

Abstract
Delay of cell cycle progression in response to double-strand DNA breaks (DSBs) is critical to allow time for DNA repair and prevent cellular transformation. Here, we show that the p38 mitogen-activated protein (MAP) kinase signaling pathway is activated in immature thymocytes along with TcRβ gene V(D)J recombination. Active p38 MAP kinase promotes a G2/M cell cycle checkpoint through the phosphorylation and activation of p53 in these cells in vivo. Inactivation of p38 MAP kinase and p53 is required for DN3 thymocytes to exit the G2/M checkpoint, progress through mitosis and further differentiate. We propose that p38 MAP kinase is activated by V(D)J-mediated DSBs and induces a p53-mediated G2/M checkpoint to allow DNA repair and prevent cellular transformation.
Keywords: G2/M checkpoint, p38 MAP kinase, p53, thymocyte development, V(D)J recombination
Introduction
During early T-cell development, bone marrow-derived T-cell progenitors enter the thymus and differentiate into immature CD4−CD8− double-negative (DN) thymocytes. DN thymocytes upregulate the expression of CD8, followed by a gradual expression of CD4 (CD8+CD4low) to become CD8+CD4+ double-positive (DP) thymocytes. DN thymocytes can be divided into four distinct populations that represent progressive stages of differentiation based on CD25 and CD44 expression. CD25− CD44+ (DN1) thymocytes represent the most immature stage. These cells upregulate CD25 to become CD25+ CD44+ cells (DN2) and then downregulate CD44 to differentiate into CD25+ CD44− (DN3) thymocytes (reviewed in Rodewald and Fehling, 1998). DN3 thymocytes undergo V(D)J recombination, a process where the V, D and J segments of the germ line β chain gene of the T-cell receptor (TcR) are rearranged to generate a functional TcRβ chain. This process is mediated by the RAG-1 and RAG-2 recombinases and involves the generation of DNA double-strand breaks (DSBs) between TcR encoding gene segments and flanking recombination signal sequences (RSS) (reviewed in Bassing et al, 2002). Repair of these DSBs by nonhomologous end joining is required for further differentiation of these cells (Blunt et al, 1995). Expression of a functional TcRβ chain and termination of V(D)J recombination is necessary for the differentiation of CD25+ CD44− (DN3) into CD25− CD44− (DN4) thymocytes.
DSBs induced by γ-radiation and DNA repair-intermediates induced by ultraviolet (UV) irradiation or chemical damage trigger a p53-mediated signaling pathway that halts the cell cycle before the onset of mitosis (G2/M checkpoint) to allow DNA repair (Levine, 1997). Once the DNA is successfully repaired, cells progress through M phase and complete the cell cycle. Delay of the cell cycle progression after DNA damage is critical to allow DNA repair and prevent cellular transformation. Although V(D)J recombination also involves the generation of DSBs, the upstream signaling pathways involved in the initiation of a potential cell cycle checkpoint and DNA repair in DN3 thymocytes are unknown.
The p38 mitogen-activated protein (MAP) kinase pathway has a significant role in mediating intracellular signaling triggered by growth factors, cytokines and environmental stress (UV, γ-radiation) (Freshney et al, 1994; Han et al, 1994; Wang et al, 2000). p38 MAP kinase is also involved in the regulation of the cell cycle in response to DNA damage in vitro (Bulavin et al, 2001). We have shown that p38 MAP kinase is highly active in DN3 but not in DN4 thymocytes and that persistent activation of p38 MAP kinase leads to thymic development arrest at the DN3 stage. Inactivation of p38 MAP kinase restores the differentiation of these cells into DN4 and DP thymocytes (Diehl et al, 2000). However, the stimuli that regulate p38 MAP kinase and the role of this pathway in DN3 thymocytes is unclear.
Here, we provide evidence indicating that the activation of the p38 MAP kinase pathway at the DN3 stage depends on V(D)J recombination events. Activation of p38 MAP kinase leads to the phosphorylation and accumulation of p53 resulting in the induction of a G2/M cell cycle checkpoint. Inactivation of p38 MAP kinase and exit from this checkpoint is required for further thymocyte differentiation. We propose that p38 MAP kinase is activated during V(D)J recombination to allow DNA repair and maintain genomic stability.
Results
In vivo activation of the p38 MAP kinase induces p53 phosphorylation and accumulation in immature thymocytes
We have previously shown that persistent activation of p38 MAP kinase in transgenic (Tg) mice expressing a constitutive active MKK6 mutant (upstream activator of p38 MAP kinase) blocks thymocyte development at the DN3 stage (Diehl et al, 2000) similar to Rag1 or Rag2 deficiencies (Mombaerts et al, 1992). Rag1−/− thymocytes are small and arrested in G0/G1 phase of cell cycle. In contrast, DN3 thymocytes from MKK6(Glu) Tg mice are large and display a phenotype characteristic of early promitotic cells (Figure 1A). Despite their phenotype, MKK6(Glu) thymocytes do not proliferate in vivo but the presence of a p38 MAP kinase inhibitor (SB203580) restores proliferation in vitro (Diehl et al, 2000), indicating that inactivation of p38 MAP kinase must occur for DN3 thymocytes to progress through cell cycle.
Figure 1.

p38 MAP kinase activation induces p53 accumulation. (A) Freshly isolated MKK6(Glu), Rag1−/− thymocytes, and a mix of both cell types (1:1) were spun, fixed, stained with Giemsa and examined under light microscopy (× 60). (B) p53 levels were determined in whole-cell extracts prepared from freshly isolated thymocytes from wild type (WT) or MKK6(Glu) transgenic (Tg) mice by Western blot. Thymocytes from WT or MKK6(Glu) (Tg) mice were cultured for 4 h in medium alone (−) or medium containing the p38 MAP kinase inhibitor SB203580 (SB) (5 μM). Whole-cell extracts were prepared and total p53 protein (C) of phosphorylated p53 (p-p53) on Ser18 or Ser389 and actin (D) levels were determined by Western blot. Data are representative of three to four independent experiments.
It has been shown that activation of p38 MAP kinase arrests cell cycle at the G2/M checkpoint by phosphorylation and stabilization of p53 in response to DNA damage in UV-irradiated fibroblasts (Bulavin et al, 1999; She et al, 2000). To test whether activation of p38 MAP kinase in thymocytes leads to the accumulation of p53 in vivo, we examined p53 levels in thymocytes from MKK6(Glu) Tg mice by Western blot. High levels of p53 were present in MKK6(Glu) thymocytes (Figure 1B). The accumulation of p53 in MKK6(Glu) thymocytes required p38 MAP kinase activity, as treatment of these cells in vitro with the selective p38 MAP kinase inhibitor SB203580 caused a rapid reduction of the p53 protein levels (Figure 1C). In addition, p53 was phosphorylated at Ser18 and Ser389 (human Ser15 and Ser392, respectively) in the MKK6(Glu) thymocytes, and the levels of phosphorylated p53 were diminished in the presence of the p38 MAP kinase inhibitor (Figure 1D). ATM and Chk1 can also phosphorylate and activate p53 in response to DNA damage (reviewed by Caspari, 2000) but no activation of ATM and Chk1 was detected in MKK6(Glu) thymocytes (Supplementary Figure S1). In correlation, no significant phosphorylation of p53 at Ser23 (human Ser20), a Chk1 target residue (Shieh et al, 1999), was observed in these thymocytes (Supplementary Figure S1). Thus, activation of p38 MAP kinase in vivo induces phosphorylation and accumulation of p53 in DN3 thymocytes.
