

Abstract
The bacterial toxin aerolysin kills cells by forming heptameric channels, of unknown structure, in the plasma membrane. Using disulfide trapping and cysteine scanning mutagenesis coupled to thiol-specific labeling on lipid bilayers, we identify a loop that lines the channel. This loop has an alternating pattern of charged and uncharged residues, suggesting that the transmembrane region has a β-barrel configuration, as observed for Staphylococcal α-toxin. Surprisingly, we found that the turn of the β-hairpin is composed of a stretch of five hydrophobic residues. We show that this hydrophobic turn drives membrane insertion of the developing channel and propose that, once the lipid bilayer has been crossed, it folds back parallel to the plane of the membrane in a rivet-like fashion. This rivet-like conformation was modeled and sequence alignments suggest that such channel riveting may operate for many other pore-forming toxins.
Keywords: β-barrel, lipid bilayers, membrane proteins, pore-forming, toxin
Introduction
Membrane proteins constitute some 30% of the human proteome, yet relatively little is still known about the structure of these proteins and how they fold and insert into the membrane. These important issues are difficult to address because the proteins of interest must first be isolated in detergents or chaotropic agents and then studied in the presence of membranes. Studies have therefore been confined mainly to two model proteins, bacteriorhodopsin and outer membrane protein A (OmpA) of Escherichia coli. A unique type of system to address folding and membrane insertion is however provided by pore-forming toxins (PFT) produced by a wide range of pathogenic bacteria. These include aerolysin from Aeromonas hydrophila (Abrami et al, 2000), α-toxin from Staphylococcus aureus (Menestrina et al, 2003) and the cholesterol-dependent toxins produced by a variety of Gram-positive bacteria (Tweten et al, 2001). These toxins are extremely intriguing because they are produced as water-soluble proteins but acquire their active state only once they have undergone a major conformational change that leads to their insertion into a lipid bilayer. A key element in this conformational change is the association into multimeric structures: either heptamers, as in the case of aerolysin and Staphylococcus α-toxin, or higher order structures (consisting of up to approximately 50 protomers) as for the cholesterol-dependent toxins. Subsequently, part of the oligomers inserts into the lipid bilayer and forms a transmembrane water-filled channel. The mechanisms that govern folding of the transmembrane region and membrane insertion are not understood at the molecular and atomic levels. Here we have focused on channel formation by aerolysin.
Aerolysin is secreted as a soluble precursor named proaerolysin, which can exist as a dimer or a monomer, and requires the removal of a C-terminal peptide by proteolysis to gain activity (Abrami et al, 2000). Upon high-affinity interaction with glycosyl-phosphatidylinositol (GPI)-anchored proteins on the plasma membrane of the target cell (Abrami et al, 2000), the protein is concentrated (Abrami and van der Goot, 1999) thereby promoting the formation of stable amphipathic heptamers that can insert into the membrane and form channels (Abrami et al, 2000). Channel formation induces ion fluxes leading to membrane depolarization and ultimately cell death (Abrami et al, 2000).
Despite the availability of the X-ray structure of the dimeric form of proaerolysin (Parker et al, 1994), the precise roles of each of its four domains (Figure 1A) have not been fully established and the mechanisms that lead to membrane insertion and channel formation remain poorly understood. Based on electron microscopy and docking experiments, models of the aerolysin channel have been proposed that suggest that domain 4 is the transmembrane region and domain 3 constitutes the mouth of the channel (Figure 1A) (Parker et al, 1994; Tsitrin et al, 2002). We were however intrigued by the presence of a loop in domain 3 (corresponding to amino acids 239–264, further called the DIII-loop) that has a striking pattern of alternating charged and none charged amino acids that could form a transmembrane β-hairpin. Alternating hydrophobic–hydrophilic residues are found in the transmembrane region of Staphylococcal α-toxin (Song et al, 1996), the proposed transmembrane regions of the protective antigen of anthrax toxin (Nassi et al, 2002), perfringolysin O (Shatursky et al, 1999), Clostridium septicum α-toxin (Melton et al, 2004) and bacterial porins (Schulz, 2002). We have previously shown that the DIII-loop in aerolysin is important because hemolytic activity is lost when this loop is covalently linked to the core of domain 3 by engineering a disulfide bridge between residues 253 and 300 (Rossjohn et al, 1998b). Recently, it has been shown that for the α-toxin of C. septicum, a toxin that shares 28% identity and 45% similarity with aerolysin but lacks domain 1 (Ballard et al, 1995), every other residue within the corresponding DIII-loop contacts a hydrophobic environment, compatible with a transmembrane β-strand configuration (Melton et al, 2004).
Figure 1.
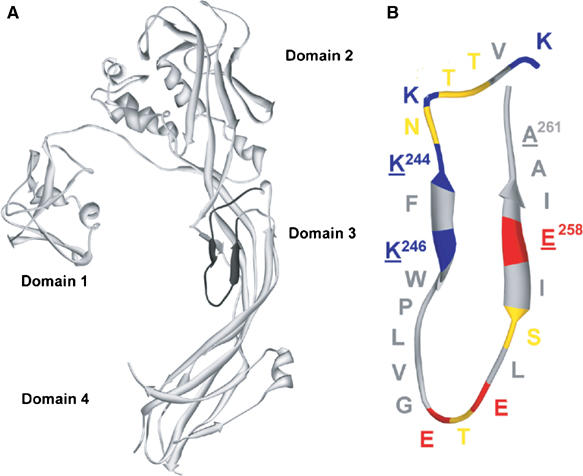
Proaerolysin structure. (A) Ribbon diagram of the proaerolysin X-ray structure (Parker et al, 1994). The DIII-loop in domain 3 (residues K238–A261) is indicated in black. (B) Schematic representation of the DIII-loop. Acidic residues are shown in red, basic residues in blue, polar uncharged residues in yellow and hydrophobic residues in gray. The alternating pattern of charged/uncharged (hydrophobic) residues is broken by a patch of five consecutive hydrophobic residues (W247–G251). Underlined residues represent residues mutated in the K244C-A261C and K246C-E258C double cysteine mutants. Figures were generated using DeepView (Guex and Peitsch, 1997) and rendered using Pov-Ray (http://www.povray.org/).
Here we have studied the topology of the DIII-loop and its functional role in channel formation. By restraining size and/or flexibility of the DIII-loop through the introduction of disulfide bridges at different positions, we first show that the DIII-loop is not required for heptamerization, but is crucial for proper membrane insertion and is directly involved in channel formation. Next, using cysteine scanning mutagenesis coupled to channel analysis using planar lipid bilayers and cysteine-specific labeling, we identify the lumen-lining residues and position the tip of the transmembrane hairpin. The hydrophobicity of this tip leads us to propose a ‘rivet'-like model for membrane anchoring of aerolysin, which also seems relevant for a number of other PFT based on sequence alignments.
