSE). CP was supported in part by L'Association Francaise Contre le Cancer. SLC and JSP were supported partly by NIH grant T32 DK07665 and AV by UW-Madison Hatch Projects #3951 and #4885.
References
- Balk J, Lill R (2004) The cell's cookbook for Fe–S clusters: recipes for fool's gold? Chem Biol Chem 5: 1044–1049 [DOI] [PubMed] [Google Scholar]
- Beaumont C, Leneuve P, Devaux I, Scoazec J-Y, Berthier M, Loiseau M-N, Grandchamp B, Bonneau D (1995) Mutation in the IRE of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet 11: 444–446 [DOI] [PubMed] [Google Scholar]
- Brazzolotto X, Timmins P, Dupont Y, Moulis JM (2002) Structural changes associated with switching activities of human IRP1. J Biol Chem 277: 11995–12000 [DOI] [PubMed] [Google Scholar]
- Brown NM, Anderson SA, Steffen DW, Carpenter TB, Kennedy MC, Walden WE, Eisenstein RS (1998) Novel role of phosphorylation in Fe–S cluster stability revealed by phosphomimetic mutations at S138 of IRP1. Proc Natl Acad Sci USA 95: 15235–15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RY, Seiser C, Schulman HM, Kuhn LC, Ponka P (1994) Regulation of TfR mRNA expression: distinct regulatory features in erythroid cells. Eur J Biochem 220: 683–692 [DOI] [PubMed] [Google Scholar]
- Chen OS, Blemings KP, Schalinske KL, Eisenstein RS (1998) Dietary iron intake rapidly influences IRPs, ferritin subunits and mitochondrial aconitase (m-acon) in rat liver. J Nutr 128: 525–535 [DOI] [PubMed] [Google Scholar]
- Chen OS, Schalinske KL, Eisenstein RS (1997) Dietary iron intake modulates the activity of IRPs and the abundance of ferritin and m-acon in rat liver. J Nutr 127: 238–248 [DOI] [PubMed] [Google Scholar]
- DeRusso PA, Philpott CC, Iwai K, Mostowski HS, Klausner RD, Rouault TA (1995) Expression of a constitutive mutant of IRP1 abolishes iron homeostasis in mammalian cells. J Biol Chem 270: 15451–15454 [DOI] [PubMed] [Google Scholar]
- Eisenstein RS (2000) IRPs and the molecular control of mammalian iron metabolism. Ann Rev Nutr 20: 627–662 [DOI] [PubMed] [Google Scholar]
- Eisenstein RS, Tuazon PT, Schalinske KL, Anderson SA, Traugh JA (1993) IRE-binding protein: phosphorylation by PKC. J Biol Chem 268: 27363–27370 [PubMed] [Google Scholar]
- Elchuri S, Oberley TD, Qi W, Eisenstein RS, Roberts LJ, Remmen HV, Epstein CJ, Huang TT (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24: 367–380 [DOI] [PubMed] [Google Scholar]
- Fillebeen C, Chahine D, Caltagirone A, Segel P, Pantopoulos K (2003) A phosphomimetic mutation at S138 renders IRP1 sensitive to iron-dependent degradation. Mol Cell Biol 23: 6973–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming MD (2002) The genetics of inherited sideroblastic anemias. Sem Hematol 39: 270–281 [DOI] [PubMed] [Google Scholar]
- Galy B, Ferring D, Benesova M, Benes V, Hentze MW (2004) Targeted mutagenesis of the murine IRP1 and IRP2 genes reveals context-dependent RNA processing differences in vivo. RNA 10: 1019–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandoni JA, Switzer RL, Makaroff CA, Zalkin H (1989) Evidence that the Fe–S cluster of B. subtilis glutamine phosphoribosylpyrophosphate amindotransferase determines stability of the enzyme to degradation in vivo. J Biol Chem 264: 6058–6064 [PubMed] [Google Scholar]
- Guo B, Phillips JD, Yu Y, Leibold EA (1995) Iron regulates the intracellular degradation of IRP2 by the proteasome. J Biol Chem 270: 21645–21651 [DOI] [PubMed] [Google Scholar]
- Hellier KD, Hatchwell E, Duncombe AS, Kew J, Hammans SR (2001) X-linked sideroblastic anaemia with ataxia: another mitochondrial disease? J Neurol Neurosurg Psychiatry 70: 65–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297 [DOI] [PubMed] [Google Scholar]