p38 MAP kinase activation promotes a G2/M cell cycle checkpoint in DN3 thymocytes
Cell division relies on the expression of cyclins that bind and activate cyclin-dependent kinases to promote cell cycle progression towards S phase and later to initiate mitosis. Association of cyclin B with the cyclin-dependent kinase Cdc2 induces Cdc2 kinase activity to promote progression through mitosis (Morgan, 1995). However, during a G2/M cell cycle checkpoint, cyclin B associates with inactive Cdc2 and these complexes accumulate in the cell, delaying the progression through mitosis (Poon et al, 1996). To investigate whether activation of p53 by p38 MAP kinase leads to a G2/M cell cycle checkpoint in thymocytes from MKK6(Glu) Tg mice, we examined cyclin B protein levels by Western blot. Despite the high rate of proliferation of DP thymocytes, very low levels of cyclin B were present in total wild type (WT) thymocytes, but higher levels of cyclin B were present in MKK6(Glu) thymocytes (Figure 2A). In addition, the levels of cyclin B in MKK6(Glu) thymocytes were higher than those in WT DN3 thymocytes (Figure 2B). Thus, activation of p38 MAP kinase leads to the accumulation of cyclin B. To determine whether cyclin B was associated with inactive Cdc2 in these cells, we examined phosphorylation of Cdc2 at Tyr15 as phosphorylation of this residue is responsible for inactivation of Cdc2 at the G2/M cell cycle checkpoint (Booher et al, 1997). Cyclin B was immunoprecipitated from whole-cell extracts and the presence of phosphorylated Cdc2 in cyclin B immunoprecipitates was determined by Western blot. Cyclin B from MKK6(Glu) thymocytes was associated with Cdc2 kinase phosphorylated at Tyr15 (Figure 2C), indicating that Cdc2 kinase is inactive in these thymocytes. Moreover, phosphorylation of cyclin B-associated Cdc2 kinase was reduced when MKK6(Glu) cells were treated with SB203580 (Figure 2C). To show that phosphorylation of Cdc2 at Tyr15 blocks its kinase activity, cyclin B was immunoprecipitated from freshly isolated MKK6(Glu) thymocytes or from MKK6(Glu) thymocytes cultured for the indicated periods of time in the presence of SB203580 and Cdc2 kinase activity was determined in vitro using histone H1 as substrate. No Cdc2 kinase activity was present in cyclin B precipitates from freshly isolated MKK6(Glu) thymocytes (Figure 2D), but SB203580 restored cyclin B-associated Cdc2 kinase activity (Figure 2D). Analysis of phospho-Cdc2 showed that the presence of a few dephosphorylated (i.e. activated) Cdc2 molecules (phospho-Cdc2/total Cdc2 ratio) was sufficient to mediate phosphorylation of a substantial number of H1 molecules (Figure 2D). These results indicate that p38 MAP kinase promotes the accumulation of inactive cyclin B–Cdc2 complexes to induce a G2/M cell cycle checkpoint.
Figure 2.

p38 MAP kinase activation induces a G2/M checkpoint in DN3 thymocytes. The levels of cyclin B and actin in total thymocytes from WT or MKK6(Glu) transgenic (Tg) mice (A) or in cell sorted WT DN3 and total MKK6(Glu) thymocytes (B) were detected by Western blot. (C) Whole-cell extracts from WT or MKK6(Glu) thymocytes cultured for 4 h in absence (−) or presence of 5 μM SB203580 (SB) were immunoprecipitated with anti-cyclin B antibody or isotype control antibody (Control), and phosphorylation of Cdc2 on Tyr15 (p-Cdc2), total Cdc2 and cyclin B levels was examined by Western blot. The heavy chain from the antibodies used for immunoprecipitation is shown (IgG). (D) Thymocytes from the MKK6(Glu) mice were cultured in the presence of SB203580 (SB) (5 μM) for the indicated periods of time. Cyclin B was immunoprecipitated and the cyclin B-associated Cdc2 kinase activity present in the immunoprecipitates was determined by an in vitro kinase assay using H1 as an exogenous substrate. Phosphorylated H1 is shown. The levels of phospho-Cdc2 (p-Cdc2) and total Cdc2 in the cyclin B immunoprecipitates were determined by Western blot. Densitometry analysis of p-Cdc2/total Cdc2 is shown. (E) Whole extracts from WT or MKK6(Glu) thymocytes (Tg) were examined for 21, p27 and actin by Western blot. (F) MKK6(Glu) thymocytes were cultured in the presence of SB203580 (5 μM) for the indicated periods of time. The levels of phospho-histone H3 (p-H3) and actin were determined by Western blot. (G) WT and MKK6(Glu) thymocytes were culture in the absence (−) or presence (SB) of SB203580 (5 μM) for 4 h. CENP-A and actin levels were determined by Western blot. (H) WT and MKK6(Glu) thymocytes were cytospun, fixed, permeabilized, stained for CENP-A (red) and examined by confocal microscopy. TOPRO (blue) was used for nuclear staining. (I) MKK6(Glu) thymocytes were cultured in the absence (−) or presence of SB203580 (5 μM) for 4 h. Cells were then cytospun, stained with Giemsa and examined under light microscopy (× 60). Cells in anaphase (A) and metaphase (M) are marked. The relative percentages of metaphase/anaphase/telophase/cytokinesis (M/A/T/C) MKK6(Glu) thymocytes in the absence or presence (SB) of SB203580 were determined in (J). Error bars represent standard errors.
The establishment of a G2/M cell cycle checkpoint by p53 is mediated by inducing transcription of genes encoding proteins such as GADD45, p21 and 14.3.3σ that directly or indirectly inactivate Cdc2 kinase (el-Deiry et al, 1993; Hermeking et al, 1997; Zhan et al, 1999). To test whether the induction of the G2/M checkpoint in the MKK6(Glu) thymocytes involves any of the p53-induced cell cycle regulators, we examined GADD45, p21 and 14.3.3σ protein levels by Western blot. The amount of 14.3.3σ protein was below detection levels in both WT and MKK6(Glu) thymocytes (data not shown). Although very low levels of GADD45 were detected, no difference was observed between WT and MKK6(Glu) thymocytes (data not shown). p21 expression, however, was increased in MKK6(Glu) thymocytes compared with WT thymocytes (Figure 2E). In contrast, the levels of the cell cycle inhibitor p27 that accumulates at the G1/S transition (Toyoshima and Hunter, 1994) were very low in the MKK6(Glu) thymocytes (Figure 2E). This suggests that p53 induces a G2/M checkpoint in the MKK6(Glu) thymocytes by upregulating p21 expression.