Results
Generation of single and double cysteine mutants
To address the role of the DIII-loop in channel formation, we first engineered disulfide bonds within the loop to restrain its size and flexibility. Based on the structure of proaerolysin, the following double mutants were designed and generated: K244C-A261C, K246C-E258C (residues underlined in Figure 1B) and W247C-I257C. In addition, single cysteine mutants were generated for all residues from T240 to E258. C-terminally histidine-tagged variants of all the mutants were produced in E. coli and purified on nickel columns. Their activity was tested by measuring the ability to lyse erythrocytes. As shown in Table I, the double cysteine mutants K244C-A261C and K246C-E258C were non-hemolytic unless reduced with β-mercaptoethanol (βMeOH), indicating that the mutant proteins were properly folded but that disulfide bridge formation occurred and that it impaired activity. The W247C-I257C mutant was always inactive irrespective of its reducing state—and therefore not further studied—most likely because the W247C mutation in itself leads to loss of activity (Table I). All other single cysteine mutants showed significant hemolytic activity (Table I).
Table 1.
Hemolytic activity, single-channel conductance and sensitivity to thiol-specific labeling of DIII-loop single cysteine mutants
| Mutant | Hemolytic activity | Single-channel conductance (pS) | Percentage of current decrease |
|
|---|---|---|---|---|
| MTSEA-biotin | MTSEA-X-biotin | |||
| WT | 11 | 449±24 | ||
| K244C-A261C | 0 | No channels | ||
| K244C-A261C+βMeOH | 9 | 374±35 | 22±4 | 34±14 |
| K246C-E258C | 0 | No channels | ||
| K246C-E258C+βMeOH | 9 | 454±80 | 84±2 | 96±1 |
| T240C | 10 | 378±23 | 31±13 | 16±10 |
| T241C | 10 | 388±32 | 0±0 | 1±1 |
| K242C | 9 | 395±26 | 26±9 | 27±13 |
| N243C | 8 | 431±31 | 0±2 | |
| K244C | 10 | 443±32 | 15±5 | 16±9 |
| F245C | 9 | 423±40 | 0±0 | |
| K246C | 9 | 422±29 | 0±1 | 0±1 |
| W247C | 0 | 452±27 | 2±1 | |
| P248C | 9 | 446±35 | 2±3 | 1±1 |
| L249C | 12 | 409±13 | 2±2 | 0± |
| V250C | 8 | 367±13 | 0±1 | 0±1 |
| G251C | 8 | 389±51 | 0±1 | 2±2 |
| E252C | 8 | 400±35 | 1±2 | 1±1 |
| T253C | 10 | 423±29 | 2±2 | 1±1 |
| E254C | 9 | 426±33 | 26±10 | 31±11 |
| L255C | 12 | 416±37 | 2±1 | |
| S256C | 10 | 431±23 | 35±6 | 24±5 |
| I257C | ND | ND | ND | |
| E258C | 9 | 428±41 | 28±9 | 34±11 |
| ΔLV | 0 | ND | ||
| ΔPLV | 0 | ND | ||
| V250D | 9 | 378±3 | ||
| L249D | 9 | 383±4 | ||
| L249C-V250D | 7 | No channels | ||
| The hemolytic activity of mutant aerolysins in comparison to WT was assayed as described in Materials and methods and expressed in the number of wells lysed after 60 min. The single-channel conductance (measured in 1 M NaCl-containing buffer at ±25 mV) for each single cysteine mutant as well as for K246C-E258C, K246C-E258C and WT aerolysin was calculated from three different experiments as a mean value of 10 single-channel opening events. The percentage of channel closure upon addition of MTSEA-biotin and MTSEA-X-biotin was calculated as (Imax−IMTS) × 100/Imax (see Figure 4A). ND: not determined. | ||||
Binding, heptamerization and membrane insertion of the disulfide-containing mutants
To investigate which step in the mode of action of aerolysin was affected by the introduced disulfide bonds, we tested whether the two double cysteine mutants were able to bind to their GPI-anchored receptors. As shown in Figure 2A, in a toxin overlay assay, both mutants recognized N-CAM and semaphorin 7, two GPI-anchored proteins present in BHK cells (Fivaz et al, 2002). Consistently, these mutants were able to bind to BHK cells (Figure 2B) and oligomerize (Figure 2C), as witnessed by Western blot analysis of extracts from toxin-treated cells. Both mutants were also able to form heptamers in solution (Figure 2D, Coomassie blue-stained gel), although oligomerization was significantly slower for the K244C-A261C mutant.
Figure 2.
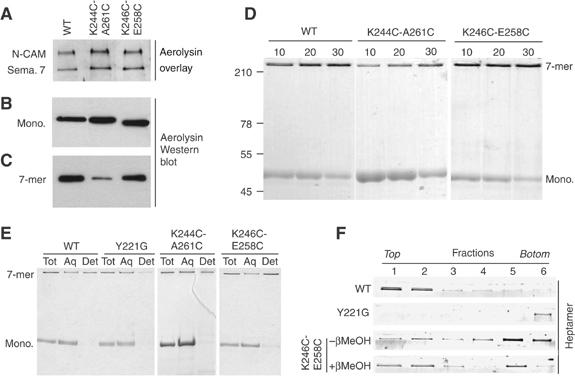
Receptor binding, heptamerization and membrane association of the disulfide-containing mutants. (A) The disulfide-containing mutants were tested in an aerolysin overlay assay on BHK cell extracts (40 μg protein per lane). Aerolysin was revealed using anti-aerolysin antibodies followed by an HRP-labeled secondary antibody. Binding to two GPI-anchored proteins, N-CAM and semaphorin 7 (sema. 7), is illustrated. (B) BHK cells were incubated with trypsin-activated WT or disulfide-containing mutant aerolysins (400 ng/ml) at 4°C for 1 h. Cell extracts (40 μg per lane), from which the nuclei were cleared by centrifugation, were analyzed by Western blotting using an anti-toxin antibody. (C) BHK cells, treated with trypsin-activated WT or mutant aerolysin as in panel B, were washed and further incubated at 37°C for 45 min. The presence of heptamers was revealed by Western blotting against aerolysin. (D) The ability of the disulfide-containing mutants to oligomerize in vitro was assayed by monitoring the appearance of the heptamer as a function of time after trypsin activation. SDS gels were revealed by Coomassie blue staining. (E) WT, Y221G and disulfide-containing mutant proaerolysins were activated with trypsin and allowed to heptamerize in vitro and then submitted to a Triton X-114 partitioning assay. Aliquots from the total sample (Tot), the aqueous (Aq) and detergent (Det) phases were analyzed by SDS–PAGE and Coomassie blue staining. (F) WT, Y221G, K246C-E258C and reduced K246C-E258C heptamer were reconstituted into proteoliposomes and submitted to separation by sucrose density flotation gradients. The six fractions of the gradient, from top to bottom, were analyzed by SDS–PAGE and Coomassie blue staining.
