

Abstract
We used a combination of genome-wide and promoter-specific DNA binding and expression analyses to assess the functional roles of Myod and Myog in regulating the program of skeletal muscle gene expression. Our findings indicate that Myod and Myog have distinct regulatory roles at a similar set of target genes. At genes expressed throughout the program of myogenic differentiation, Myod can bind and recruit histone acetyltransferases. At early targets, Myod is sufficient for near full expression, whereas, at late expressed genes, Myod initiates regional histone modification but is not sufficient for gene expression. At these late genes, Myog does not bind efficiently without Myod; however, transcriptional activation requires the combined activity of Myod and Myog. Therefore, the role of Myog in mediating terminal differentiation is, in part, to enhance expression of a subset of genes previously initiated by Myod.
Keywords: muscle differentiation, Myod, Myogenin, regulatory network
Introduction
The scope of genes differentially expressed during myogenesis has been studied by expression array analyses in regenerating muscle (Yan et al, 2003; Zhao et al, 2003), differentiating C2C12 myoblasts (Moran et al, 2002; Delgado et al, 2003; Shen et al, 2003; Tomczak et al, 2004), and in Myod-mediated differentiation of mouse embryonic fibroblasts (MEFs) (Bergstrom et al, 2002), and genome-wide transcription factor binding profiling has been used to assemble regulatory networks controlling myogenesis (Blais et al, 2005), but the relative roles of Myod and Myog in the regulatory network of muscle gene expression have not been directly addressed. Four related factors comprise the myogenic bHLH family of transcription factors: Myf5, Myod, Mrf4, and Myog (Sabourin and Rudnicki, 2000; Pownall et al, 2002; Buckingham et al, 2003). Genetic studies indicate that Myod and Myf5 are necessary to specify the skeletal muscle lineage (Rudnicki et al, 1993), whereas Myog appears to have a critical role in the terminal differentiation of the specified muscle cells (Hasty et al, 1993; Nabeshima et al, 1993) and Mrf4 combines attributes of both a specification factor and a differentiation factor (Kassar-Duchossoy et al, 2004). Protein motifs conserved in Myod and Myf5 are necessary to initiate the expression of a subset of genes critical for the myogenic program (Gerber et al, 1997; Bergstrom and Tapscott, 2001), including the initiation of the Myog gene itself, and the ability to initiate the expression of a specific set of genes might account, at least in part, for their roles in specifying the myogenic lineages. Indeed, myogenesis is impaired when the Myog coding region is substituted for the Myf5 coding region in mice (Wang and Jaenisch, 1997), indicating that differences in the protein sequence confer either specification or differentiation functions. It remains unclear, however, whether Myod and Myog have different functional roles at a common set of genes, or whether they have similar activation function but are recruited to an overlapping but distinct set of regulatory elements. A recent study using chromatin immunoprecipitation (ChIP) to assess Myod and Myog binding to an array of approximately 4700 promoters demonstrated that Myod and Myog recognize distinct but overlapping targets (Blais et al, 2005) and provides a basis for investigating the functional roles of Myod and Myog at these targets.
One plausible model is that Myod activates one set of promoters and that Myog independently activates a distinct but overlapping set of promoters. This model is supported by the observation that either Myod or Myog can activate a common set of muscle promoters in transient transfection assays and that some promoter elements respond specifically to Myod or Myog (Yutzey et al, 1990; Asakura et al, 1993). Arguing against this model is the fact that Myod and Myog have similar consensus DNA binding sequences; however, promoter-specific activity might be achieved through associated sites for other regulatory factors and the demonstration that non-DNA binding domains likely recruit Myod to specific promoters (Berkes et al, 2004) raises the possibility that Myog might be targeted to its own set of differentiation-specific promoters by a similar mechanism. Therefore, some data support the model that Myod and Myog act relatively independently at an overlapping set of promoters.
An alternative model is that Myod and Myog have distinct functions that occur sequentially at individual promoters. For example, Myod might initiate chromatin remodeling at some genes for later activation by Myog. This model is supported by the observation that the Helix 3 domains of Myod and Myog have the potential for distinct functions: the Helix 3 domain of Myod is necessary to interact with the Pbx/Meis complex but does not function as an activation domain, whereas the Helix 3 domain of Myog has activation function but does not functionally substitute for the Helix 3 domain in Myod (Bergstrom and Tapscott, 2001; Berkes et al, 2004). In both of these models, Myod and Myog might have a largely overlapping set of binding sites and target genes; however, the first model predicts that the majority of the overlapping set of targets would be regulated by either Myod or Myog alone, whereas the second model makes the activity of Myog largely dependent on an initial function of Myod.
We used a combination of genome-wide and promoter-specific DNA binding and expression analyses to assess the functional roles of Myod and Myog in regulating the program of skeletal muscle gene expression. Our findings indicate that Myod and Myog bind a similar set of target genes and have distinct regulatory functions at these genes. At early targets of Myod, Myod is sufficient for near full expression. In contrast, at late expressed genes, Myod initiates regional histone modification but is not sufficient for gene expression. Myog does not bind efficiently without Myod, perhaps because it does not efficiently initiate histone modifications, and transcriptional activation requires the combined activity of Myod and Myog. These findings demonstrate that Myod and Myog function at sequential steps in regulating a set of genes: Myod is necessary to induce chromatin modifications at the late loci prior to Myog binding and activation functions of Myog are necessary for the full expression of these late genes. Therefore, the role of Myog in mediating terminal differentiation is, in part, to enhance expression of a subset of genes previously initiated by Myod.
Results
In order to determine the genes directly bound by Myod and Myog, we used ChIP with antisera to Myod and Myog and a custom self-printed microarray containing 1000 bp of DNA surrounding the start site of ∼13 000 mouse promoters. Cultured MEFs were derived from mice that were double knockouts for Myod and Myf5 so that the expression of these endogenous genes would not interfere with our analysis and these were stably transduced with a Myod-estrogen receptor hormone binding domain fusion protein (MDER cells) (Hollenberg et al, 1993). In these MDER cells, synchronous myogenic differentiation can be induced by switching the cells from growth medium to differentiation medium (low mitogen medium with the addition of β-estradiol; see Materials and methods).