Phosphorylation of histone H3 on Ser10 allows chromatin condensation in proliferating cells undergoing mitosis (Mahadevan et al, 1991). To confirm that the MKK6(Glu) thymocytes were arrested at the G2/M checkpoint instead of being proliferating cells in mitosis, we determined the levels of phosphorylated histone H3 (p-H3). No p-H3 was detected in freshly isolated MKK6(Glu) thymocytes (Figure 2F). However, treatment of these cells with the p38 MAP kinase inhibitor SB203580 in vitro induced histone H3 phosphorylation (Figure 2F), indicating that p38 MAP kinase inhibition promotes exit out of the G2/M checkpoint and progression through mitosis. Another parameter that allows distinction between cells in G2/M and cells in the interphase is the presence of increased levels of centromeric proteins. CENP-A is a variant of histone H3 and replaces this protein in the nucleosome core of centromeric chromatin at the inner plate of the kinetochore (Howman et al, 2000). Increased levels of CENP-A were observed in MKK6(Glu) thymocytes relative to WT thymocytes by Western blot (Figure 2G) and by immunofluorescence and confocal microscopy (Figure 2H) analyses. The levels of CENP-A in MKK6(Glu) thymocytes diminished when these cells were treated with the p38 MAP kinase inhibitor in vitro (Figure 2G). Accordingly, we could detect a significant number of cells that have progressed from early prophase to metaphase, anaphase and other phases of the mitosis (telophase and cytokinesis) in SB203580-treated MKK6(Glu) thymocytes (Figure 2I and J). Collectively, these results show that activation of p38 MAP kinase in vivo induces a G2/M cell cycle checkpoint at the DN3 stage and that inactivation of p38 MAP kinase is required to exit this cell cycle checkpoint for further differentiation of these cells.
p53 is required to sustain the G2/M cell cycle checkpoint induced by p38 MAP kinase activation
To determine whether p53 is required to sustain the G2/M checkpoint induced by p38 MAP kinase, p53−/− males were crossed with p53+/−MKK6(Glu) females, but no p53−/−MKK6(Glu) mice were identified in over more than a hundred screened littermates, despite the normal litter size. Thus, we examined the effect of p38 MAP kinase activation in p53 haplo-insufficient mice (p53+/−). In contrast to p53−/− mice, p53+/− mice do not develop tumors, they contain about half of p53 protein levels and have been extensively used (Gottlieb et al, 1997; Pani et al, 2002). We therefore examined whether the reduction of p53 levels (Figure 3A) in thymocytes from p53+/−MKK6(Glu) mice could restore cell cycle progression of these cells. Flow cytometry analysis of cell forward scatter showed that a large proportion of thymocytes from the p53+/−MKK6(Glu) mice were smaller than thymocytes from p53+/+MKK6(Glu) mice (Figure 3B), indicating that some cells were able to exit the promitotic stage. Accordingly, decreased levels of p21 were observed in p53+/−MKK6(Glu) thymocytes compared to those observed in p53+/+MKK6(Glu) thymocytes (Figure 3C).
Figure 3.

Induction of G2/M checkpoint by p38 MAP kinase is mediated by p53. (A) Whole-cell extracts were prepared from freshly isolated thymocytes from p53+/+-WT, p53+/−-WT, p53+/+-MKK6(Glu) transgenic (Tg) or p53+/−-MKK6(Glu) Tg mice and levels of p53 and actin were determined by Western blot. (B) Thymocytes were obtained from p53+/+-MKK6(Glu) (p53+/+ Tg) and p53+/−-MKK6(Glu) (p53+/− Tg) mice and cell size was determined by examining forward light scatter by flow cytometry. (C) Whole-cell extracts were prepared from the mice described in (A) and p21 and actin levels were determined by Western blot. (D) Thymocytes from the indicated mice were stained for CD4, CD8, CD25 and CD44. Numbers indicate the relative percentages of the specific populations in total thymocytes (central panels) or in the gated populations (right and left panels).
As we have previously described (Diehl et al, 2000), most thymocytes from adult p53+/+ MKK6(Glu) mice display a CD8+CD4lowCD25+CD44− phenotype characteristic of fetal immature thymocytes and only a few truly CD4+CD8+ DP thymocytes were present in these mice (Figure 3D and Table I). Similarly, mature single CD4+ and single CD8+ (CD25-) thymocytes were almost undetectable in p53+/+MKK6(Glu) Tg mice (Figure 3D and Table I). In contrast, the percentage of a CD4+CD8+ CD25− (truly DP thymocytes) thymocytes in p53+/−MKK6(Glu) mice was significantly higher (Figure 3D and Table I), although it remained lower than the percentage of DP in WT mice (Figure 3D and Table I). The fraction of mature single CD4+ and single CD8+ (CD25-) thymocytes also increased in p53+/−MKK6(Glu) mice, indicating that p53 haploinsuficiency partially restored cell cycle and differentiation of thymocytes in MKK6(Glu) mice. In addition, the total number of thymocytes in p53+/−MKK6(Glu) mice was also lower than in p53+/+MKK6(Glu) mice (Table I). This was not the result of increased cell death as the levels of active caspase 3 and cleaved PARP-1 were similar in p53+/+MKK6(Glu) and p53+/−MKK6(Glu) thymocytes (Supplementary Figure S2). These results demonstrate that activation of the p38 MAP kinase induces a G2/M cell cycle checkpoint mediated by p53 and that inactivation of p53 is required for exiting the checkpoint and differentiation into DP thymocytes.
Table 1.
p53 haploinsuficiency partially restores MKK6(Glu) thymocyte phenotype
| WT |
Tg |
|||
|---|---|---|---|---|
| p53+/+ | p53+/− | p53+/+* | p53+/−* | |
| % DPa | 83.6±8.4 | 78.3±15.6 | 7.2±2.1 | 20.7±7.2 |
| % CD4 SPa | 8.4±0.5 | 7.7±1.6 | 1.1±0.2 | 2.5±0.3 |
| % CD8 SPa | 9.0±0.2 | 9.1±0.3 | 0.1±0.1 | 2.3±0.9 |
| % CD25+ CD44−b | 3.5±1.4 | 2.8±0.37 | 80.3±3.4 | 52.8±9.2 |
| Total cell numberc | 95.5±3.5 | 103±8.0 | 203±5.7 | 35.3±8.5 |
| *The standard error of the mean is shown (n=7). The differences in the mean values for the percentage of DP (P=0.0015), CD4 SP (P=0.0132), CD8 SP (P=0.0168) and CD25+CD44− (P=0.0012) and the total cell number (P=0.0145) between p53+/+-MKK6(Glu) and p53+/−-MKK6(Glu) mice were statistically significant. | ||||
| aThymocytes were obtained from p53+/+-MKK6(Glu) (p53+/+ Tg) and p53+/−-MKK6(Glu) (p53+/− Tg) mice and stained for CD4, CD8, CD25 and CD44. The percentage of DP (CD4+CD8+), CD4+ single positive (SP), and CD8+ SP thymocytes was determined in the CD25- population. | ||||
| bCD25 expression was analyzed by flow cytometry in the total thymocyte population. | ||||
| cTotal number of thymocytes (106) in p53+/+-WT, p53+/−-WT, p53+/+-MKK6(Glu) transgenic (Tg) or p53+/−-MKK6(Glu) transgenic mice. | ||||
DN3 but not DN4 thymocytes undergo a G2/M checkpoint