The ability of the disulfide-containing mutants to interact specifically with GPI-anchored proteins and to heptamerize suggests that the overall structure of the toxin was not grossly altered by the introduced disulfide bonds. In the normal course of events, concomitant to heptamerization, hydrophobic patches become exposed, leading to membrane insertion of aerolysin and channel formation. Owing to its amphipathic nature, the heptamer partitions into the detergent phase after solubilization in Triton X-114 (Bordier, 1981), as we have previously shown (Tsitrin et al, 2002). When similar experiments were performed on the disulfide-containing mutants, both mutant heptamers were found in the detergent phase, indicating the presence of hydrophobic surfaces (Figure 2E, Coomassie blue-stained gel). A previously characterized single point aerolysin mutant, Y221G, was used as a control, as this mutant forms heptamers that are soluble (Tsitrin et al, 2002).
Focusing on the K246C-E258C mutant, we next tested whether the heptamers could be reconstituted into proteoliposomes, as can be performed for the wild-type (WT) channel (Cabiaux et al, 1997). Heptamers were generated in solution and mixed with liposomes and detergent. The detergent was subsequently removed using Bio-beads and the liposomes were separated from free toxin by floatation on density gradients. As a negative control, we again used the Y221G heptamers, which indeed did not associate with the liposomes and remained at the bottom of the gradient—where the sample was initially loaded—in marked contrast to WT heptamers, which were used as a positive control (Figure 2F). A significant fraction of the K246C-E258C heptamers clearly floated in the gradient, demonstrating that they were bound/inserted to liposomes. However, they were found throughout the gradient, indicating that they associated with lipids less efficiently than WT heptamers, and were thus partially released during the separation procedure. Membrane association was significantly increased when the K246C-E258C mutant was reduced by βMeOH before the generation of heptamers.
Channel formation by the double cysteine mutants requires disulfide reduction
As the disulfide-containing mutants were able to bind to their receptors, heptamerize and expose hydrophobic patches enabling them to interact, albeit less efficiently, with lipids, the simplest explanation for their complete lack of hemolytic activity is that they are unable to form channels. We therefore analyzed their behavior using planar lipid bilayers, an extremely sensitive technique that permits the analysis of single-channel events. For all experiments, the toxin was preactivated with trypsin and added to the cis side of the bilayer setup. When approximately 10 channel events were detected, the cis chamber was perfused (with 5–10 times the chamber volume) to remove the toxin remaining in the solution. Whereas addition of WT aerolysin led to a stepwise increase in membrane current (not shown), as previously observed (Wilmsen et al, 1990), addition of either of the disulfide mutants did not, even after 1 h (not shown). Subsequently adding a reducing agent to the cis chamber was insufficient to trigger channel formation. However, when the mutants were preincubated with βMeOH before the addition to the bilayer chamber, then channels readily formed (Figure 3), and had a single-channel conductance similar to that of WT channels (Table I). These observations suggest that the disulfide bond was accessible to reducing agents in the monomer/dimer form but not in the membrane-associated heptameric form, and more importantly that, once the bond was broken, WT-like channels were formed.
Figure 3.
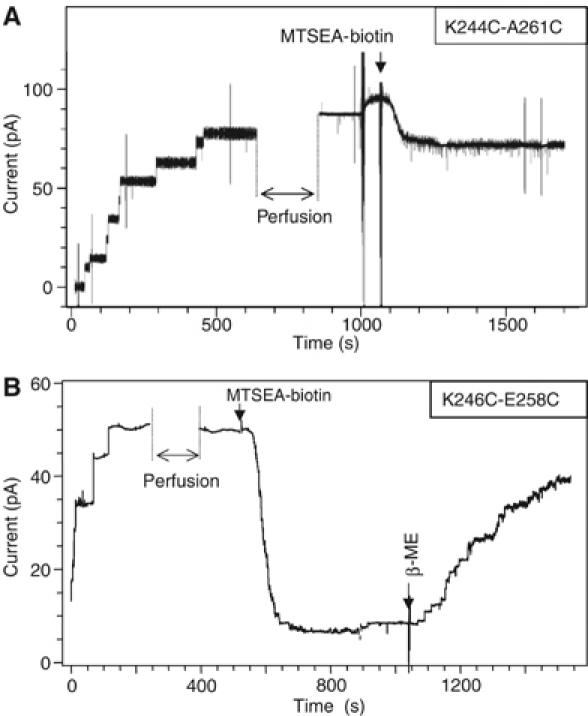
Channel analysis of the disulfide-containing mutants. The K244C-A261C (A) and K246C-E258C (B) mutants were incubated with βMeOH, activated with trypsin and added to the cis chamber of the bilayer setup, after having generated an EggPC:DOPE (1:1) membrane between the two chambers. After 5–10 channels had formed, the cis chamber was perfused as indicated. At the time indicated by an arrow, MTSEA-X-biotin was added to the trans chamber at 200 nM. For both K244C-A261C (A) and K246C-E258C (B), a decrease in current was observed, an effect that could be reversed by the addition of βMeOH as shown for K246C-E258C mutant (B).
We next investigated whether the free sulfhydryl groups resulting from disulfide bridge reduction were accessible to labeling with thiol-reactive probes and whether this labeling would affect the channel properties. Addition of the thiol-specific reagent MTSEA-biotin to the trans chamber led to a small decrease (variable from 30 to 50% from one experiment to another) in current for the K244C-A261C mutant (Figure 3A) and a drastic decrease (93%) for the K246C-E258C mutant (Figure 3B), indicating that for both mutants at least one of the cysteines was accessible to labeling and located in the lumen of the channel. The inhibitory effect of MTSEA-biotin could be reversed by the addition of βMeOH, as illustrated for K246C-E258C (Figure 3B). For both mutants, the transmembrane current was reduced, irrespective of whether the MTSEA-biotin was added to the trans or the cis side of the bilayer. Also, current decrease showed little sensitivity toward the spacer length between MTSEA and the biotin moiety (using MTSEA-biotin, MTSEA-X-biotin or MSTEA-XX-biotin) as well as toward the concentration of the MTS reagent (not shown). The negatively charged MTSES in contrast did not lead to a current decrease, for either of the mutants. Taking into account these observations, all subsequent labeling experiments were performed with MTSEA-biotin and/or MTSEA-X-biotin, at a single MTS concentration.
Cysteine scanning analysis of the channel structure
We next analyzed the ability of all the single cysteine mutants to form channels. As described above for the disulfide-containing mutants, single cysteine mutants were preactivated with trypsin, treated with βMeOH (pretreatment with a reducing agent often increases the reactivity of cysteines) and added to the cis chamber of the bilayer setup. All mutants formed channels with a single conductance similar to that of WT channels (Table I). After about 5–10 channels had formed, the toxin remaining in solution was perfused out of the chamber. MSTEA-(X)i-biotin was then added to the trans chamber and the reduction in current was monitored, as illustrated for the E258C mutant in Figure 4A and for other mutants in Supplementary Figure S1. Changes in current upon the addition of MTS probes (MSTEA-biotin as well as MSTEA-X-biotin) were observed for cysteines at positions 240, 242 and 244 and then 254, 256 and 258 (Table I and Figure 4B). In contrast, no significant changes in current were observed for cysteines at the alternating positions, that is, 241, 243 and 245, and then 253 and 255 (Table I and Figure 4B), consistent with their orientation toward the lipid bilayer, which would render them inaccessible to labeling.