Genome-wide analysis of Myod and Myog promoter binding
ChIP assays for genome-wide analysis of Myod and Myog promoter binding were performed 36 h after the induction of differentiation, a time when Myod has activated expression of the Myog gene and both transcription factors are present. Compared to input DNA, ChIP with Myod antiserum resulted in enrichment of 201 target promoters using a stringent P-value cutoff of 0.001, and ChIP with Myog antiserum showed enrichment of 176 target genes at this threshold (Figure 1A), each representing approximately 2% of promoters featured on the array. Previous experience suggests that this cutoff minimizes false positives while still capturing a substantial fraction of true binding events (Odom et al, 2004). The Myod and Myog target genes belong to a variety of functional classes, including many muscle-specific genes, genes encoding signaling and receptor proteins, and transcription and chromatin remodeling factors (Supplementary Table S1), consistent with our prior expression array study showing that Myod activates the expression of genes from many functional classes (Bergstrom et al, 2002). When the number of bound genes associated with particular gene ontology terms was compared to the total number of genes in those categories on the array, genes associated with muscle development and contraction were significantly enriched among Myod and Myog targets (Supplementary Table S2).
Figure 1.
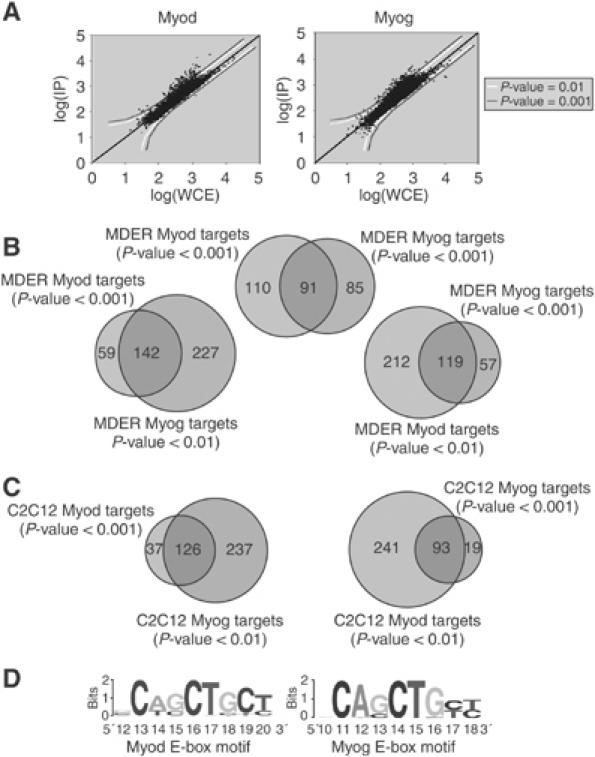
Myod and Myog bind to a similar set of promoters. (A) Representative scatter plots showing enrichment of Myod- and Myog-bound promoters by genome-wide location analysis. Confidence thresholds of P=0.001 and P=0.01 are shown. (B) Venn diagrams indicating the degree of overlap between Myod and Myog targets in MDER cells. The overlap increases when confidence thresholds are lowered to include more targets. (C) Venn diagrams indicating the degree of overlap between Myod and Myog targets in C2C12 myotubes differentiated for 36 h. (D) E-box motifs identified from lists of Myod- and Myog-bound promoters.
Included among the observed targets of Myod and Myog in the MDER cells were previously identified muscle-specific genes such as ckm (Jaynes et al, 1988) and chrng (Liu et al, 2000), as well as a number of transcription factors and chromatin regulators. Some of these, such as Mef2c (Molkentin and Olson, 1996) and Idb1 (Benezra et al, 1990), are well-known regulators of myogenesis. Others have not been previously tied to myogenic differentiation and these targets may represent novel potentiators of myogenesis and act as amplifiers of differentiation. Foxp1, for instance, has been identified as a key regulator of other biological processes, but does not have a clearly defined role in muscle differentiation (Wang et al, 2004; Banham et al, 2005).
Physical connections are also observed between Myod and/or Myog and components of a number of signaling pathways known to either promote or inhibit myogenesis. Calmodulin-dependent signaling has been shown to activate myogenesis (Olson and Williams, 2000; Xu et al, 2002; Friday et al, 2003) and genes encoding components of this pathway (Calm1, Camk1g) are bound by Myod and Myog. Conversely, elements of the TGF-β pathway (Tieg1, Tgfbr2), as well as Stat3, Mdm2, and Hdac9, all of which function to inhibit myogenesis (Brennan et al, 1991; Fiddler et al, 1996; Zhang et al, 2001; Kataoka et al, 2003; Zhu et al, 2004), are directly bound by Myod and/or Myog. Pbxip1, an inhibitor of Pbx binding to DNA (Abramovich et al, 2000; Berkes et al, 2004), is also a target of Myod and Myog. Also included among these targets are the putative promoter regions for four miRNA genes present on the array, which may represent RNA modulators of myogenic differentiation.
Myod and Myog bind the same promoters
A strong overlap between Myod and Myog targets was observed, and this overlap extended beyond the highest-confidence targets when the threshold for comparison was expanded (Figure 1B). Setting a threshold results in a somewhat arbitrary division of genes into ‘bound' and ‘not bound' categories, and the true overlap between Myod and Myog target sets may be even greater than that depicted. Individual gene confirmation supports this notion: for example, Urod, Myo10, and Gys1 were identified as targets of Myog but not Myod at a stringent high-confidence threshold, whereas promoter-specific PCR on the same DNA input samples showed that Myod was bound to both the Myo10 and Gys1 promoter (data not shown). To confirm that the overlap between Myod and Myog targets was not an attribute peculiar to the MDER MEFs, we performed similar ChIP studies in the C2C12 cell line, a widely used mouse myoblast cell line (Yaffe and Saxel, 1977). Genome-wide location analysis of Myod and Myog in differentiated C2C12 myotubes confirmed the large overlap in target genes seen in the MDER cells (Figure 1C). These results were also in general agreement with those reported in a recently published study (Blais et al, 2005) using a microarray containing promoters for 4700 mouse genes to define genomic targets of Myod and Myog in C2C12 myotubes differentiated for 96 h (Supplementary Figure S1).
The similarity of Myod and Myog target genes was further supported by an analysis of DNA sequence motifs associated with either Myod or Myog target genes. We searched the bound promoter sequences represented on the array for enriched motifs using the program MEME, and retrieved essentially the same E-box motifs for both Myod-bound promoters and for Myog-bound promoters (Figure 1D). These motifs agree with the consensus 5′-CANNTG-3′ preferred binding site for Myod identified by in vitro selection and indicate that the same motif is associated with both Myod- and Myog-regulated genes (Blackwell and Weintraub, 1990; Huang et al, 1996). E-boxes were not recovered when shuffled promoter sequences or promoters not identified as bound by Myod or Myog were used as inputs. The frequency of this specific canonical E-box in particular promoter sequences varied from zero to six, with a mean of 1.2±0.8 present in promoter sequences bound by Myod, and 1.2±0.9 present in promoter sequences bound by Myog. Binding of Myod and Myog to promoters that do not contain this specific canonical E-box sequence might be through other E-box sequences or through non-canonical sequences, such as occurs at the non-canonical binding site for Myod adjacent to the Pbx site in the Myog promoter (Berkes et al, 2004).