If the establishment of a G2/M cell cycle checkpoint is associated with V(D)J recombination, large cells with a phenotype similar to the MMK6(Glu) thymocyte phenotype should be present in WT DN3 thymocytes. Flow cytometry analysis of WT thymocytes showed that some DN3 thymocytes have an increased cell size compared with DN4 thymocytes that have already completed recombination (Figure 4A). Moreover, WT DN3 thymocytes contained higher levels of active p38 MAP kinase than WT DN4 thymocytes (Figure 4B). In correlation, the levels of p53 were also higher in DN3 thymocytes (Figure 4C). To demonstrate that the presence of active p38 and p53 in DN3 thymocytes was associated with the presence of cells at the G2/M cell cycle checkpoint in this population, we examined the hallmarks of this cell cycle checkpoint. Although it is well known that DN4 thymocytes are highly proliferating, cyclin B levels were substantially higher in DN3 thymocytes than in DN4 thymocytes (Figure 3D). Furthermore, the levels of phospho-Cdc2 were also increased in DN3 thymocytes compared with DN4 thymocytes (Figure 4D). Consistent with the presence of p53 in DN3 thymocytes, the levels of p21 were also elevated in these cells relative to the levels in DN4 thymocytes (Figure 4D). In contrast to p21, the levels of the G0/G1 cell cycle inhibitor p27 were higher in DN4 thymocytes than in DN3 thymocytes (Figure 4D). To further confirm the presence of a G2/M cell cycle checkpoint specifically in DN3 cells, we examined the levels of p-H3. In correlation with the high rate of proliferation of DN4 thymocytes, p-H3 was present in this population, but it was practically undetectable in DN3 thymocytes despite the elevated levels of cyclin B (Figure 4E). The elevated levels of p-H3 in DN4 relative to DN3 thymocytes was further demonstrated by intracellular staining and flow cytometry analysis (Figure 4F). The levels of p-H3 in DN3 thymocytes were comparable with those in mature single CD4+ thymocytes that do not undergo proliferation (Figure 4F). Together, these results demonstrate the presence of cells in G2/M cell cycle checkpoint within the DN3 population that are absent in the DN4 population, although DN4 cells may have a higher rate of proliferation. To determine whether the subset of large cells in the DN3 population (Figure 4A) represents cells undergoing the G2/M cell cycle checkpoint, the levels of active p38 MAP kinase and p53 in the small and large DN3 thymocytes from WT mice were compared. Large DN3 thymocytes contained higher levels of p53 and active p38 MAP kinase than small DN3 thymocytes (Figure 4G). In addition, whereas 99% of small DN3 thymocytes were in G0/G1, almost 60% of the large DN3 thymocytes were in G2/M as determined by propidium iodide (PI) staining (Supplementary Figure S3), confirming that large DN3 thymocytes represent cells in G2/M cell cycle checkpoint.
Figure 4.

DN3 thymocytes undergo cell cycle arrest at the G2/M checkpoint. (A) WT thymocytes were stained and the cell size (based on forward scatter) of DN3 and DN4 thymocytes was determined by flow cytometry. The levels of phospho-p38 MAP kinase (p-p38) and total p38 MAP kinase (p38) (B), p53 and actin (C), CyB, p21, phospho-Cdc2 (p-Cdc2), p27 and actin (D), and phospho-histone H3 (pH3) and actin (E) in WT DN3 and DN4 thymocytes were determined by Western blot. (F) Intracellular staining for phospho-H3 in WT DN3 (filled histogram), DN4 (thick line) and single CD4+ (thin line) thymocytes was examined by flow cytometry. (G) Intracellular staining for phospho-p38 MAP kinase (p-p38) or p53 in WT DN3 thymocytes were determined by flow cytometry gating in the CD25+ CD44− CD4– small (DN3S) or large (DN3L) thymocytes. Isotype control antibodies (Iso) on the DN3S population are shown. (H) Thymocytes from dominant-negative p38 MAP kinase Tg (dnp38) or wild-type (WT) mice were stained as above and cell size was determined in the DN3 populations based on forward light scatter by flow cytometry. Numbers indicate the percentage of cells of large size (indicated gate) in the DN3 population from WT and dnp38 Tg mice. This is a representative experiment and (I) shows the mean percentage of large-sized cells in the DN3 populations among four independent experiments (n=4). Error bars represent standard error. (J) Levels of p53 in the DN3 thymocytes from WT and dnp38 Tg mice were determined by flow cytometry.
To examine whether the induction of the G2/M cell cycle checkpoint in DN3 thymocytes requires p38 MAP kinase activation, we compared the cell size of DN3 thymocytes from WT mice with the size of DN3 thymocytes from Tg mice expressing a dominant-negative p38 MAP kinase mutant (dnp38) under the control of the proximal Lck promoter. (Diehl et al, 2000). This mutant inhibits approximately 50% of the endogenous p38 MAP kinase activity. Analysis of forward scatter by flow cytometry in DN3 thymocytes showed that dnp38 Tg mice contained a significantly reduced fraction of large DN3 thymocytes compared with WT mice (Figure 4H and I), indicating that there are fewer DN3 thymocytes undergoing G2/M cell cycle arrest. In correlation, the p53 protein levels in DN3 thymocytes from the dnp38 Tg mice were lower than in DN3 thymocytes from WT mice (Figure 4J). Thus, during normal thymocyte development the activation of p38 MAP kinase at the DN3 stage induces a p53-mediated G2/M cell cycle checkpoint.
p38 MAP kinase is activated by DSBs generated by V(D)J recombination
During V(D)J recombination DSBs are generated by RAG1 and RAG2 in order to recombine the TcRβ chain. DSBs are then repaired by the DNA repair machinery in order to generate an in-frame TcRβ chain (Lieber, 1999). p38 MAP kinase is activated by DNA damage generated after UV or chemical agents exposure in vitro (Bulavin et al, 1999). The above results indicate that p38 MAP kinase induces a p53-dependent G2/M cell cycle checkpoint in DN3 thymocytes undergoing V(D)J recombination. We therefore tested whether the activation of the p38 MAP kinase pathway at the DN3 stage was caused by DSBs generated during V(D)J recombination. Owing to the absence of RAG 1, DN3 thymocytes from Rag1−/− mice do not undergo V(D)J recombination (Mombaerts et al, 1992). We examined the levels of active p38 MAP kinase in freshly isolated Rag1−/− or WT DN3 thymocytes. The levels of active p38 MAP kinase in Rag1−/− DN3 thymocytes were lower than in WT DN3 thymocytes (Figure 5A). In correlation, lower levels of p53 were also observed in Rag1−/− DN3 thymocytes and p21 protein could not be detected in these cells (Figure 5B).
Figure 5.
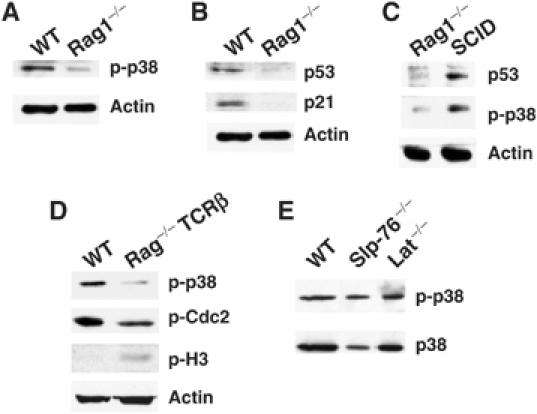
Activation of p38 MAP kinase in DN3 thymocytes requires V(D)J recombination. Whole-cell extracts were prepared from freshly isolated wild-type (WT) DN3 thymocytes or Rag1−/− thymocytes and the levels of p-p38 (A), p53 and p21(B) were determined by Western blot. (C) The levels of p-p38, p53 and actin in Rag1−/− or SCID thymocytes were determined by Western blot. (D) The levels of p-p38, p-Cdc2, p-H3 and actin in WT and Rag−/−-TcRβ DN3 thymocytes were determined by Western blot. (E) The levels of p-p38 MAP kinase and total p38 MAP kinase in WT, Slp-76−/− or LAT−/− DN3 thymocytes were determined by Western blot.