Figure 4.
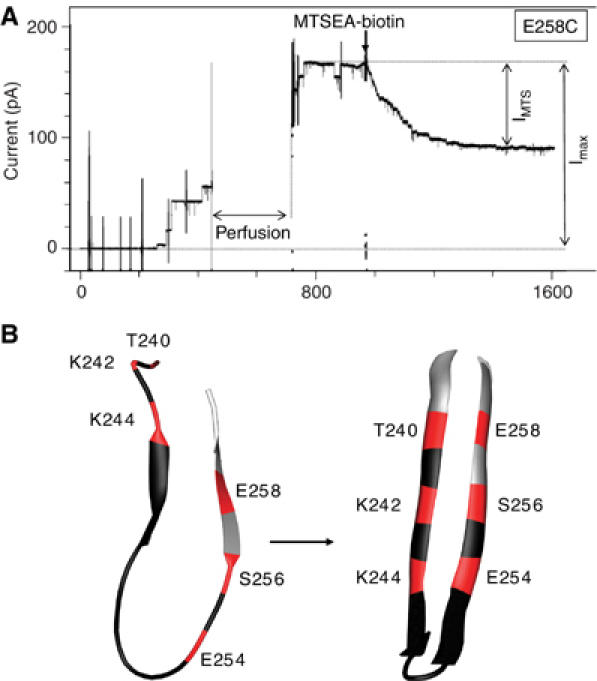
Effect of MTSEA labeling on the single cysteine mutants. (A) The E258C single cysteine mutant was incubated with βMeOH and activated with trypsin and added to the cis chamber of the bilayer setup. After formation of some channels, free toxin was removed by perfusion of the cis chamber, as indicated. At the time indicated by an arrow, MTSEA-X-biotin was added to the trans chamber at 200 nM. (B) Schematic diagram illustrating the positions in the DIII-loop where cysteine residues were sensitive to thiol labeling (red) or insensitive (black). On the left panel, the loop is shown with the conformation it has in the crystal structure of the proaerolysin dimer (Parker et al, 1994). On the right panel, the loop is shown in the β-hairpin configuration as modeled in the current work (see Figure 6A and B).
When cysteine mutants corresponding to the central continuous stretch 246–252 were analyzed, there was no alternating behavior: the conductance of all mutant channels was insensitive to MTSEA-biotin addition (Table I and Figure 4B). The central position of this region combined with the break in the alternating behavior toward MTSEA-biotin labeling suggests that it is not part of the transmembrane β-hairpin but rather forms the loop between the two β-strands (Figure 4B). This hairpin loop is expected to be on the trans side of the membrane and thus accessible to labeling. The lack of effect of MTSEA-biotin on channel conductance could be due to the fact that the biotin molecules when added to the hairpin loops are too small to obstruct the channel. With the aim of increasing the size of the labels, we removed excess MSTEA-(X)i-biotin from the bilayer chambers by perfusion and then added avidin or streptavidin, which are large molecules that bind biotin with extremely high affinity. We however did not observe any effect of avidin/streptavidin for any of the mutants with cysteines at positions 246–252, strongly suggesting that these residues were in fact inaccessible to labeling with MSTEA-(X)i-biotin.
Modification of the WPLVG sequence
The above cysteine scanning mutagenesis experiments identify the WPLVG sequence as the center of the transmembrane hairpin. This sequence, which is absolutely conserved among aerolysins of all Aeromonas species (Gurcel et al, 2006), is very hydrophobic, raising the possibility that it could drive membrane insertion, especially considering that bringing seven sequences together in the heptamer would generate a 35-residue patch.
To address the importance of the hydrophobic WPLVG stretch in membrane insertion, we first generated a WPLVG deletion mutant. This mutant failed to fold, in hindsight not surprisingly considering the numerous contacts this sequence has with the rest of the proaerolysin molecule: Trp-247 is involved in an aromatic ring stacking interaction with Phe-404 and its indole nitrogen is involved in a hydrogen bond with Tyr-304; Pro-248 is involved in van der Waals interactions with Phe-184; finally, Val-250 forms van der Waals contacts with Phe-184, His-186, Val-189 and Tyr-304. The inability of the ΔWPLVG mutant to fold suggests that exposure of the core of domain 3, which is normally covered by the loop (Figure 1A), leads to instability in solution.
We next analyzed the effects of introducing a charged residue in the middle of the WPLVG sequence. Leu-249 and Val-250 were changed to aspartic acid in single point mutants, leading to a 75% loss in hemolytic activity (corresponding to three wells difference in Table I), despite the fact that binding to erythrocytes (Figure 5A) and oligomerization (Figure 5B) were not significantly altered. The mutants were also able to expose hydrophobic patches upon heptamerization, as illustrated by their ability to partition into the detergent phase of Triton X-114 in the oligomeric form (shown for V250D in Figure 5C). Channels could be observed in lipid bilayers, with normal conductances (Table I), albeit only when adding 10-fold higher concentrations of toxin to the bilayer chamber. Altogether, these observations suggest that the introduction of a charged residue (times 7 in the heptamer) impairs crossing the lipid bilayer, that is, the step that separates heptamerization from the final transmembrane channel, but that once formed the channel has a normal conductance.
Figure 5.
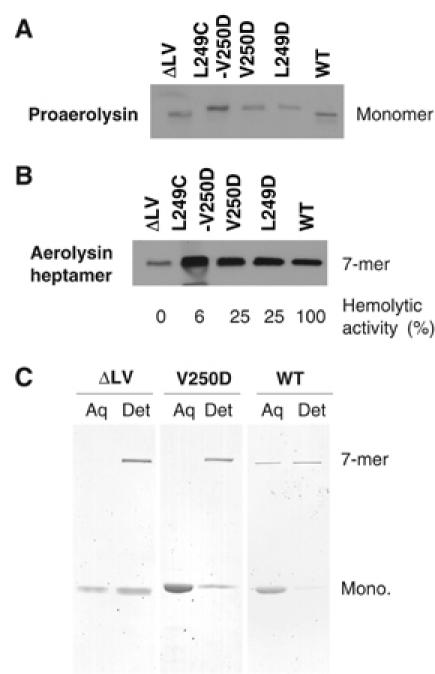
Mutagenesis of the hairpin LV tip does not prevent aerolysin binding and heptamerization. WT or mutant (L249D, V250D, L249C-V250D and DLV) proaerolyins were incubated for 10 min at room temperature with erythrocyte ghosts and analyzed by Western blotting with anti-PA antibodies (A). (B) Following proaerolysin binding, erythrocytes were treated with trypsin for 10 min at room temperature to convert proaerolysin to aerolysin, and then with a 10-fold excess of trypsin inhibitor. Oligomerization was allowed to proceed for 60 min and samples were analyzed by Western blotting against the toxin. (C) WT and mutants were activated with trypsin for 20 min at room temperature followed by addition of trypsin inhibitor, and allowed to oligomerize for 60 min. Samples were submitted to Triton X-114 partitioning. Aqueous (Aq) and detergent (Det) phases were analyzed by SDS–PAGE and Coomassie blue staining.