In order to determine whether binding of Myod or Myog was associated with recruitment of RNA polymerase II (RNA Pol II) and gene transcription, genome-wide location analysis was performed on the initiation form of RNA Pol II in the MDER cells before and after differentiation. When the Myod and Myog binding data were compared to the Pol II binding data, Myod and Myog target genes were significantly enriched among genes that showed an increase in Pol II occupancy during the course of differentiation (Figure 2), indicating that binding of Myod and Myog was generally associated with gene activation.
Figure 2.
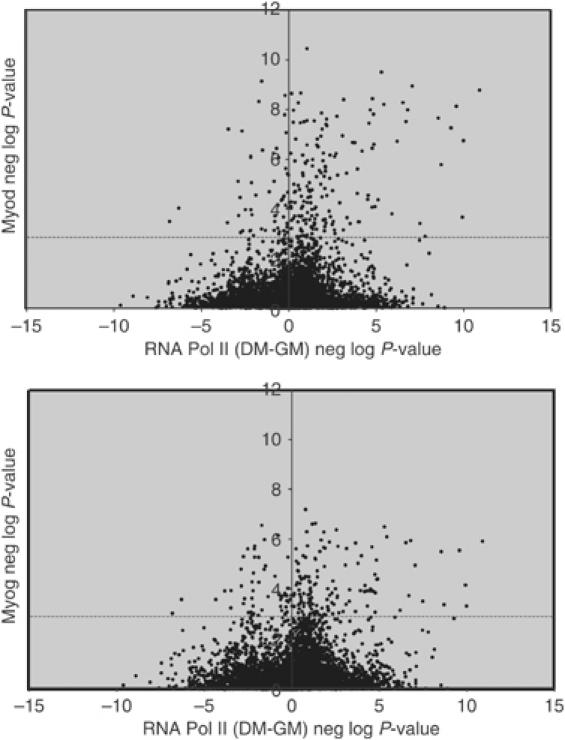
Myod and Myog are associated with genes that RNA Pol II is recruited to during differentiation. Genome-wide location analysis was performed on the initiating form of RNA Pol II in MDER cells maintained in growth medium (GM) or differentiated for 36 h (DM). The x-axis represents the difference in the negative log of the P-value for RNA Pol II binding in DM versus GM, so that points on the right side of the axis show an increase in Pol II occupancy during the course of differentiation. The y-axis indicates the negative log of the P-value for either Myod or Myog binding, so that points higher on the axis are more likely to be bound by Myod or Myog. The dashed line indicates the 0.001 P-value significance threshold for Myod or Myog binding. There is a statistically significant (P-value of 5.3 × 10−8 for Myod, 9.9 × 10−5 for Myog) enrichment of points in the upper right-hand quadrant of the graphs (bound by Myod/Myog and increase in Pol II occupancy upon shift to DM).
The genome-wide ChIP studies demonstrated that Myod and Myog have a large number of shared target genes and those targets have similar E-box binding sites. To determine the role of each factor in gene regulation, we developed a cell line in which the activity of Myog and Myod can be assessed relatively independently. We have not been able to generate a functional fusion protein between Myog and a hormone binding domain similar to the MDER. Therefore, we introduced a constitutively expressed Myog into the MDER MEFs (hereafter referred to as MDER-Myog cells). We used a high-titer retrovirus to create a highly complex polyclone to limit clonal variation. RNA and Western analysis confirmed that, in the absence of virally driven Myog, Myog RNA and protein were undetectable prior to Myod induction, were barely detectable following 12 h of Myod induction, and had relatively high abundance at 24 and 48 h following induction (Figure 3A and B). The amount of the virally driven Myog RNA and protein at 12, 24, and 48 h in differentiation medium without β-estradiol induction was comparable to the peak levels of endogenous Myog RNA and protein at 24 and 48 h of Myod induction. Therefore, we can assess the activity of Myog in the absence of Myod activity when it is expressed at levels similar to that attained by the endogenous Myog. Western analysis also shows that the amount of Myod protein is relatively constant during the 48 h induction period in the induced cells (Figure 3B); however, the Myod protein is less stable in the uninduced cells and relatively hypophosphorylated. Using the MDER and MDER-Myog cells, we can assess the activity of Myog in the absence of Myod throughout the time course, the activity of Myod in the absence of Myog at the early time points, and the effect of the combined activity of Myod and Myog at the early time points.
Figure 3.
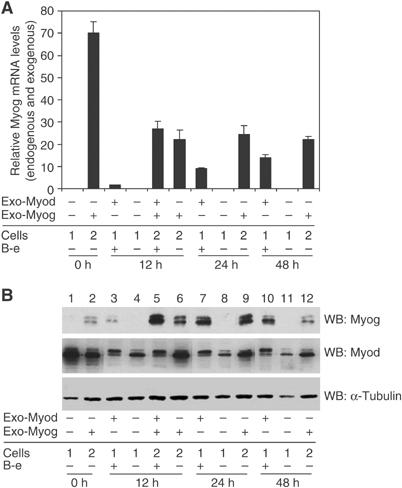
Exogenous Myog is expressed at levels comparable to endogenous Myog at 24 h of differentiation. MDER (Cells 1) or MDER-Myog (Cells 2) cells were induced for the indicated time in DM in the presence or absence of β-estradiol (B-e). MDER was activated by the addition of β-estradiol. Exo-Myod and Exo-Myog indicates whether the cells were expressing exogenous Myod or Myog. (A) Four independent RNA samples were isolated at the indicated time and the total amount of Myog transcripts in each sample was determined by real-time PCR using a probe against the Myog coding region. The relative expression levels were normalized to Timm17b in the same samples. (B) Cell lysates collected at the indicated time were subjected to SDS–PAGE and immunoblotted with anti-Myog, anti-Myod, or anti-α-tubulin antibodies. The multiple bands in Myod and Myog blots are consistent with known phosphorylation of Myod and Myog.