Similar to Rag1−/− mice, thymocyte development in the severe combined immunodeficiency (SCID) mice is arrested at the DN3 stage. SCID thymocytes lack DNA-dependent protein kinase (DNA-PK) activity because of mutations in the DNA-pk encoding genes (Blunt et al, 1995). Because DNA-PK is a key component of the nonhomologous-end joining machinery, SCID thymocytes are unable to repair Rag-mediated DSBs (Lieber et al, 1988) resulting in impaired TcRβ chain expression and a developmental arrest. Thus, in contrast with Rag1−/− thymocytes, unresolved V(D)J coding ends accumulate in thymocytes from SCID mice. To determine whether the accumulation of DSBs in SCID thymocytes induced the activation of p38 MAP kinase, we compared the levels of active p38 MAP kinase in Rag1−/− and SCID thymocytes by Western blot. SCID thymocytes contained higher levels of active p38 MAP kinase than Rag1−/− thymocytes (Figure 5C). In agreement with previously published data (Guidos et al, 1996), SCID thymocytes also contained higher p53 levels than Rag1−/− thymocytes (Figure 5C).
Unlike WT DN3 thymocytes, Rag−/− thymocytes have an impaired proliferation capacity (Hoffman et al, 1996). Thus, it is possible that the activation of p38 MAP kinase in WT DN3 thymocytes results from the presence of random DNA breaks generated in proliferating cells instead of RAG-mediated DNA breaks. To eliminate this possibility, we determined the levels of active p38 MAP kinase in DN3 thymocytes from Rag-deficient mice expressing a Tg TcRβ chain (Rag-TcRβ). Owing to the expression of the TcRβ chain, these thymocytes proliferate and differentiate into DP thymocytes, but they lack RAG activity and therefore RAG-induced DNA DSBs (Shinkai et al, 1993). Lower levels of active p38 MAP kinase were present in Rag-TcRβ DN3 thymocytes compared with those found in WT DN3 thymocytes (Figure 5D). Furthermore, the levels of Cdc2 phosphorylated on Tyr15 (inactive) were also lower in Rag-TcRβ DN3 thymocytes compared with those observed in WT DN3 thymocytes. In contrast, we found higher levels of p-H3 in Rag-TcRβ DN3 thymocytes than in WT DN3 thymocytes (Figure 5D), indicating that Rag-TcRβ DN3 thymocytes undergo more rapid proliferation probably due to the absence of cell cycle checkpoint. Thus, random DNA breaks generated during cell proliferation are not responsible for the activation of the p38 MAP kinase in WT DN3 thymocytes. In addition, as pre-TcR is functional in Rag-TcRβ DN3 thymocytes, these results indicate that pre-TcR signals in the absence of V(D)J recombination are not sufficient to activate p38 MAP kinase in DN3 thymocytes. To further demonstrate that activation of p38 MAP kinase is independent of pre-TcR in DN3 thymocytes, we examined the levels of active p38 MAP kinase in DN3 thymocytes from the Slp-76 or LAT-deficient mice. Although thymocytes from these mice recombine TcR-β chain and contain pre-TcR, they are arrested at the DN3 stage because of the lack of pre-TcR signaling (Clements et al, 1998; Zhang et al, 1999). Despite the absence of pre-TcR signaling, DN3 thymocytes from the Slp-76−/− or the LAT−/− mice contained similar levels of active p38 MAP kinase to those found in WT DN3 thymocytes (Figure 5E). Together, these results demonstrate that activation in p38 MAP kinase in DN3 thymocytes requires V(D)J-mediated DSBs, but does not require pre-TcR signaling.
V(D)J-generated DSBs are present in post-G1 DN3 thymocytes
Together, these results demonstrate that DSBs generated by V(D)J recombination are required for the activation of p38 MAP kinase and the induction of a G2/M cell cycle checkpoint at the DN3 stage. It has been shown that most of the DSBs generated during V(D)J recombination occur in G1 and are repaired at the G1/S checkpoint (Schlissel et al, 1993; Li et al, 1996). To determine whether the unresolved Rag-induced DSBs in WT DN3 thymocytes might induce the G2/M checkpoint, we assayed V(D)J DSBs at the TcRβ locus by ligation-mediated PCR (LM-PCR) (Schlissel et al, 1993). We isolated DN3 thymocytes from WT mice by cell sorting and stained them with PI (Supplementary Figure S4). We then sorted and collected DN3 thymocytes with G0/G1 and S/G2/M DNA content. To confirm the purity of the sorted populations, a portion of cells was restained with PI and reanalyzed by fluorescence-activated cell sorting (FACS) (Supplementary Figure S4). Sorted G1/G0 and S/G2/M DN3 thymocytes were immediately lysed to extract genomic DNA and performed LM-PCR as previously described (Schlissel et al, 1993). As a control, we used genomic DNA from WT total thymocytes (positive control) and liver (negative control), as well as genomic DNA from MKK6(Glu), SCID and RAG1−/− thymocytes. Equal amounts of linker-ligated DNA were assayed for DSBs at RSS 5′ of the Dβ1 and Dβ2 gene segments (indicative of V-to-DJβ rearrangement) (Figure 6A). As shown in previous studies, specific PCR products for 5′ Dβ1 and Dβ2 RSS were present in total WT thymocytes and SCID thymocytes, but were absent in liver cells and Rag-deficient thymocytes (Figure 6B). PCR products corresponding to the 5′ of Dβ1 and Dβ2 broken ends were also detected in both WT G1 and S/G2/M DN3 thymocytes (Figure 6B). In addition, we also detected Dβ1 and Dβ2 DSBs in MKK6(Glu) thymocytes (Figure 6B).
Figure 6.

DN3 thymocytes in G2/M contain RAG-mediated DSBs. (A) The schematic diagram represents the TcRβ genomic locus (not drawn to scale). D and J gene segments (boxes) are flanked by consensus RSS containing 12-bp (open triangles) or 23-bp (black triangles) spacers. A cryptic RSS containing a 23-bp was also identified at the 3′ of the Dβ2 gene segment (gray triangle). Thick bars represent the LM-PCR products using primers specific for the 5′ of Dβ1 or Dβ2 and for the linkers bound to the broken ends. Thin bars represent the probes used for Southern blot analysis of the PCR products. (B) LM-PCR was performed using DNA from total WT thymocytes (T), WT liver cells (L), WT G0/G1 DN3 thymocytes (G1), WT S/G2/M DN3 thymocytes (G2/M), MKK6(Glu) DN3 thymocytes, SCID DN3 thymocytes and Rag1−/− DN3 thymocytes. PCR products were resolved by Southern blot analysis using 32p-labeled locus-specific primers (Dβ-1, Dβ-2). (C) The p53 gene was amplified from the same amount of DNA used in the LM-PCR reactions (B), and PCR products were separated on agarose gels and visualized by ethidium bromide staining.