When both Leu-249 and Val-250 were modified, to cysteine and aspartic acid respectively, channel formation in lipid bilayers was completely abrogated (Table I) and hemolytic activity was reduced by 94%, despite normal binding and oligomerization at the surface of the erythrocytes (Figure 5A and B), altogether again indicating that membrane insertion was impaired.
Deleting these two residues was similarly deleterious because the ΔLV mutant was hemolytic inactive, despite normal binding to erythrocytes (Figure 5A) and the exposure of some hydrophobic surface (Figure 5C). This mutant could only be produced in small amounts, most of it coming out of solution during the production in E. coli as for the full WPLVG deletion. This altered stability might also explain why the ΔLV mutant shows an oligomerization defect (Figure 5B). This can however not account for the loss in hemolytic activity and channel formation if one compares the ΔLV mutant to the K244C-A261C mutant, which also oligomerizes more slowly (Table I and Figure 2).
Modeling of the aerolysin β-barrel
The pattern observed by the cysteine scanning mutagenesis in the aerolysin transmembrane hairpin tip differs from the pattern seen in the case of other PFT, such as anthrax protective antigen where the β-hairpin is fully accessible to labeling (Nassi et al, 2002). As mentioned above, the aerolysin hairpin loop is composed of hydrophobic residues (W247PLVG), the side chains of which are unlikely—based on energetic considerations—to dip into the cytoplasmic milieu once on the trans side of the membrane. They are instead likely to curl back into the lipid bilayer, hence conferring a ‘rivet-like' conformation to the hairpin loop. Such a side-chain configuration would be consistent with the insensitivity to thiol labeling of cysteines at positions 246–252.
We next modeled this transmembrane rivet-like conformation of the DIII-loop. The Staphylococcal α-toxin β-barrel, which is also heptameric (Song et al, 1996), was used as an initial template. Residues V239–F245 of the aerolysin DIII-loop were matched with the Y118–V124 β-strand in α-toxin and residues T253–A260 to the α-toxin G134–S141 β-strand. This was repeated seven times (chains A–G), generating the circular heptameric structure. Next, to model the hairpin loop, we screened a loop database distributed with DeepView (Guex and Peitsch, 1997), searching for a loop with the following characteristics: (i) an overall rivet-like shape, (ii) hydrophobic side chains that all point in the same direction and (iii) steric fit into the heptamer ensemble structure. The L113–C117 loop of chain A of the metalloendopeptidase from Crotalus atrox (PDB code 1ATL; Zhang et al, 1994) satisfied these three requirements and was therefore used as a template for modeling residues W247–G251 of aerolysin. The loop was then anchored at positions K246 and E252 to the antiparallel strands forming the β-barrel and the resulting model was energy minimized. In the resulting model, the hairpin loops fold back, somewhat perpendicular to the barrel axis (Figure 6A). Repeated seven times, the loops assemble into a propeller-like structure (as seen from the top; Figure 6B) leading to a rivet-like anchoring (as seen from the side; Figure 6A). Within each loop, the hydrophobic side chains are oriented toward the core of the bilayer and the two charged side chains, K246 and E252, snorkel toward the polar head groups. When mutated to cysteine, the side chains at positions 246 and 252 might however also orient themselves toward the lipid bilayer, a possibility that would explain why the K246C and E252C channels are insensitive to thiol labeling (Table I and Figure 4B).
Figure 6.

The rivet model of the transmembrane aerolysin β-barrel. Our model of the aerolysin transmembrane β-barrel is shown as a ribbon diagram from the side (A). One of the β-hairpins is highlighted in dark gray and the side chains are shown as balls and sticks. The membrane is shown as a semitransparent gray box. The rivet model as seen from the top (B) with the channel lining residues space-filled. Acidic residues are shown in red, basic residues in blue, polar uncharged residues in yellow and hydrophobic residues in gray. In this rivet model, the tip of each loop folds back, with the hydrophobic residues pointing up toward the core of the bilayer and the flanking charged residues snorkeling down toward the polar head groups. (C) Ribbon diagram of a side view of the Staphylococcal transmembrane β-barrel (Song et al, 1996). One of the β-hairpins is highlighted in dark gray and the side chains are shown as balls and sticks. (D) Space-filled model of the Staphylococcal transmembrane β-barrel (the cap and rim domains were omitted) viewed from the top, that is, the side where the cap domain is located. (E) Ribbon diagram of the β-barrel of the bacterial outer membrane iron transporter FhuA (Ferguson et al, 1998; Locher et al, 1998) (PDB code: 1BY3). The plug domain was omitted for clarity. Side chains for hairpins 9 and 10 are shown as balls and sticks using the same color code as in panel A. The hairpins 9 and 10 of FhuA were extracted from the structure to better illustrate side-chain conformations (F). Similar types of loops were found in osmoporin (Dutzler et al, 1999) (1OSM) (G) and in maltoporin loop (Meyer et al, 1997) (2MPR) (H).
Interestingly, when examining the known structures of β-barrel forming transmembrane proteins, we found that the rivet-like configuration of β-hairpin loops, that is, loop orientation perpendicular to the β-barrel axis, can be found in porins such as maltoporin (Meyer et al, 1997), osmoporin (Dutzler et al, 1999), OmpF (Cowan et al, 1992), sucrose-specific porin (Forst et al, 1998) and phosphoporin (Cowan et al, 1992) and the outer membrane iron transporter FhuA (Ferguson et al, 1998; Locher et al, 1998). This configuration is however not found in all hairpin loops within these structures, but merely in one or two, as illustrated for FhuA (Figure 6E and F), osmoporin (Figure 6G) and maltoporin (Figure 6H).
Discussion
Proaerolysin is an L-shaped molecule with an unusual loop protruding from domain 3 (Figure 1A). We here show that this DIII-loop plays numerous roles in the life of this PFT: (1) it is essential for proper folding of the monomeric/dimeric protoxin, (2) although it is not required for the heptamerization process itself, it drives membrane insertion owing to the presence of a central hydrophobic stretch of five residues W247PLVG, (3) it lines the aerolysin channel, consistent with previous observations on Clostridium α-toxin showing that alternating residues in the corresponding loop interacted with lipids (Melton et al, 2004), and (4) finally we propose that the W247PLVG hydrophobic hairpin tip serves to anchor the aerolysin β-barrel in the membrane in a rivet-like fashion.