To determine the genes activated by Myod and Myog, we induced Myod activity, harvested independent triplicate sets of RNA at representative times after induction, and analyzed expression levels using the Affymetrix MOE 430A expression arrays that contain 22 626 features representing approximately 12 822 genes. Consistent with our previous studies, the induction of Myod activity increased the expression of a broad range of genes using a relatively stringent criteria of a two-fold change and a false discovery rate (FDR) <0.05. At 12 h following Myod induction, when there is very little endogenous Myog present (see Figure 3B, lane 3), 184 genes were increased relative to time zero cells and uninduced cells at 12 h. At 24 and 48 h following Myod induction, when endogenous Myog is abundant (see Figure 3B, lanes 7 and 10), 389 and 678 genes, respectively, were induced (Figure 4A). Comparison of the gene expression results with global binding data indicated that of the 167 genes identified as Myod targets by global ChIP that were also present on the Affymetrix arrays, 78 showed a 1.5-fold or greater change in expression with an FDR <0.05 at some point during the course of differentiation (P=3.8 × 10−15). Likewise, of the 131 Myog targets in the global ChIP assay that are represented on the Affymetrix arrays, 57 showed a 1.5-fold or greater change in expression with an FDR <0.05 during the course of differentiation (P=6.5 × 10−10). For genes that are targets of both Myod and Myog, there is an even stronger correlation between binding and changes in expression with 41 of 65 target genes that change significantly in expression (P=3.7 × 10−14).
Figure 4.
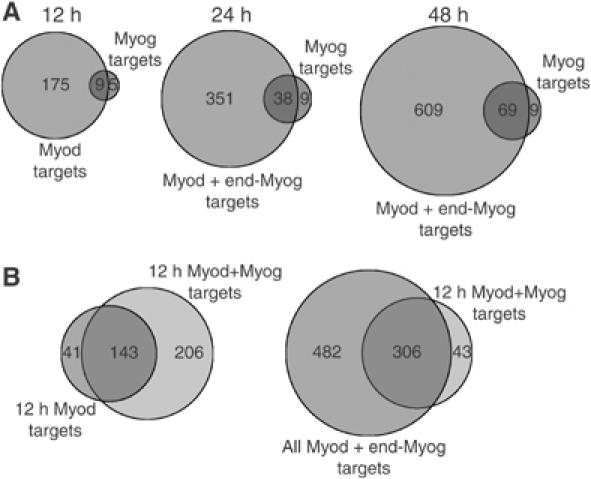
Myod and Myog cooperate to activate a set of genes normally expressed late in differentiation. (A) Myog activates a subset of Myod-activated genes. Venn diagrams show the overlap of Myod- and Myog-activated genes at different time points. At 12 h, Myod alone activates a substantially larger number of genes than Myog alone, whereas at the 24 and 48 h time points, the combination of Myod and endogenous Myog activates a larger number of genes than Myog alone. (B) Precocious expression of Myog together with Myod shifts the expression of normally late-activated genes to an earlier time point. Venn diagrams show that the 12 h Myod+Myog-activated genes encompass the set of genes activated by Myod alone at 12 h (left panel) and are a subset of genes activated by Myod at 12, 24, or 48 h (right panel) (note that 24 and 48 h Myod targets are activated in the presence of endogenous Myog).
By shifting the MDER-Myog cells to differentiation medium without β-estradiol, we can assess genes activated by Myog in the absence of Myod, when Myog protein is present at levels comparable to the peak levels of endogenous Myog during Myod-induced differentiation (see Figure 3B, compare lanes 6 and 9 with lanes 7 and 10). In comparison to Myod induction, few genes were increased by Myog alone. Using the same criteria as for Myod (FDR <0.05 and a two-fold change in expression), Myog increased 14 genes at 12 h, 47 genes at 24 h, and 78 genes at 48 h. At each time point, most of the genes activated by Myog were also activated by Myod (Figure 4A). Therefore, induction of Myod activates approximately eight-fold more genes than Myog alone, and the genes activated by Myog are a subset of genes activated by Myod. The fact that the Myog-activated genes are a subset of Myod-activated genes might be due to the induction of Myog expression by Myod at the 24 and 48 h time points.
As noted above, one of the targets of Myod is Myog, which is initially expressed between 12 and 24 h following Myod induction. Therefore, the late Myod targets, genes activated at 24 and 48 h in this study, are activated in the presence of both Myod and Myog. We sought to determine whether Myog cooperates with Myod to activate some of these late target genes. To do this, we compared the induction of genes by Myod at 12 h in the MDER cells, a time point with very little expression of the endogenous Myog, to genes activated by Myod in the MDER-Myog cells that constitutively express Myog. In comparison to the 184 genes activated by Myod alone at 12 h, in the presence of Myog a total of 349 genes were activated by the combination of Myod and Myog, representing most of the 12 h Myod targets and an additional ∼200 genes not activated by Myod alone at 12 h. Almost all of these ∼200 genes were expressed in the MDER cells at the later time points of 24 and 48 h when endogenous Myog is expressed (Figure 4B). Therefore, Myog appears to cooperate with Myod to regulate the expression of a subset of genes normally expressed later in the program of Myod-mediated differentiation. Furthermore, the precocious expression of Myog can shift the expression of these genes to an earlier time point following Myod induction.
Myod-dependent histone acetylation and binding of Myod and Myog
To determine the mechanisms of Myod and Myog activities, we focused our analyses on a promoter activated early following Myod induction (Itga7, activated before 12 h), an intermediate gene (Myog, activated between 12 and 24 h), and three late genes (Tnnc2, Myh3, and Mylpf, activated between 24 and 48 h). We used real-time RT–PCR and Northern analysis to confirm RNA expression and assessed histone acetylation and the binding of Myod and Myog by ChIP. At the early activated gene, RT–PCR showed that Myod robustly activated gene expression at 12 h, whereas constitutive expression of Myog induced a low baseline activation at time zero but did not further induce gene expression at 12 h of low serum induction, and the combination of Myod and Myog at 12 h was just a little more than additive (Figure 5A). ChIP showed a substantial induction of H3 and H4 acetylation by Myod induction, whereas Myog did not induce histone acetylation and the combination of Myod and Myog did not enhance the degree of acetylation beyond that attained by Myod alone. ChIP with antisera to Myog and Myod revealed very little enhancement of Myog binding at either 12 h alone or in the presence of Myod, whereas Myod showed robust binding following induction with β-estradiol and low serum. Therefore, at this early gene, only the binding of Myod is associated with increased histone acetylation and is alone sufficient for nearly full activation of expression.
Figure 5.
