

Abstract
The protein phosphatase 2A (PP2A) acts on several kinases in the extracellular signal-regulated kinase (ERK) signaling pathway but whether a specific holoenzyme dephosphorylates ERK and whether this activity is controlled during mitogenic stimulation is unknown. By using both RNA interference and overexpression of PP2A B regulatory subunits, we show that B56, but not B, family members of PP2A increase ERK dephosphorylation, without affecting its activation by MEK. Induction of the early gene product and ERK substrate IEX-1 (ier3) by growth factors leads to opposite effects and reverses B56-PP2A-mediated ERK dephosphorylation. IEX-1 binds to B56 subunits and pERK independently, enhances B56 phosphorylation by ERK at a conserved Ser/Pro site in this complex and triggers dissociation from the catalytic subunit. This is the first demonstration of the involvement of B56-containing PP2A in ERK dephosphorylation and of a B56-specific cellular protein inhibitor regulating its activity in an ERK-dependent fashion. In addition, our results raise a new paradigm in ERK signaling in which ERK associated to a substrate can transphosphorylate nearby proteins.
Keywords: ERK, phosphatases, PP2A, substrates
Introduction
Extracellular signal-regulated kinases (ERKs) are central to signaling by growth factors. Their activation involves a cascade of phosphorylation events initiated by stimulation of Ras and ending by MAPK kinases (MEK1/2)-mediated dual phosphorylation of ERK1/2 at neighboring Thr and Tyr residues in the activation loop (reviewed in Pearson et al, 2001). The ERK pathway is implicated in diverse cellular processes including proliferation, differentiation and survival. This variety of biological responses is determined by the cell-specific combination of downstream substrates and by differences in the magnitude and kinetics of ERK signaling (Marshall, 1995; Murphy et al, 2002). The length of time that ERKs are active depends, notably, on the nature of the stimuli, the cooperation of several pathways downstream of Ras, the localization of the kinases or the presence of scaffold proteins (Pearson et al, 2001). The net ERK activation is also dependent on the levels between kinases and phosphatases. Since dephosphorylation of either of the Thr or Tyr residue results in ERK inactivation, phosphatases with dual, Ser/Thr or Tyr specificities (reviewed in Keyse, 2000), or a complex of the latter two (Wang et al, 2003) can regulate ERK dephosphorylation and generate a fine tuning of ERK signal. Notably, Tyr- and Ser/Thr phosphatases, which are constitutively expressed, are involved in the rapid initial steps of ERK inactivation, whereas some dual specificity phosphatases (DUSP or MKP), which are early gene products, control the late phases. Interestingly, ERK itself regulates its own dephosphorylation by MKPs and Tyr phosphatases. For example, the association of MKP-3 with ERK enhances its phosphatase activity (Camps et al, 1998); phosphorylation of the Tyr phosphatases STEP and PTP-SL by ERK is required for ERK inactivation (Pulido et al, 1998), while phosphorylation of MKP-1 either positively or negatively controls its stability (Brondello et al, 1999; Lin et al, 2003). Although protein phosphatase 2A (PP2A) has long been identified as the major Ser/Thr phosphatase involved in ERK inactivation (Alessi et al, 1995), no data are available on a possible regulation of this activity during growth factor stimulation.