The present observations, combined with data available in the literature, suggest the following more detailed sequence of events leading to channel formation. In proaerolysin, the DIII-loop is largely unstructured, containing only two very short antiparallel β-strands (Figure 4B). Upon proteolytic removal of the C-terminal activation peptide, conformational changes are transmitted throughout the protein (Cabiaux et al, 1997), all the way to domain 2, which is implicated in the initiation of the oligomerization process (Buckley et al, 1995). Heptamerization brings in close proximity the DIII-loops, the seven central W247PLVG251 regions of which generate a large 35-residue hydrophobic patch. In vivo, heptamerization occurs in very close proximity of the host plasma membrane, because the toxin, via domain 2 (MacKenzie et al, 1999; Hong et al, 2002), binds to the glycan core of GPI-anchored proteins (Abrami et al, 2000), just above the lipid head groups. The heptameric hydrophobic patch will therefore spontaneously insert into the membrane but clearly be insufficient to cross the lipid bilayer. Indeed, a hairpin formed by five residues is far too short to span a bilayer. Initial membrane insertion therefore still occurs in the two double disulfide-containing mutants but is aborted at that stage. Complete membrane insertion requires elongation of the two short antiparallel β-strands, formed by residues 244–246 and 257–259, and sliding of the strands one with respect to the other (Figure 4B), a motion that would be blocked in the disulfide-containing mutants. Strand elongation and assembly with neighboring strands in the heptamer would lead to the formation of a β-barrel with a hydrophobic exterior, thus providing the required hydrophobicity and length to span the entire bilayer. In this scenario, the hydrophobic tip would not only initiate, and be essential for, membrane insertion, as shown here, but also ensure coupling between barrel folding and membrane insertion, as proposed for the outer membrane bacterial porin OmpA (Tamm et al, 2001; Kleinschmidt and Tamm, 2002). Finally, the exact positioning and thus anchoring of the barrel, with respect to the bilayer, would be fixed by the rivet-like configuration of the seven hairpin loops (Figure 5A and B). Trp-247 probably plays a crucial role, considering the acknowledged role of tryptophanes in fixing the position of membrane proteins with respect to the lipid head group–acyl boundary and the fact that W248C is the only point mutation in the DIII-loop that completely abrogates hemolytic activity (Table I). Finally, the rivet configuration would also ensure that de-insertion never occurs, which might explain at least in part the irreversible nature of aerolysin channel formation in living cells (Abrami et al, 2000).
The rivet model we propose here for the aerolysin β-barrel is significantly different from the Staphylococcal α-toxin β-barrel (Figure 6C and D). Staphylococcal α-toxin forms a perfectly cylindrical structure from one side of the membrane to the other (Figure 6C). Barrel anchoring is however ensured, not by a rivet-type mechanism, but by the presence of three charged residues on the trans side of the membrane. This anchoring device might however be less efficient allowing target cells to repair the lesions (Valeva et al, 2000). The two transmembrane β-barrels also differ in terms of channel lumen. As shown by the present study and illustrated in Figure 6B, the lumen of the aerolysin channel is highly charged (K242, K244, K246, E254, E258 repeated seven times), whereas the β-strands of the α-toxin barrel are free of any charges. In terms of evolution of the transmembrane β-barrels in PFT, it is of interest that charges are observed either in the barrel walls and not in the tip or in the tip but then not in the walls, consistent with the notion that during the translocation process charge shielding is a limiting factor.
A number of studies have proposed alignments between the putative transmembrane regions of Staphylococcal α-toxin, aerolysin and a variety of other pore-forming proteins rich in β-secondary structure (Rossjohn et al, 1998a; Cole et al, 2004; Melton et al, 2004; Mancheno et al, 2005; Sher et al, 2005). These alignments are based not on sequence similarity, but on the more or less rigorous alternating pattern of polar/non-polar residues compatible with a transmembrane β-barrel configuration. These alignments generally proposed that the transmembrane region of aerolysin channel is composed of residues 238–267 and that the tip is formed by residues E252–T253–E254 (Mancheno et al, 2005). Based on our identification of the β-hairpin turn, we now propose a revised alignment (Figure 7). Interestingly, we found that most PFT, in particular C. septicum α-toxin (Gordon et al, 1997), enterolobin (Sousa et al, 1994), Clostridium perfringens epsilon toxin (Cole et al, 2004), the Laetiporus sulphureus hemolytic lectin (Mancheno et al, 2005), and cholesterol-dependent toxins (PFO, SLO, LLO, PPLY, ILY; Tweten et al, 2001) all have a stretch of hydrophobic residues separating the two predicted transmembrane β-strands, as well as charged residues within these strands (Figure 7). Therefore, the Staphylococcal α-toxin β-hairpin seems to be an exception, even among the Staphylococcal PFT, which share extensive sequence similarity. Staphylococcal leukotoxins, such as LukM and LukS-I for example, indeed also have a stretch of hydrophobic residues in the middle of the predicted β-hairpin transmembrane domains. It is therefore tempting to speculate that membrane anchoring of a variety of PFT occurs via a rivet-like configuration, as proposed here for aerolysin. Also the importance of the hydrophobic tip in driving membrane insertion of aerolysin and the absence of such a tip in Staphylococcal α-toxin suggest that, once unraveled, the mechanisms by which these β-barrels form, penetrate and cross the lipid bilayer might show interesting differences.
Figure 7.

Alignments of putative transmembrane domains of different β-barrel PFT. The DIII-loop of aerolysin was aligned manually with the corresponding regions in C. perfringens epsilon-toxin (a toxin with a similar three-dimensional structure to aerolysin) (Cole et al, 2004), Clostridial α-toxin (Clost. α-toxin) and enterolobin, three Staphylococcal PFT (α-toxin, LukM, LukS-I) and the protective antigen (PA) of anthrax (Nassi et al, 2002) as well as the two transmembrane domains (TM1 and TM2) of five cholesterol-dependent cytolysins (PFO: perfringolysin; SLO: streptolysin; LLO: listeriolysin; PLY: pneumolysin; ILY: intermedilysin; Tweten et al, 2001). Alignments were generated based on the alternating pattern of polar and hydrophobic residues. Hydrophobic residues are shown in gray, charged residues in red and other residues are left uncolored.
Materials and methods
Reagents
The thiol-specific reagents MSTEA-biotin (2-((biotinoyl)amino)ethyl methanethiosulfonate), MTSEA-X-biotin (2-((6-((biotinoyl)amino)hexanoyl)amino) ethyl methanethiosulfonate) and MTSEA-XX-biotin (2-((6-((6-((biotinoyl)amino) hexanoyl)amino)hexanoyl)amino)ethyl methanethiosulfonate) were purchased from Sigma (St Louis), Triton X-100 Ultra Pure was from Pierce Chemical Co. (Rockford, IL).
Mutagenesis and protein purification
The AerA gene was amplified by PCR using oligonucleotides, leading to the introduction of NcoI (in 5′) and XhoI (in 3′) restriction sites (5′-ACATGCCATGGCAGAGCCCGTCTAT-3′ and 5′-AAGCTCGAGTTGATTGGCAGCAGG-3′), and introduced into the pET22b vector (Novagen), leading to the expression of C-terminally His-tagged toxin in the bacterial periplasm. Site-directed mutagenesis was performed using the QuickChange® Site-directed Mutagenesis kit (Stratagene) according to the supplier's instructions. WT and mutant toxins were expressed in E. coli (BL21DE3 pLysS, Promega) and purified using Nicklec columns (Tsitrin et al, 2002).