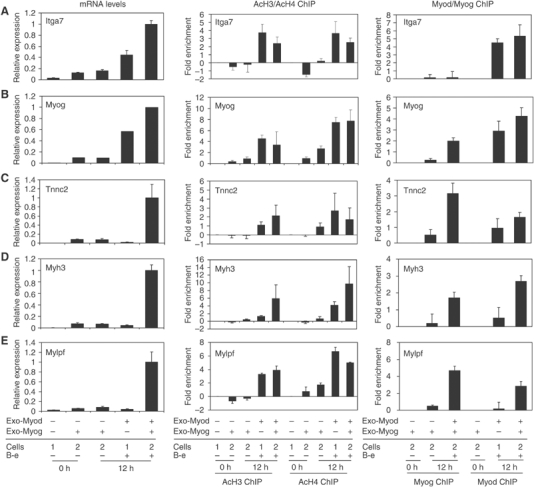
Myod and Myog have distinct and sequential roles in regulating late gene expression. Cells expressing MDER (Cells 1) or MDER+Myog (Cells 2) were induced for the indicated time as described in Figure 3. Exo-Myod and Exo-Myog indicate whether the exogenous Myod or Myog is expressed in the cells. For each gene studied, the left panel shows the mRNA levels quantified by quantitative real-time PCR (A, C–E) or Northern blot (B), the middle panel shows the ChIP results performed with acetyl-H3 and acetyl-H4 antisera, and the right panel shows the ChIP results with Myod and Myog antisera. The relative expression levels of real-time PCR were normalized to the amount of Timm17b mRNA in the same samples, and the data shown are the means±s.e. of reactions from four independent samples. The endogenous Myog mRNA levels were detected by Northern blot with a probe against the 3′-UTR region, and the same blot reprobed with 18S rRNA was used as a loading control. For the ChIP assays, pancreatic amylase 2 (Amy2) was used as an internal control in multiplex PCR for the indicated promoters. The fold enrichments were calculated relative to the amount of input chromatin and the basal histone acetylation or Myod or Myog binding at the 0 h time was subtracted to determine fold change relative to uninduced cells. The error bars indicate standard errors of the mean for the particular experiments performed in triplicate.
At the Myog gene, a gene activated at an intermediate time point, Myog alone showed a marginal induction of gene expression, a low level of histone acetylation, and marginal binding at the Myog promoter, whereas Myod alone robustly induced gene expression and showed robust binding and histone acetylation (Figure 5B). Similar to the early gene, the combination of Myod and Myog had a roughly additive effect on expression and did not substantially alter the degree of Myod-induced acetylation; however, in the presence of Myod, the amount of bound Myog increased substantially, perhaps reflecting greater chromatin accessibility due to the histone acetylation induced by Myod. In contrast, the presence of Myog did not alter the amount of Myod binding to the Myog promoter.
At the normally late-activated Tnnc2, Mylpf, and Myh3 genes, neither Myog nor Myod alone substantially activated gene expression at 12 h, whereas the combination synergistically induced high levels of expression (Figure 5C–E). Interestingly, although Myod alone did not induce expression of these genes, it was sufficient to induce histone acetylation. At these late genes, the binding of Myog was largely dependent on the presence of Myod, and the binding of Myod was also enhanced by the presence of Myog. Therefore, Myod is sufficient to initiate binding and histone modifications at these late genes but the formation of a stable complex and gene expression requires the additional activity of Myog.
Discussion
These data support distinct roles for Myod and Myog at a common set of promoters: Myod initiates histone modifications and is sufficient for expression of early genes but requires combined activity with Myog at late genes, whereas Myog does not efficiently activate very many genes in the absence of Myod but acts synergistically with Myod on a set of genes normally expressed late in the program of myogenic differentiation. Even when constitutively expressed in GM, Myog does not substantially activate muscle gene expression in the absence of Myod (see 0 h time point in the first panels of Figure 5). The dependence of Myog on the activity of Myod could reflect the relatively inefficient binding of Myog to the late genes in the absence of Myod, perhaps because the Myod-mediated histone modifications are critical to permit the formation of a stable DNA binding complex at these promoters.
Our demonstration that Myod and Myog have distinct roles at a common set of target genes extends an emerging model of the transcriptional regulation of the complex program of skeletal muscle differentiation. As noted above, genetic studies have assigned a role for Myod and Myf5 in the specification of the skeletal muscle lineage and a role for Myog in the differentiation of the specified muscle cells. We have previously shown that protein motifs conserved in Myod and Myf5, the H/C and Helix 3 domains, interact with the Pbx/Meis homeodomain protein complex that resides at a subset of Myod-regulated genes, and that mutation of these domains prevents the initial recruitment of Myod to these target promoters (Berkes et al, 2004). Myod has been shown to complex with the HATs p300 and PCAF and its recruitment to endogenous promoters is correlated with histone acetylation (Puri et al, 1997; Bergstrom et al, 2002). In addition, Myod can recruit the Swi/Snf complex in a p38-dependent manner (Simone et al, 2004). At the Myog promoter, the recruitment of Myod through protein interactions with the Pbx/Meis complex can initiate chromatin remodeling prior to the formation of a stable interaction with its cognate E-box binding site (de La Serna et al, 2005). Therefore, the role of Myod in the specification of the muscle lineage can be understood by its ability to find its target genes within a native chromatin context and initiate chromatin remodeling to make factor binding sites available, including its own E-box sites.
In contrast, the role of Myog as a differentiation factor has not been as well characterized. Previously, we demonstrated that the Helix 3 domain of Myog cannot functionally substitute for the Helix 3 domain of Myod, but instead had activity consistent with a more classical activation domain (Bergstrom and Tapscott, 2001). Specifically, the Myog Helix 3 domain activated transcription of a reporter gene when fused to the Gal4 DNA binding domain, whereas the Myod Helix 3 was a very poor transcriptional activator. This led us to propose a model where Myod and Myog might have distinct and sequential roles at specific promoters: Myod initiates chromatin remodeling but is not sufficient for gene expression, whereas Myog can only bind at genes previously initiated by Myod and brings a new activation domain to the promoter. However, at that time, it was not possible to assess whether Myog might have functions equivalent to Myod at its own set of promoters, and that the Myog Helix 3 might permit Myog to initiate genes that Myod could not. Our current study shows that Myod and Myog largely bind to the same set of genes, and that the activity of Myog as a transcription factor is facilitated by the initiating activity of Myod at the majority of Myog-regulated target genes.
It is interesting that at the early and intermediate targets of Myod (Itga7 and Myog), the expression of Myog has a relatively small, additive effect on the level of transcription, or at least RNA abundance, indicating that the activation functions of Myod are sufficient for this set of promoters. We have previously shown that the regulatory E-boxes in the Myog promoter are not accessible to restriction endonucleases in fibroblasts prior to the expression of Myod, whereas they become accessible within 12 h after Myod expression, roughly coincident with Myod-mediated histone modifications. Similar to the late genes, Myog does not efficiently bind to its own promoter without the activity of Myod, nor does it induce substantial histone H3 acetylation. In contrast to the late genes, however, the constitutive expression of Myog had only an additive effect on the level of Myog expression at 12 h of Myod induction. It should be emphasized, however, that the requirement for Myog to mediate terminal differentiation was most pronounced in vivo (Nabeshima et al, 1993) and it is possible that some signaling pathways or cofactors that are not active in our tissue culture model would increase the dependence of the Myog promoter for autoregulation, or make it resistant to differentiation-related repression.