To confirm that the LM-PCR products corresponded to dsDNA breaks at the 5′ of Dβ1 and Dβ2 RSS, we subjected the various PCR products to cloning and DNA sequence analysis. Three clones for each band and each sample were sequenced. The results (Supplementary Figure S5) confirmed that the fragment corresponding in size to Dβ1 was indeed the 5′ region of the Dβ1 segment and contained the specific 5′ RSS. The faint band of higher molecular weight detected in G1/G0 DN3 thymocytes was nonspecific. The two well-defined bands detected in the Dβ2 assay also contained the region 5′ of the Dβ2 segment (Supplementary Figure S5). The lower band of the expected size (E) present in all the samples corresponded to the DSB at the 5′ RSS of Dβ2 (Supplementary Figure S5). The upper band corresponded to a cryptic RSS located at 3′ of the Dβ2 gene segment (Figure 6A and Supplementary Figure S5). Interestingly, this band was present in total thymocytes and G1/G0 DN3 thymocytes but it was undetectable in S/G2/M DN3 thymocytes probably because it had already been repaired. To verify that similar amounts of DNA were used in each LM-PCR, we performed PCR for p53 genomic DNA as an internal control. Similar levels of p53 genomic amplification were obtained in all samples including liver and RAG-deficient thymocytes where no LM-PCR products were detected (Figure 6C). These results demonstrate that although some DSBs are repaired at the G1/S checkpoint in DN3 thymocytes, S/G2/M DN3 thymocytes contain RAG-mediated DSBs that could induce a G2/M checkpoint.
If V(D)J recombination is upstream of p38 MAP kinase and the initiation of the cell cycle checkpoint in DN3 thymocytes, persistent activation of this pathway in Rag1−/− mice should be able to activate p53 and induce a G2/M cell cycle checkpoint in the absence of V(D)J recombination. To activate p38 MAP kinase in Rag1−/− thymocytes, Rag1−/− mice were crossed with the MKK6(Glu) Tg mice. Flow cytometry analysis revealed that similar to Rag1−/− and MKK6(Glu) thymocytes, MKK6(Glu)-Rag1−/− thymocytes were arrested at the DN3 stage (CD25+CD44−) (Figure 7A). However, in contrast to Rag1−/− thymocytes that were small and arrested at G0/G1, MKK6(Glu)-Rag1−/− thymocytes were large (Figure 7B) and promitotic (data not shown). Furthermore, MKK6(Glu)-Rag1−/− thymocytes contained elevated levels of p53, p21 and cyclin B compared with Rag1−/− thymocytes as determined by Western blot (Figure 7C). Interestingly, whereas thymocytes from the Rag1−/− mice are unable to survive in vivo and thymi of these mice are very small, DN3 thymocytes from MKK6(Glu)-Rag1−/− mice survived and accumulated in vivo (Figure 7D). Thus, DSBs generated during V(D)J recombination are required for p38 MAP kinase activation, but once this pathway is activated, it can trigger the G2/M cell cycle checkpoint without DSBs. Collectively, these results show that the activation of p38 MAP kinase is downstream of DNA damage in the signaling cascade leading to p53 activation and the induction of a G2/M cell cycle checkpoint during V(D)J recombination.
Figure 7.
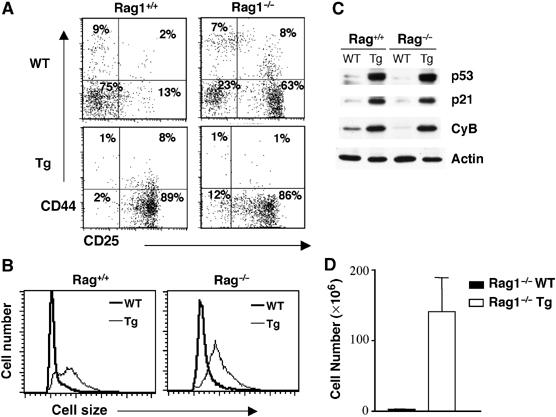
Activation of the p38 MAP kinase pathway in Rag-deficient thymocytes promotes a G2/M cell cycle checkpoint. (A) FACS profiles represent the distribution and percentage of CD25−CD44+, CD25+CD44+, CD25+CD44− and CD25−CD44− populations in total thymocytes isolated from WT-Rag1−/−, MKK6(Glu) transgenic (Tg)-Rag1+/+ and Tg-Rag1−/− mice or in gated DN thymocytes from WT-Rag1+/+ mice. CD4 and CD8 profiles are shown in Supplementary Figure S6. (B) Thymocytes were isolated from the mice described in (A) and cell size was analyzed by flow cytometry. (C) The levels of p53, p21, cyclin B and actin were determined by Western blot. (D) Total thymocyte numbers in Rag1−/− WT or Rag1−/− MKK6(Glu) Tg mice. The average and standard error of the mean are shown (n=6).
Discussion
Although V(D)J recombination is advantageous because it ensures maximal diversity of the TcR repertoire, it is also a risk for host cells as genomic aberrations resulting from un- or mis-repaired DNA breaks may lead to malignancy (Nacht et al, 1996; Difilippantonio et al, 2000). Thus, an efficient DNA repair mechanism and the presence of a cell cycle checkpoint to delay cell division while DNA repair occurs is essential. Although it has been shown that RAG-induced DNA breaks are repaired at the G1/S checkpoint (Schlissel et al, 1993), the establishment of a G2/M cell cycle checkpoint in DN3 thymocytes in response to DSBs resulting from V(D)J recombination has not been formally demonstrated. Here, we show for the first time that activation of p38 MAP kinase in response to DSBs induces a p53-dependent G2/M checkpoint in developing DN3 thymocytes.
In fibroblasts, phosphorylation of p53 by p38 MAP kinase promotes the stabilization of p53 and cell cycle arrest at the G2/M checkpoint in response to DNA damage after UV or gamma irradiation in vitro (Bulavin et al, 1999; Huang et al, 1999). Here, we demonstrate that activation of p38 MAP kinase in vivo leads to phosphorylation and accumulation of p53 and induction of a G2/M cell cycle checkpoint. In addition to the p53-dependent pathway, it has been recently shown that p38 MAP kinase can also promote cell cycle arrest at the G2/M checkpoint in vitro by inactivating the Cdc25 phosphatase, resulting in accumulation of inactive Cdc2 and cell cycle arrest (Bulavin et al, 2001). It is therefore possible that, together with p53, Cdc25 may also participate in the establishment of the G2/M checkpoint induced by p38 MAP kinase in DN3 thymocytes.
It has been proposed that pre-TcR signals can also activate p38 MAP kinase (Mulroy and Sen, 2001). However, our results indicate that pre-TcR signals are not required as long as V(D)J recombination takes place (Figure 5). Because DSBs generated by DNA-damaging agents promote p38 MAP kinase activation in vitro (Wang et al, 2000; Bulavin et al, 2001), we propose that the presence of DSBs generated by of V(D)J recombination in DN3 thymocytes is the main stimulus that activates the p38 MAP kinase pathway in vivo. This model is supported by the presence of active p38 MAP kinase in WT and SCID DN3 thymocytes, but not in WT DN4, Rag1−/− and RAG-deficient-TcRβ chain Tg thymocytes. Although p38 MAP kinase is clearly activated in response to DNA damage in vitro, the molecular mechanisms remain largely unknown. ATM kinase has been suggested to be responsible for the activation of the p38 MAP kinase pathway after DNA damage (Wang et al, 2000). Whether ATM plays a role in the activation of p38 MAP kinase in response to RAG-induced DSBs during V(D)J recombination remains to be determined. However, no difference in ATM activation was observed between DN3 and DN4 thymocytes (data not shown) arguing that activation of p38 MAP kinase in DN3 thymocytes is likely independent of ATM.