Assays
Hemolytic activity was measured as previously described (Buckley et al, 1995). The number of lysed wells was monitored after 60 min. Cell surface binding and oligomerization, and binding to GPI-anchored proteins by toxin overlay were measured as described (Abrami et al, 1998). Briefly, WT or mutant proaerolysin was activated with trypsin for 10 min at room temperature and then added to BHK cells at a concentration of 20 ng/ml for 1 h at 4°C followed by 10 min at 37°C to allow oligomerization. Cells were then washed, lysed using a syringe and centrifuged to remove the nuclei. The remaining cell extracts were analyzed by SDS–PAGE using conventional Laemmli sample buffer, blotted onto nitrocellulose membranes and analyzed by Western blotting using anti-aerolysin polyclonal antibodies.
In vitro oligomerization was evaluated by activating WT or mutant proaerolysin (200 μg/ml), in 150 mM NaCl and 20 mM HEPES (pH 7.4), with 10 μg/ml trypsin for 10 min at room temperature followed by 1 h at 4°C.
Erythrocyte ghosts were prepared by three rounds of osmotic shock by performing a five-fold dilution in 10 mM Tris (pH 8) followed by membrane pelleting by high-speed centrifugation.
Triton X-114 phase separation
Triton X-114 partitioning experiments were adapted from Bordier (1981) as described (Tsitrin et al, 2002). Briefly, proaerolysin, WT or mutant, in 150 mM NaCl and 20 mM HEPES (pH 7.4) was activated with trypsin for 20 min at room temperature. A 10-fold excess trypsin inhibitor was added and oligomerization was allowed to proceed for 1 h. The sample was then dialyzed against 20 mM HEPES (pH 7.4), to further promote oligomerization at low ionic strength. Triton X-114 (Merck, Germany), purified as described (Bordier, 1981), was then added to reach 1%. After 20 min incubation at 4°C (to obtain a single phase), the sample was shifted to 37°C for 5 min, to induce phase separation, and submitted to centrifugation for 3 min at 13 000 r.p.m. The aqueous phase was recovered. The detergent phase was resuspended to reach 1% Triton X-114 and resubmitted to a round of phase separation. Aqueous and detergent phases were analyzed by SDS–PAGE followed by Coomassie blue staining.
Formation of proteoliposomes
These experiments were performed as described (Cabiaux et al, 1997). Briefly, proaerolysin was activated in vitro and allowed to heptamerize. The toxin (0.4 mg/ml) in 150 mM NaCl and 20 mM HEPES (pH 7.4) was mixed at a protein/lipid ratio of 1/7 (w:w) with the following lipid composition: 50% PC (egg lecithin)/50% DOPE (w:w) mix in 1% (v/v) octyl-POE. After 30 min, the detergent was removed using Bio-Beads (Bio-Rad) and sucrose was added to the sample to reach 43% (w/w). A discontinuous flotation gradient was prepared with successive layers of 35 and 8% sucrose in 150 mM NaCl and 20 mM HEPES (pH 7.4) and centrifuged at 200 000 g for 2 h at 16°C. Six fractions of 400 μl were then collected and their protein content precipitated before loading on a 10% acrylamide SDS gel.
Planar lipid bilayer
Planar lipid bilayer experiments were performed as described previously (Wilmsen et al, 1990). The bilayer was formed by painting a solution of 50% PC (egg lecithin)/50% DOPE (w:w) in n-decane (40 mg/ml) on an aperture (d=150 μm, pretreated with the same solution) in a delrin cuvette separating two chambers, each containing 1 ml of 1 M NaCl, 5 mM CaCl2 and 10 mM HEPES (pH 7) and agar bridge connection (1 M KCl) to Ag/AgCl electrodes (Warner Instrument Corp., Hamden, CT). Currents were recorded at either ±25 mV in a Faraday cage, amplified (BC-525c, Warner Instrument Corp.), low-pass filtered with an eight-pole Bessel filter at 4 kHz (LPF-8, Warner Instrument Corp.), A-D converted (DigiData 1200, Axon Instruments, Union City, CA) and sampled at 10 kHz by computer (pClamp 8.0, Axon Instruments). Samples (1–5 μg of pre-oligomerized toxin) were added to the cis chamber and the toxin was removed by perfusion with 5–10 chamber volumes of the same buffer after channel formation (5–20 channels). All the conductance measurements were performed at room temperature. Cysteine-specific reagents were prepared at 20 mM stock solutions and added to either the cis or the trans side at concentrations varying between 100 and 200 nM.
Modeling the barrel structure
A homology model of the aerolysin β-barrel was generated using the aerolysin sequence as a target (accession no. P09167) and the S. aureus α-toxin β-barrel (PDB code: 7AHL; Song et al, 1996) as a template. Target to template alignment was carried out in DeepView (Guex and Peitsch, 1997). The loop was next modeled using the loop database distributed with DeepView. The resulting model was energy minimized during 100 steps of Steepest Descent simulations using the GROMOS96 force field (van Gunsteren et al, 1996) as implemented in DeepView (Guex and Peitsch, 1997).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
We thank Franc Pattus (Ecole Supérieure de Biotechnologie de Strasbourg, UPR9050CNRS) for helping us with the preliminary bilayer experiments, Christine Lucain for generating mutant toxins and Sarah Thurnheer and Marie-Claire Velluz for technical assistance. This work was supported by grants from the Swiss National Science Foundation, the Société Académique de Genève, the Fondation Boninchi and the EMBO Young Investigator Program to GvdG and the National Health and Medical Research Council of Australia to MWP. Finally, PP benefited from a fellowship from the Roche Research foundation and then from FEBS.