It is at the normally late-activated genes in our model system of Myod-mediated myogenic differentiation, the genes activated between 24 and 48 h after Myod induction, that Myog has its most pronounced synergistic activity with Myod. At these late genes, Myod initiates histone modifications and binds to the regulatory elements in the absence of Myog; however, the bound Myod is not sufficient to mediate target gene transcription. Similarly, at the early 12 h time points, Myog alone is unable to mediate transcription at these late target genes. This could be due to inefficient binding in the absence of Myod, perhaps because Myog is much less effective at mediating histone modifications, or because a combination of the Myod and Myog activation functions is necessary at these promoters, or a combination of both mechanisms.
Myogenesis occurs relatively normally in Myod-null mice; however, the combined inactivation of both Myod and Myf5 results in the absence of myogenic specification in the somites and limbs (Rudnicki et al, 1993). The domains of Myod required to initiate chromatin remodeling at some loci are conserved in Myf5 and Mrf4, but not in Myog (Bergstrom and Tapscott, 2001). Therefore, in the absence of Myod, either Myf5 or Mrf4 might initiate chromatin remodeling and function cooperatively with Myog to mediate the expression of this set of genes. In addition, the initial chromatin remodeling is likely to affect the efficiency of Myog binding but might not be an absolute requirement for Myog activity. Indeed, overexpression of Myog can activate the expression of many of these genes and the role of Myod might be mainly to establish tight temporal control over gene expression, rather than an absolute requirement for gene expression.
It is interesting that Myod is sufficient to activate early gene expression but not sufficient to activate the promoters of late genes. We have previously shown that DNA binding and transcriptional activation are separable functions in Myod and proposed that interaction with other factors recruited to the promoter is necessary to expose the activation function of Myod (Davis et al, 1990; Weintraub et al, 1991). Myod will not activate expression from a single binding site but requires paired sites or binding of an adjacent transcription factor such as Pbx (Weintraub et al, 1990; Knoepfler et al, 1999), indicating that either homophilic or heterophilic interactions are necessary for the activation domain of Myod to function. Our current findings are consistent with this original model and suggest that the early activated promoters have sequences and factors necessary for Myod to function as a transcriptional activator, whereas the later genes require the subsequent binding of other transcription factors, such as myogenin. Expression of Myod also results in the decline of transcript levels for some genes and it remains to be determined if this is due to promoter-specific recruitment of repressors or to the activation of miRNA or siRNA from separate loci.
The combination of genome-wide factor binding and gene expression analyses together with promoter-specific profiling of regulatory events is revealing the molecular circuitry of complex biological processes (Lee et al, 2002; Harbison et al, 2004). This study shows that Myod and Myog participate in a feed-forward regulatory network, in which Myod transcriptionally activates the expression of Myog, and Myog subsequently cooperates with Myod to activate expression of another set of genes. The intrinsic time delay of a feed-forward circuit results in an obligate temporal patterning of gene expression, as demonstrated in this study when pre-expression of Myog resulted in the shift of normally late expressed genes to an earlier time following the induction of Myod activity. We previously demonstrated that Myod generates a similar feed-forward circuit involving the Mef2D transcription factor and the p38 MAP kinase pathway to temporally pattern a subset of normally late-expressed genes (Penn et al, 2004). The fact that Myog also participates in a feed-forward circuit with Myod suggests that this mechanism might be broadly used in patterning muscle gene expression and likely indicates that feed-forward mechanisms will be shown to regulate a large variety of complex responses to single initiating events. Ultimately, it will be interesting to determine how these circuits evolve and the possible functional relationship among the different components of feed-forward circuits that utilize a common master regulatory factor, such as Myod, together with different co-effector factors, such as Myog and Mef2D.
Materials and methods
Cell culture
Mouse embryo Myf-5/Myod null fibroblasts transduced with pBABE-MDER (Bergstrom et al, 2002) (MDER cells) were maintained in DME with 10% bovine calf serum (Hyclone). MDER-Myog cells were developed by infecting MDER cells with LXSH-Myog retrovirus. Control cells were infected with LXSH empty vector. Differentiation was induced at confluence in DME with 1% horse serum, 10 μg/ml insulin, and 10 μg/ml transferrin (DM). MDER was activated in DM plus 10−7 M β-estradiol. C2C12 cells were grown in DMEM with 20% fetal bovine serum (Gibco), and differentiation was induced at confluence in DM for 36 h.
Genome-wide location analysis
Location analysis experiments were performed essentially as described by Odom et al (2004). Briefly, ∼0.5–1.0 × 108 MDER or C2C12 cells were crosslinked with 1% formaldehyde at room temperature. Crosslinked cells were washed and harvested, pellets were sonicated, and the resulting chromatin fragments were immunoprecipitated overnight at 4°C. Crosslinks were reversed and enriched DNA fragments amplified by ligation-mediated PCR (LM-PCR) and fluorescently labeled using Cy5-dUTP (Amersham). A reference unenriched sample of DNA was also LM-PCR amplified and labeled with Cy3-dUTP, and the two pools of labeled DNA were mixed and hybridized to mouse 13K promoter arrays. A single-array error model (Lee et al, 2002) was used to determine P-values for enrichment of individual features on the array. For ChIP experiments directed against RNA Pol II, a set of intergenic control spots were used as the basis for an error model to describe confidence thresholds for the association of RNA Pol II with transcriptional start sites, as median normalization was not appropriate owing to the large number of enriched features. Data presented in this paper are the result of triplicate experiments. For MDER cells, anti-Myod antisera (Tapscott et al, 1988), anti-Myog F5D (Santa Cruz Biotechnology), and anti-RNA Pol II 8WG16 antibodies were used for triplicate experiments, and results were confirmed using anti-Myod sc-760 (Santa Cruz Biotechnology) and anti-Myog sc-576 (Santa Cruz Biotechnology) in singleton experiments. For C2C12 cells, anti-Myod sc-760, anti-Myog sc-576, and anti-RNA Pol II 8WG16 were used. Array data are available at NCBI GEO (www.ncbi.nlm.nih.gov/geo/), accession GSE3858.