- Hirling H, Henderson BR, Kühn LC (1994) Mutational analysis of the (4Fe–4S)-cluster converting iron regulatory factor from its RNA binding form to c-acon. EMBO J 13: 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T-T, Yasunami M, Carlson EJ, Gillispie AM, Reaume AG, Hoffman EK, Chan PH, Scott RW, Epstein CJ (1997) Superoxide-mediated cytotoxicity in Sod-deficient fetal fibroblasts. Arch Biochem Biophys 344: 424–432 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Kato M, Hori H, Ishimori K, Kirisako T, Tokunaga F, Iwai K (2005) Involvement of the heme regulatory motif in heme-mediated ubiquitination and degradation of IRP2. Mol Cell 19: 171–181 [DOI] [PubMed] [Google Scholar]
- Kennedy MC, Emptage MH, Dryer J-L, Beinert H (1983) The role of iron in the activation–inactivation of aconitase. J Biol Chem 258: 11098–11105 [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R III, Grinberg A, Love P, Tresser N, Rouault TA (2001) Targeted deletion of the gene encoding IRP2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27: 209–214 [DOI] [PubMed] [Google Scholar]
- Leibold EA, Munro HN (1988) Cytoplasmic protein binds in vitro to a conserved sequence in the 5′ UTR of ferritin H- and L-chain mRNAs. Proc Natl Acad Sci USA 85: 2171–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HH, Merchant S (1995) Degradation of plastocyanin in copper-deficient Chlamydomonas reinhardtii. J Biol Chem 270: 23504–23510 [DOI] [PubMed] [Google Scholar]
- Maier CM, Chan PH (2002) Role of Sod in oxidative damage and neurodegenerative disorders. Neuroscientist 8: 323–334 [DOI] [PubMed] [Google Scholar]
- Mascotti DP, Rup D, Thach RE (1995) Regulation of iron metabolism: translational effects mediated by iron, heme, and cytokines. Annu Rev Nutr 15: 239–261 [DOI] [PubMed] [Google Scholar]
- Mettert EL, Kiley PJ (2005) ClpXP-dependent proteolysis of FNR upon loss of its O2-sensing [4Fe–4S] cluster. J Mol Biol 354: 220–232 [DOI] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, Iwai K, LaVaute T, Brazzolotto X, Berger UV, Land W, Olivarerre-Wilson H, Grinberg A, Love P, Rouault TA (2004a) Genetic ablations of IRP1 and 2 reveal why IRP2 dominates iron homeostasis. EMBO J 23b: 386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyron-Holtz EG, Ghosh MC, Rouault TA (2004b) Mammalian tissue O2 levels modulate IRP activities in vivo. Science 306: 2087–2090 [DOI] [PubMed] [Google Scholar]
- Missirilis F, Hu J, Kirby K, Hilliker AJ, Rouault TA, Phillips JP (2003) Compartment-specific protection of Fe–S proteins by superoxide. J Biol Chem 278: 47365–47369 [DOI] [PubMed] [Google Scholar]
- Mok H, Jelinek J, Pai S, Cattanach BM, Prchal JT, Youssoufian H, Schumacher A (2004) Disruption of ferroportin 1 regulation causes dynamic alterations in iron homeostasis and erythropoiesis in polycythaemia mice. Development 131: 1859–1868 [DOI] [PubMed] [Google Scholar]
- Neonaki M, Graham DC, White KN, Bomford A (2001) Down-regulation of liver IRP1 in haemochromatosis. Biochem Soc Trans 30: 726–728 [DOI] [PubMed] [Google Scholar]
- O'Farrell PH (1975) High resolution 2D electrophoresis of proteins. J Biol Chem 250: 4007–4021 [PMC free article] [PubMed] [Google Scholar]
- Oliveira L, Drapier JC (2000) Down-regulation of IRP1 gene expression by nitric oxide. Proc Natl Acad Sci USA 97: 6550–6555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagon RA, Bird TD, Detter JC, Pierce I (1985) Hereditary sideroblastic anaemia and ataxia: an X-linked recessive disorder. J Med Genet 22: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitula JS, Deck KM, Clarke SL, Anderson SA, Vasanthakumar A, Eisenstein RS (2004) Selective inhibition of the citrate-to-isocitrate reaction of c-acon by phosphomimetic mutation of S711. Proc Natl Acad Sci USA 101: 10907–10912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondarré C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han A-P, Medlock A, Kutok J, Anderson SA, Eisenstein RS, Fleming MD. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic Fe–S cluster biogenesis. submitted [DOI] [PubMed]