The fact that RSS-associated dsDNA breaks have been preferentially observed in cells in G0/G1 (Schlissel et al, 1993; Li et al, 1996) has lead to the conclusion that V(D)J recombination occurs only during the G0/G1 phase of the cell cycle and that DNA damage is repaired exclusively at the G1/S cell cycle checkpoint (Schlissel et al, 1993; Guidos et al, 1996). Our results show that WT DN3 thymocytes in S/G2/M phases of the cell cycle contain signal end DSBs at the TcRβ locus. These signal end DSBs can trigger the G2/M checkpoint to allow signal joint formation and convert them into inert products (Hiom et al, 1998; Roth and Craig, 1998), as linear double-stranded DNA can easily integrate randomly and compromise genomic stability. Indeed, interchromosomal transposition of signal ends in human T cells has been recently described (Messier et al, 2003). Although there is no direct evidence that signal ends induce the same cellular response as coding ends, most of the DNA repair machinery recognizes DSBs regardless of whether they are in the chromosomes. It is therefore logical that signal ends can also induce a G2/M cell cycle checkpoint.
Thus, we propose that V(D)J recombination at the DN3 stage of thymocyte development triggers activation of the p38 MAP kinase pathway. Activated p38 MAP kinase promotes a G2/M cell cycle checkpoint through the phosphorylation and accumulation of p53 to allow DNA repair of deleterious mutations produced during V(D)J recombination (Figure 8). Although activation of the p38/p53 pathway at the DN3 stage may be important for genomic stability, subsequent inactivation of p38 MAP kinase and termination of the G2/M checkpoint is also required for DN3 thymocytes to progress through mitosis, downregulate CD25 and differentiate into DN4 thymocytes.
Figure 8.
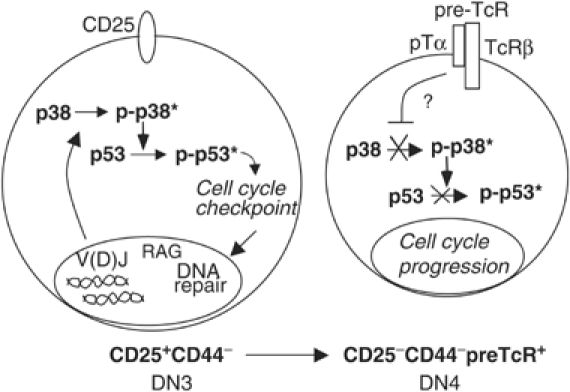
Activation of p38 MAP kinase by V(D)J recombination leads to the establishment of a p53-dependent G2/M cell cycle checkpoint to allow repair of RAG-mediated DSBs.
Materials and methods
Mice
The MKK6(Glu) Tg and dominant-negative p38 MAP kinase Tg mice have been previously described (Diehl et al, 2000). Rag1−/−, p53−/− and SCID mice were purchased from Jackson Laboratory. Rag2−/−/TcRβ chain Tg mice were purchased from Taconic Laboratory. SLP-76−/− and LAT−/− mice have been previous described. Procedures that involved mice were approved by institutional guidelines for animal care.
Cell surface staining and cell sorting
The distribution of different thymocyte populations was examined after cell surface staining with anti-CD4, CD8, CD25 (Pharmingen), and anti-CD44-PE (Caltag) antibodies by flow cytometry (LSR II, BD). The DN3 and DN4 populations were purified by cell sorting (FacsAria, BD) after four-color staining. To isolate G1 and S/G2/M DN3 thymocytes, total thymocytes were stained (CD4, CD8, CD25 and CD44), DN3 thymocytes were sorted, collected and stained with PI and sorted again based on DNA content. Post sorted G0/G1 and S/G2/M DN3 thymocytes were then collected and lysed for isolation of genomic DNA. Detailed gates, steps and purity of the sorting are described in Supplementary Figure S4. To examine cell cycle in small and large DN3 thymocytes, total thymocytes were stained for four colors as described above, large and small DN3 were sorted based on the forward scatter, collected and stained with PI to examine the DNA content. p53 intracellular staining was performed after cell surface staining using anti-p53-PE antibody (Pharmingen) as recommended. For phosphorylated p38 MAP kinase intracellular staining, thymocytes were stained for cell surface markers, fixed with paraformaldehyde, permeabilized with 0.05% saponin and stained with an anti-phospho-p38 (Thr180/Tyr182) antibody (Cell Signaling) followed by anti-Rabbit-FITC antibody (Santa Cruz Biotechnology). For detection of phosphorylated histone H3, cells were fixed, permeabilized (0.1% Tween) and stained using the FITC-anti-phospho-H3 (Cell Signaling). Cell size was determined by flow cytometry and analysis of forward light scatter.
Western blot
Whole-cell extracts were prepared in Triton lysis buffer as previously described (Pedraza-Alva et al, 1996). Cyclin B1 was immunoprecipitated from whole-cell extracts with anti-cyclin B1 antibody (Santa Cruz Biotechnology). Immunoprecipitated proteins or total cell extracts were resolved by SDS–PAGE, transferred to membranes and immunostained with anti-p53 (Novocastra), anti-p21Waf1, anti-p27Kip, anti-cyclin B1, anti-Cdc2, anti-actin (Santa Cruz Biotechnology), anti-phosphorylated Cdc2 (Tyr15), anti-phosphorylated p38 (Thr180/Tyr182), anti-phosphorylated p53 (Ser15 or Ser392) anti-p38 (Cell Signaling), anti-phosphorylated histone H3 (Ser10) and anti-CENP-A (Upstate Biotech) antibodies.
Cdc2 kinase assay
Cyclin B1 precipitates were washed twice with lysis buffer, twice with kinase buffer (20 mM Tris pH 7.5, 7.5 mM MgCl2, 1 mM DTT) and incubated at 30°C for 30 min in 25 μl of kinase buffer containing 10 μCi (γ 32P)-ATP (3000 Ci/mmol, Amersham), 100 μM ATP and 1 μg of histone H1 (Worthington) as exogenous substrate. The reaction was mixed with an equal volume of 2 × SDS–PAGE loading buffer. Proteins were separated by SDS–PAGE and 32P-labeled histone H1. Levels of phospho-histone H1 were determined by phosphoimaging on a FX molecular imager (Bio-Rad).
Ligation-mediated-PCR
WT G0/G1 and S/G2/M DN3 thymocytes were isolated by cell sorting, DNA was purified from sorted cells and LM-PCR was performed as previously described (Schlissel et al, 1993). The sequence of the TcRβ locus-specific oligonucleotide PCR primers and probes used for LM-PCR has been previously published (Hempel et al, 1998). The control p53 amplification was performed using the following primers: OIMRO336: 5′-ATAGGTCGGCGGTTCAT-3′ and OIMRO337: 5′-CCCGAGTATCTGGAAGACAG-3′. The PCR products were separated on 1.5% agarose gel and detected by ethidium bromide staining. The LM-PCR bands were cut from the gel and the DNA was extracted using the Qiaex II gel extraction kit (Qiagen) following the manufacturer's instructions. The DNA was then ligated directly into the TOPO A/T plasmid (Invitrogen) and transformed into bacteria. DNA was isolated and sent for sequencing using the universal M13 primers.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Acknowledgments
We thank Karen A Fortner and Oliver Dienz for helpful discussions and critical reading of the manuscript. This work was supported by Grant R01 AI051454 (M Rincón) from the National Institute of Health. G Pedraza-Alva was partially supported by the Sistema Nacional de Investigadores/CONACYT, México.