Mouse 13K promoter array
A custom self-printed mouse promoter microarray containing ∼13 000 features generally spanning 750 bp upstream to 250 bp downstream of transcription start sites was designed and manufactured by a process analogous to that for previously described human promoter microarrays (Odom et al, 2004; Guenther et al, 2005). A set of intergenic control probes were designed by identifying the largest gaps between predicted genes and designing probes to the largest of these gaps.
Motif analysis
Probe sequences spotted on the promoter array that were identified as Myod or Myog targets in MDER cells with a P-value <0.001 were input into the program MEME (Bailey and Elkcan, 1994), with search parameters set to look for enriched motifs between 5 and 10 nucleotides long assuming sites were present once per sequence or not at all. As controls for motifs specific to Myod/Myog targets, both shuffled Myod/Myog-bound probe sequences and a set of 200 random probe sequences from the array were input into MEME using the same search parameters. E-box motifs identified by MEME were input into the program WebLogo to generate sequence logos.
Western and Northern analyses
MDER and Myog proteins were detected using rabbit anti-Myod (Tapscott et al, 1988) and mouse anti-Myog antibodies (F5D). Northern blot was performed by standard techniques using a Myog probe described previously (Bergstrom et al, 2002).
Quantitative real-time PCR
Real-time PCRs were performed according to the manufacturer's instructions (Applied Biosystems). The probe and primer sets are described in Supplementary Table S3. The relative expression levels were normalized to those of Timm17b in the same samples.
Chromatin immunoprecipitation
ChIP was performed as described previously (Bergstrom et al, 2002). Antibodies used for ChIP were as follows: anti-Myod (Tapscott et al, 1988), anti-Myog (Santa Cruz), anti-acetyl H3 (UBI), and anti-acetyl H4 (UBI). PCR primers used for detecting Cdh15, Myog, Tnnc2, Des, Mylf, Myh3, and Amy2 loci were described before (Penn et al, 2004). PCR primers used for detecting Itga7 locus are listed in Supplementary Table S3. PCR products were stained with SYBR Gold (Molecular Probes) and quantified using a Typhoon scanner (Molecular Dynamics). Real-time probes and primers to Myh3, Mylpf, and Amy2 are in Supplementary Table S3.
Affymetrix gene expression arrays
Hybridization samples were prepared according to the Affymetrix GeneChip Expression Analysis Manual (Affymetrix, Santa Clara, CA) using 10 μg of total RNA and hybridized to Affymetrix mouse MOE 430A arrays. The PM (perfect match) probe intensities were corrected by robust multiarray average, normalized by quantile normalization, and summarized by medianpolish using the Affy package of Bioconductor. The comparison of global gene expression profiles was made using the LIMMA package of Bioconductor. Array data are available at NCBI GEO, accession GSE3858.
Supplementary Material
Figure S1
Supplementary Figure 1
Table S1
Table S2
Table S3
Acknowledgments
We thank DT Odom and the Whitehead Microarray Facility for production of the promoter arrays, the Whitehead Institute Bioinformatics Group and Elizabeth Herbolsheimer for computational support, and J Delrow and the FHCRC Genomics Facility. This work was supported by NIH AR45113 (SJT), NIH CA74841 (CK), and NIH GM069400 and HG002668 (RAY). YC was supported by an FHCRC Interdisciplinary Training grant sponsored by Amgen and a Fellowship from the Merck Research Laboratories. RMK was supported by an American Cancer Society postdoctoral fellowship and LAB by NRSA postdoctoral fellowship CA094664.
References
- Abramovich C, Shen WF, Pineault N, Imren S, Montpetit B, Largman C, Humphries RK (2000) Functional cloning and characterization of a novel nonhomeodomain protein that inhibits the binding of PBX1–HOX complexes to DNA. J Biol Chem 275: 26172–26177 [DOI] [PubMed] [Google Scholar]
- Asakura A, Fujisawa-Sehara A, Komiya T, Nabeshima Y (1993) MyoD and myogenin act on the chicken myosin light-chain 1 gene as distinct transcriptional factors. Mol Cell Biol 13: 7153–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkcan C (1994) Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. Menlo Park, CA: AAAI Press [Google Scholar]
- Banham AH, Connors JM, Brown PJ, Cordell JL, Ott G, Sreenivasan G, Farinha P, Horsman DE, Gascoyne RD (2005) Expression of the FOXP1 transcription factor is strongly associated with inferior survival in patients with diffuse large B-cell lymphoma. Clin Cancer Res 11: 1065–1072 [PubMed] [Google Scholar]
- Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H (1990) The protein Id: a negative regulator of helix–loop–helix DNA binding proteins. Cell 61: 49–59 [DOI] [PubMed] [Google Scholar]
- Bergstrom DA, Penn BH, Strand A, Perry RL, Rudnicki MA, Tapscott SJ (2002) Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell 9: 587–600 [DOI] [PubMed] [Google Scholar]
- Bergstrom DA, Tapscott SJ (2001) Molecular distinction between specification and differentiation in the myogenic basic helix–loop–helix transcription factor family. Mol Cell Biol 21: 2404–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkes CA, Bergstrom DA, Penn BH, Seaver KJ, Knoepfler PS, Tapscott SJ (2004) Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell 14: 465–477 [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Weintraub H (1990) Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 250: 1104–1110 [DOI] [PubMed] [Google Scholar]
- Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD (2005) An initial blueprint for myogenic differentiation. Genes Dev 19: 553–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Edmondson DG, Li L, Olson EN (1991) Transforming growth factor beta represses the actions of myogenin through a mechanism independent of DNA binding. Proc Natl Acad Sci USA 88: 3822–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F (2003) The formation of skeletal muscle: from somite to limb. J Anat 202: 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Cheng PF, Lassar AB, Weintraub H (1990) The MyoD DNA binding domain contains a recognition code for muscle-specific gene activation. Cell 60: 733–746 [DOI] [PubMed] [Google Scholar]
- de La Serna IL, Ohkawa Y, Berkes CA, Bergstrom DA, Dacwar CS, Tapscott SJ, Imbalzano AN (2005) MyoD targets chromatin complexes to the myogenin locus prior to forming a stable DNA-bound complex. Mol Cell Biol 25: 3997–4009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado I, Huang X, Jones S, Zhang L, Hatcher R, Gao B, Zhang P (2003) Dynamic gene expression during the onset of myoblast differentiation in vitro. Genomics 82: 109–121 [DOI] [PubMed] [Google Scholar]
- Fiddler TA, Smith L, Tapscott SJ, Thayer MJ (1996) Amplification of MDM2 inhibits MyoD-mediated myogenesis. Mol Cell Biol 16: 5048–5057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday BB, Mitchell PO, Kegley KM, Pavlath GK (2003) Calcineurin initiates skeletal muscle differentiation by activating MEF2 and MyoD. Differentiation 71: 217–227 [DOI] [PubMed] [Google Scholar]