References
- Abrami L, Fivaz M, Glauser P-E, Parton RG, van der Goot FG (1998) A pore-forming toxin interact with a GPI-anchored protein and causes vacuolation of the endoplasmic reticulum. J Cell Biol 140: 525–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Fivaz M, van Der Goot FG (2000) Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol 8: 168–172 [DOI] [PubMed] [Google Scholar]
- Abrami L, van der Goot FG (1999) Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J Cell Biol 147: 175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard J, Crabtree J, Roe BA, Tweten RK (1995) The primary structure of Clostridium septicum alpha-toxin exhibits similarity with that of Aeromonas hydrophila aerolysin. Infect Immun 63: 340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordier C (1981) Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem 256: 1604–1607 [PubMed] [Google Scholar]
- Buckley JT, Wilmsen HU, Lesieur C, Schultze A, Pattus F, Parker MW, van der Goot FG (1995) Protonation of His-132 promotes oligomerization of the channel-forming toxin Aerolysin. Biochemistry 34: 16450–16455 [DOI] [PubMed] [Google Scholar]
- Cabiaux V, Buckley JT, Wattiez R, Ruysschaert J-M, Parker MW, van der Goot FG (1997) Conformational changes in aerolysin during the transition from the water-soluble protoxin to the membrane channel. Biochemistry 36: 15224–15232 [DOI] [PubMed] [Google Scholar]
- Cole AR, Gibert M, Popoff M, Moss DS, Titball RW, Basak AK (2004) Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat Struct Mol Biol 11: 797–798 [DOI] [PubMed] [Google Scholar]
- Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP (1992) Crystal structures explain functional properties of two E. coli porins. Nature 358: 727–733 [DOI] [PubMed] [Google Scholar]
- Dutzler R, Rummel G, Alberti S, Hernandez-Alles S, Phale P, Rosenbusch J, Benedi V, Schirmer T (1999) Crystal structure and functional characterization of OmpK36, the osmoporin of Klebsiella pneumoniae. Struct Fold Des 7: 425–434 [DOI] [PubMed] [Google Scholar]
- Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W (1998) Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282: 2215–2220 [DOI] [PubMed] [Google Scholar]
- Fivaz M, Vilbois F, Thurnheer S, Pasquali C, Abrami L, Bickel P, Parton R, van der Goot F (2002) Differential sorting and fate of endocytosed GPI-anchored proteins. EMBO J 21: 3989–4000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forst D, Welte W, Wacker T, Diederichs K (1998) Structure of the sucrose-specific porin ScrY from Salmonella typhimurium and its complex with sucrose. Nat Struct Biol 5: 37–46 [DOI] [PubMed] [Google Scholar]
- Gordon VM, Benz R, Fujii K, Leppla SH, Tweten RK (1997) Clostridium septicum alpha-toxin is proteolytically activated by furin. Infect Immun 65: 4130–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18: 2714–2723 [DOI] [PubMed] [Google Scholar]
- Gurcel L, Iacovache I, van der Goot FG (2006) Aerolysin and related Aeromonas toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins, Alouf JE, Freer JH (eds) London: Academic Press (in press) [Google Scholar]
- Hong Y, Ohishi K, Inoue N, Kang JY, Shime H, Horiguchi Y, van der Goot FG, Sugimoto N, Kinoshita T (2002) Requirement of N-glycan on GPI-anchored proteins for efficient binding of aerolysin but not Clostridium septicum a-toxin. EMBO J 21: 5047–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt JH, Tamm LK (2002) Secondary and tertiary structure formation of the beta-barrel membrane protein OmpA is synchronized and depends on membrane thickness. J Mol Biol 324: 319–330 [DOI] [PubMed] [Google Scholar]
- Locher KP, Rees B, Koebnik R, Mitschler A, Moulinier L, Rosenbusch JP, Moras D (1998) Transmembrane signaling across the ligand-gated FhuA receptor: crystal structures of free and ferrichrome-bound states reveal allosteric changes. Cell 95: 771–778 [DOI] [PubMed] [Google Scholar]
- MacKenzie CR, Hirama T, Buckley JT (1999) Analysis of receptor binding by the channel-forming toxin aerolysin using surface plasmon resonance. J Biol Chem 274: 22604–22609 [DOI] [PubMed] [Google Scholar]
- Mancheno JM, Tateno H, Goldstein IJ, Martinez-Ripoll M, Hermoso JA (2005) Structural analysis of the Laetiporus sulphureus hemolytic pore-forming lectin in complex with sugars. J Biol Chem 280: 17251–17259 [DOI] [PubMed] [Google Scholar]
- Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK (2004) The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J Biol Chem 279: 14315–14322 [DOI] [PubMed] [Google Scholar]
- Menestrina G, Dalla Serra M, Comai M, Coraiola M, Viero G, Werner S, Colin DA, Monteil H, Prevost G (2003) Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett 552: 54–60 [DOI] [PubMed] [Google Scholar]
- Meyer JE, Hofnung M, Schulz GE (1997) Structure of maltoporin from Salmonella typhimurium ligated with a nitrophenyl-maltotrioside. J Mol Biol 266: 761–775 [DOI] [PubMed] [Google Scholar]
- Nassi S, Collier RJ, Finkelstein A (2002) PA(63) channel of anthrax toxin: an extended -barrel. Biochemistry 41: 1445–1450 [DOI] [PubMed] [Google Scholar]
- Parker MW, Buckley JT, Postma JPM, Tucker AD, Leonard K, Pattus F, Tsernoglou D (1994) Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 367: 292–295 [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Feil SC, Mckinstry WJ, van der Goot FG, Buckley JT, Parker MW (1998a) Aerolysin—a paradigm for membrane insertion of beta-sheet protein toxins. J Struct Biol 121: 92–100 [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Raja SM, Nelson KL, Feil SC, van der Goot FG, Parker MW, Buckley JT (1998b) Movement of a loop in domain 3 of aerolysin is required for channel formation. Biochemistry 37: 741–746 [DOI] [PubMed] [Google Scholar]
- Schulz GE (2002) The structure of bacterial outer membrane proteins. Biochim Biophys Acta 1565: 308–317 [DOI] [PubMed] [Google Scholar]
- Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK (1999) The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99: 293–299 [DOI] [PubMed] [Google Scholar]
- Sher DJ, Fishman Y, Zhang M, Lebendiker M, Gaathon A, Mancheno JM, Zlotkin E (2005) Hydralysins: a new category of beta-pore-forming toxins in cnidaria. Characterization and preliminary structure–function analysis. J Biol Chem 280: 22847–22855 [DOI] [PubMed] [Google Scholar]
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of Staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 274: 1859–1866 [DOI] [PubMed] [Google Scholar]
- Sousa MV, Richardson M, Fontes W, Morhy L (1994) Homology between the seed cytolysin enterolobin and bacterial aerolysins. J Protein Chem 13: 659–667 [DOI] [PubMed] [Google Scholar]
- Tamm LK, Arora A, Kleinschmidt JH (2001) Structure and assembly of beta-barrel membrane proteins. J Biol Chem 276: 32399–32402 [DOI] [PubMed] [Google Scholar]
- Tsitrin Y, Morton CJ, El Bez C, Paumard P, Velluz MC, Adrian M, Dubochet J, Parker MW, Lanzavecchia S, Van Der Goot FG (2002) Conversion of a transmembrane to a water-soluble protein complex by a single point mutation. Nat Struct Biol 9: 729–733 [DOI] [PubMed] [Google Scholar]
- Tweten RK, Parker MW, Johnson AE (2001) The cholesterol-dependent cytolysins. Curr Top Microbiol Immunol 257: 15–33 [DOI] [PubMed] [Google Scholar]
- Valeva A, Walev I, Gerber A, Klein J, Palmer M, Bhakdi S (2000) Staphylococcal alpha-toxin: repair of a calcium-impermeable pore in the target cell membrane. Mol Microbiol 36: 467–476 [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, Scott WRP, Tironi IG (1996) Biomolecular Simulation: The GROMOS 96 Manual and User GuideBiomolecular Simulation: The GROMOS 96 Manual and User Guide. ETH Zürich, Zürich: Hochschulverlag AG [Google Scholar]
- Wilmsen HU, Pattus F, Buckley JT (1990) Aerolysin, a hemolysin from Aeromonas hydrophila, forms voltage-gated channels in planar bilayers. J Membr Biol 115: 71–81 [DOI] [PubMed] [Google Scholar]
- Zhang D, Botos I, Gomis-Ruth FX, Doll R, Blood C, Njoroge FG, Fox JW, Bode W, Meyer EF (1994) Structural interaction of natural and synthetic inhibitors with the venom metalloproteinase, atrolysin C (form d). Proc Natl Acad Sci USA 91: 8447–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2