References
- Bassing CH, Swat W, Alt FW (2002) The mechanism and regulation of chromosomal V(D)J recombination. Cell 109 (Suppl): S45–S55 [DOI] [PubMed] [Google Scholar]
- Blunt T, Finnie NJ, Taccioli GE, Smith GC, Demengeot J, Gottlieb TM, Mizuta R, Varghese AJ, Alt FW, Jeggo PA, Jackson SP (1995) Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80: 813–823 [DOI] [PubMed] [Google Scholar]
- Booher RN, Holman PS, Fattaey A (1997) Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem 272: 22300–22306 [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ Jr (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411: 102–107 [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, Fornace AJ Jr (1999) Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J 18: 6845–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T (2000) How to activate p53. Curr Biol 10: R315–R317 [DOI] [PubMed] [Google Scholar]
- Clements JL, Yang B, Ross-Barta SE, Eliason SL, Hrstka RF, Williamson RA, Koretzky GA (1998) Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science 281: 416–419 [DOI] [PubMed] [Google Scholar]
- Diehl NL, Enslen H, Fortner KA, Merritt C, Stetson N, Charland C, Flavell RA, Davis RJ, Rincon M (2000) Activation of the p38 mitogen-activated protein kinase pathway arrests cell cycle progression and differentiation of immature thymocytes in vivo. J Exp Med 191: 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404: 510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J (1994) Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell 78: 1039–1049 [DOI] [PubMed] [Google Scholar]
- Gottlieb E, Haffner R, King A, Asher G, Gruss P, Lonai P, Oren M (1997) Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J 16: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS (1996) V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev 10: 2038–2054 [DOI] [PubMed] [Google Scholar]
- Han J, Lee JD, Bibbs L, Ulevitch RJ (1994) A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265: 808–811 [DOI] [PubMed] [Google Scholar]
- Hempel WM, Stanhope-Baker P, Mathieu N, Huang F, Schlissel MS, Ferrier P (1998) Enhancer control of V(D)J recombination at the TCRbeta locus: differential effects on DNA cleavage and joining. Genes Dev 12: 2305–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B (1997) 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell 1: 3–11 [DOI] [PubMed] [Google Scholar]
- Hiom K, Melek M, Gellert M (1998) DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell 94: 463–470 [DOI] [PubMed] [Google Scholar]
- Hoffman ES, Passoni L, Crompton T, Leu TM, Schatz DG, Koff A, Owen MJ, Hayday AC (1996) Productive T-cell receptor beta-chain gene rearrangement: coincident regulation of cell cycle and clonality during development in vivo. Genes Dev 10: 948–962 [DOI] [PubMed] [Google Scholar]
- Howman EV, Fowler KJ, Newson AJ, Redwards S, MacDonald AC, Kalitis P, Choo KH (2000) Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA 97: 1148–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Ma WY, Maxiner A, Sun Y, Dong Z (1999) p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J Biol Chem 274: 12229–12235 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Li Z, Dordai DI, Lee J, Desiderio S (1996) A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity 5: 575–589 [DOI] [PubMed] [Google Scholar]
- Lieber MR (1999) The biochemistry and biological significance of nonhomologous DNA end joining: an essential repair process in multicellular eukaryotes. Genes Cells 4: 77–85 [DOI] [PubMed] [Google Scholar]
- Lieber MR, Hesse JE, Lewis S, Bosma GC, Rosenberg N, Mizuuchi K, Bosma MJ, Gellert M (1988) The defect in murine severe combined immune deficiency: joining of signal sequences but not coding segments in V(D)J recombination. Cell 55: 7–16 [DOI] [PubMed] [Google Scholar]
- Mahadevan LC, Willis AC, Barratt MJ (1991) Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell 65: 775–783 [DOI] [PubMed] [Google Scholar]
- Messier TL, O'Neill JP, Hou SM, Nicklas JA, Finette BA (2003) In vivo transposition mediated by V(D)J recombinase in human T lymphocytes. EMBO J 22: 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE (1992) RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68: 869–877 [DOI] [PubMed] [Google Scholar]
- Morgan DO (1995) Principles of CDK regulation. Nature 374: 131–134 [DOI] [PubMed] [Google Scholar]
- Mulroy T, Sen J (2001) p38 MAP kinase activity modulates alpha beta T cell development. Eur J Immunol 31: 3056–3063 [DOI] [PubMed] [Google Scholar]
- Nacht M, Strasser A, Chan YR, Harris AW, Schlissel M, Bronson RT, Jacks T (1996) Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev 10: 2055–2066 [DOI] [PubMed] [Google Scholar]
- Pani L, Horal M, Loeken MR (2002) Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev 16: 676–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza-Alva G, Merida LB, Burakoff SJ, Rosenstein Y (1996) CD43-specific activation of T cells induces association of CD43 to Fyn kinase. J Biol Chem 271: 27564–27568 [DOI] [PubMed] [Google Scholar]
- Poon RY, Jiang W, Toyoshima H, Hunter T (1996) Cyclin-dependent kinases are inactivated by a combination of p21 and Thr-14/Tyr-15 phosphorylation after UV-induced DNA damage. J Biol Chem 271: 13283–13291 [DOI] [PubMed] [Google Scholar]
- Rodewald HR, Fehling HJ (1998) Molecular and cellular events in early thymocyte development. Adv Immunol 69: 1–112 [DOI] [PubMed] [Google Scholar]
- Roth DB, Craig NL (1998) VDJ recombination: a transposase goes to work. Cell 94: 411–414 [DOI] [PubMed] [Google Scholar]
- Schlissel M, Constantinescu A, Morrow T, Baxter M, Peng A (1993) Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev 7: 2520–2532 [DOI] [PubMed] [Google Scholar]
- She QB, Chen N, Dong Z (2000) ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem 275: 20444–20449 [DOI] [PubMed] [Google Scholar]
- Shieh SY, Taya Y, Prives C (1999) DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J 18: 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai Y, Koyasu S, Nakayama K, Murphy KM, Loh DY, Reinherz EL, Alt FW (1993) Restoration of T cell development in RAG-2-deficient mice by functional TCR transgenes. Science 259: 822–825 [DOI] [PubMed] [Google Scholar]
- Toyoshima H, Hunter T (1994) p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell 78: 67–74 [DOI] [PubMed] [Google Scholar]
- Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J (2000) Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol 20: 4543–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Antinore MJ, Wang XW, Carrier F, Smith ML, Harris CC, Fornace AJ Jr (1999) Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene 18: 2892–2900 [DOI] [PubMed] [Google Scholar]
- Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, Grinberg A, Tsay HC, Jacobs HM, Kessler CM, Long EO, Love PE, Samelson LE (1999) Essential role of LAT in T cell development. Immunity 10: 323–332 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6