- Gerber AN, Klesert TR, Bergstrom DA, Tapscott SJ (1997) Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev 11: 436–450 [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA (2005) Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci USA 102: 8603–8608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364: 501–506 [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, Cheng PF, Weintraub H (1993) Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc Natl Acad Sci USA 90: 8028–8032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Blackwell TK, Kedes L, Weintraub H (1996) Differences between MyoD DNA binding and activation site requirements revealed by functional random sequence selection. Mol Cell Biol 16: 3893–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaynes JB, Johnson JE, Buskin JN, Gartside CL, Hauschka SD (1988) The muscle creatine kinase gene is regulated by multiple upstream elements, including a muscle-specific enhancer. Mol Cell Biol 8: 62–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassar-Duchossoy L, Gayraud-Morel B, Gomes D, Rocancourt D, Buckingham M, Shinin V, Tajbakhsh S (2004) Mrf4 determines skeletal muscle identitiy in Myf5:MyoD double-mutant mice. Nature 431: 466–471 [DOI] [PubMed] [Google Scholar]
- Kataoka Y, Matsumura I, Ezoe S, Nakata S, Takigawa E, Sato Y, Kawasaki A, Yokota T, Nakajima K, Felsani A, Kanakura Y (2003) Reciprocal inhibition between MyoD and STAT3 in the regulation of growth and differentiation of myoblasts. J Biol Chem 278: 44178–44187 [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Bergstrom DA, Uetsuki T, Dac-Korytko I, Sun YH, Wright WE, Tapscott SJ, Kamps MP (1999) A conserved motif N-terminal to the DNA-binding domains of myogenic bHLH transcription factors mediates cooperative DNA binding with pbx-Meis1/Prep1. Nucleic Acids Res 27: 3752–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804 [DOI] [PubMed] [Google Scholar]
- Liu S, Spinner DS, Schmidt MM, Danielsson JA, Wang S, Schmidt J (2000) Interaction of MyoD family proteins with enhancers of acetylcholine receptor subunit genes in vivo. J Biol Chem 275: 41364–41368 [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN (1996) Combinatorial control of muscle development by basic helix–loop–helix and MADS-box transcription factors. Proc Natl Acad Sci USA 93: 9366–9373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JL, Li Y, Hill AA, Mounts WM, Miller CP (2002) Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol Genomics 10: 103–111 [DOI] [PubMed] [Google Scholar]
- Nabeshima YK, Hanaoka M, Hayasaka M, Esumi E, Li S, Nonaka I (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364: 532–535 [DOI] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA (2004) Control of pancreas and liver gene expression by HNF transcription factors. Science 303: 1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN, Williams RS (2000) Calcineurin signaling and muscle remodeling. Cell 101: 689–692 [DOI] [PubMed] [Google Scholar]
- Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, Tapscott SJ (2004) A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev 18: 2348–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pownall ME, Gustafsson MK, Emerson CP Jr (2002) Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 18: 747–783 [DOI] [PubMed] [Google Scholar]
- Puri PL, Sartorelli V, Yang XJ, Hamamori Y, Ogryzko VV, Howard BH, Kedes L, Wang J Y, Graessmann A, Nakatani Y, Levrero M (1997) Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1: 35–45 [DOI] [PubMed] [Google Scholar]
- Rudnicki MA, Schnegelsberg PN, Stead RH, Braun T, Arnold HH, Jaenisch R (1993) MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75: 1351–1359 [DOI] [PubMed] [Google Scholar]
- Sabourin LA, Rudnicki MA (2000) The molecular regulation of myogenesis. Clin Genet 57: 16–25 [DOI] [PubMed] [Google Scholar]
- Shen X, Collier JM, Hlaing M, Zhang L, Delshad EH, Bristow J, Bernstein HS (2003) Genome-wide examination of myoblast cell cycle withdrawal during differentiation. Dev Dyn 226: 128–138 [DOI] [PubMed] [Google Scholar]
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL (2004) p38 pathway targets SWI–SNF chromatin remodeling complex to muscle-specific loci. Nat Genet 36: 738–743 [DOI] [PubMed] [Google Scholar]
- Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB (1988) MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 242: 405–411 [DOI] [PubMed] [Google Scholar]
- Tomczak KK, Marinescu VD, Ramoni MF, Sanoudou D, Montanaro F, Han M, Kunkel LM, Kohane IS, Beggs AH (2004) Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J 18: 403–405 [DOI] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW (2004) Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development 131: 4477–4487 [DOI] [PubMed] [Google Scholar]
- Wang Y, Jaenisch R (1997) Myogenin can substitute for Myf5 in promoting myogenesis but less efficiently. Development 124: 2507–2513 [DOI] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Lockshon D, Lassar A (1990) MyoD binds cooperatively to two sites in a target enhancer sequence: occupancy of two sites is required for activation. Proc Natl Acad Sci USA 87: 5623–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Dwarki VJ, Verma I, Davis R, Hollenberg S, Snider L, Lassar A, Tapscott SJ (1991) Muscle-specific transcriptional activation by MyoD. Genes Dev 5: 1377–1386 [DOI] [PubMed] [Google Scholar]
- Xu Q, Yu L, Liu L, Cheung CF, Li X, Yee SP, Yang XJ, Wu Z (2002) p38 mitogen-activated protein kinase-, calcium-calmodulin-dependent protein kinase-, and calcineurin-mediated signaling pathways transcriptionally regulate myogenin expression. Mol Biol Cell 13: 1940–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Saxel O (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270: 725–727 [DOI] [PubMed] [Google Scholar]
- Yan Z, Choi S, Liu X, Zhang M, Schageman JJ, Lee SY, Hart R, Lin L, Thurmond FA, Williams RS (2003) Highly coordinated gene regulation in mouse skeletal muscle regeneration. J Biol Chem 278: 8826–8836 [DOI] [PubMed] [Google Scholar]
- Yutzey KE, Rhodes SJ, Konieczny SF (1990) Differential trans-activation associated with the muscle regulatory factors MyoD1, myogenin, and MRF4. Mol Cell Biol 10: 3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Olson EN (2001) The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci USA 98: 7354–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Seo J, Wang Z, Wang Y, Shneiderman B, Hoffman EP (2003) In vivo filtering of in vitro expression data reveals MyoD targets. C R Biol 326: 1049–1065 [DOI] [PubMed] [Google Scholar]
- Zhu S, Goldschmidt-Clermont PJ, Dong C (2004) Transforming growth factor-beta-induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ Res 94: 617–625 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Supplementary Figure 1
Table S1
Table S2
Table S3