- Raskind WH, Wijsman E, Pagon RA, Cox TC, Bawden MJ, May BK, Bird TD (1991) X-linked sideroblastic anemia and ataxia: linkage to phosphoglycerate kinase at Xq 13. Am J Hum Genet 48: 335–341 [PMC free article] [PubMed] [Google Scholar]
- Rotig A, de-Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial Fe–S protein deficiency in Friedreich ataxia. Nat Genet 17: 215–217 [DOI] [PubMed] [Google Scholar]
- Roy A, Solodovnikova A, Nicholson T, Antholine W, Walden WE (2003) A novel eukaryotic factor for cytosolic Fe–S assembly. EMBO J 22: 4826–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalinske KL, Anderson SA, Tuazon PT, Chen OS, Eisenstein RS (1997) The Fe–S cluster of IRP1 modulates accessibility of RNA binding and phosphorylation sites. Biochem 36: 3950–3958 [DOI] [PubMed] [Google Scholar]
- Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, Procaccio V, Patel M, Drapier JC, Koenig M, Puccio H (2005) Friedreich ataxia: the oxidative stress paradox. Hum Mol Genet 14: 463–474 [DOI] [PubMed] [Google Scholar]
- Starzynski RR, Lipinski P, Drapier J-C, Diet A, Smuda E, Barthlomiejczyk T, Gralak MA, Kruszweski M (2005) Down-regulation of IRP1 activities and expression in Sod1 knockout mice is not associated with alterations in iron metabolism. J Biol Chem 280: 4207–4212 [DOI] [PubMed] [Google Scholar]
- Stehling O, Elasser HP, Bruckel B, Muhlenhoff U, Lill R (2004) Fe–S protein maturation in human cells: evidence for a function of frataxin. Hum Mol Genet 13: 3007–3015 [DOI] [PubMed] [Google Scholar]
- Strain J, Lorenz CR, Bode J, Garland S, Smolen GA, Ta DT, Vickery LE, Culotta VC (1998) Suppressors of SOD1 deficiency in S. cerevisiae. Identification of proteins predicted to mediate Fe–S cluster assembly. J Biol Chem 273: 31138–31144 [DOI] [PubMed] [Google Scholar]
- Teixeira S, Kühn C (1991) Post-transcriptional regulation of the TfR and 4F2 antigen heavy chain mRNA during growth activation of spleen cells. Eur J Biochem 202: 819–826 [DOI] [PubMed] [Google Scholar]
- Testa U, Ku"hn L, Petrini M, Quaranta MT, Pelosi E, Peschle C (1991) Differential regulation of IRE-binding protein(s) in cell extracts of activated lymphocytes versus monocytes–macrophages. J Biol Chem 266: 13925–13930 [PubMed] [Google Scholar]
- Testa U, Petrini M, Quaranta MT, Pelosi-Testa E, Mastroberardino G, Camagna A, Boccoli G, Sargiacomo M, Isacchi G, Cozzi A, Arosio P, Peschle C (1989) Iron up-modulates the expression of TfRs during monocyte–macrophage maturation. J Biol Chem 264: 13181–13197 [PubMed] [Google Scholar]
- Thomson AM, Rogers JT, Leedman PJ (2000) Thyrotropin-releasing hormone and epidermal growth factor regulate IRP binding in pituitary cells via PKC-dependent and -independent signaling pathways. J Biol Chem 275: 31609–31615 [DOI] [PubMed] [Google Scholar]
- Tong WH, Rouault T (2000) Distinct Fe–S assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J 19: 5692–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K (2002) Conditional derepression of ferritin synthesis in cells expressing a constitutive IRP1 mutant. Mol Cell Biol 22: 4638–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmidt B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Zon LI (2005) Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature 436: 1035–1039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material