PP2A is involved in a broad range of cellular processes, including signal transduction, transcriptional regulation and control of the cell cycle (reviewed in Janssens and Goris, 2001). This diversity of functions is conferred by a diversity of regulatory subunits, the combination of which can give rise to over 50 different forms of PP2A. The PP2A holoenzyme is a heterotrimer that consists of a core dimer, composed of a scaffold (A) and a catalytic (C) subunit that associates with a variety of regulatory (B) subunits. The B subunits have been divided into gene families named B (or PR55), B′ (or B56 or PR61) and B′′(or PR72). Each family comprises several members. For example, five distinct mammalian genes encode members of the B56 family, called B56α, β, γ, δ and ɛ, generating at least eight isoforms (McCright et al, 1996). In addition to B, B′ and B′′ subunits, certain tumor antigens encoded by polyoma viruses associate with the A/C core (Pallas et al, 1990). The different families of B subunits bind to overlapping regions on the A subunit and their association to the core enzyme is mutually exclusive (Ruediger et al, 1999). The B subunits determine substrate specificity and subcellular localization. For example, the B family targets PP2A to microtubules (Sontag et al, 1995), while the B56 family mediates PP2A functions in the Wnt/β−catenin signaling cascade and interacts with cyclin G (Seeling et al, 1999; Okamoto et al, 2002). Thus, the ratio of various heterotrimers in a cell is expected to play a fundamental role in the regulation of the diverse activities of PP2A. Modifications in the association of the B subunits with the A/C heterodimer may result from fluctuation in the phosphorylation and/or methylation levels of the C or B subunits (Ogris et al, 1997; Usui et al, 1998; Janssens and Goris, 2001). However, little is known about the upstream signals regulating these modifications.
IEX-1 (or ier3) is an early-response gene involved in survival and proliferation, which is rapidly induced by growth factors, viral infections or chemical carcinogens (Wu, 2003). We previously isolated IEX-1 as a substrate of ERK and showed that it has a dual role in ERK signaling: on the one hand, IEX-1 acquires antiapoptotic functions upon phosphorylation by ERK; on the other hand, by binding to active ERK through a DEF type of docking site (Jacobs et al, 1999), IEX-1 behaves as a positive regulator of ERK activation, prolonging ERK signal in response to various growth factors (Garcia et al, 2002). In the hematopoietic cell line UT7, induction of the IEX-1 protein by TPO is required to sustain ERK signal. IEX-1 has no effect on MEK or the p38 and JNK MAPKs (Garcia et al, 2002). We show here that IEX-1 prevents the dephosphorylation of ERK on its Thr residue, suggesting that it may inhibit ERK inactivation by PP2A. We found that PP2A containing the B56, but not the B, family of regulatory subunits are involved in ERK dephosphorylation. IEX-1 inhibits B56-PP2A activity by allowing ERK-mediated phosphorylation of the B56 subunits at a Ser/Pro site conserved in all B56 members, and thereby dissociation of B56 from A/C. This is the first identification of the involvement of B56-PP2A in ERK inactivation and of the control of its activity by a nonviral protein and by the ERK pathway. Our results also underscore a new mechanism of ERK regulation in which a substrate, by associating with the active kinase, favors the phosphorylation of another protein.
Results
IEX-1 enhances ERK activity by preventing its dephosphorylation on threonine
To test whether IEX-1 potentiates ERK activation by altering its dephosphorylation, CHO cells were stimulated with FCS and the phosphorylation of ERK was measured at various times following starvation by using an antibody that recognizes pERK phosphorylated on both Thr and Tyr residues (pTpY-ERK, Figure 1A). Expression of IEX-1 greatly delayed the rapid disappearance of pERK levels occurring upon removal of the stimulus but had no effect on pMEK. This suggests that IEX-1 inhibits the action of endogenous phosphatases on ERK. To identify which type of phosphatase was involved, we used antibodies that recognize ERK phosphorylated either on Thr (pT-ERK) or on Tyr (pY-ERK) only. Whereas the levels of pT-ERK decreased rapidly upon stimulus removal, ERK dephosphorylation on Tyr was much slower. IEX-1 had no effect on the levels of pY-ERK, but it markedly slowed down the decay of pT-ERK. Likewise, in CHO-EpoR cells in which Epo stimulation induces a transient ERK response (Garcia et al, 2002), IEX-1 expression prolonged the levels of pT-ERK, while it did not change the pY-ERK signal (Figure 1B). Thus, IEX-1 decreases ERK dephosphorylation by a S/T phosphatase.
Figure 1.
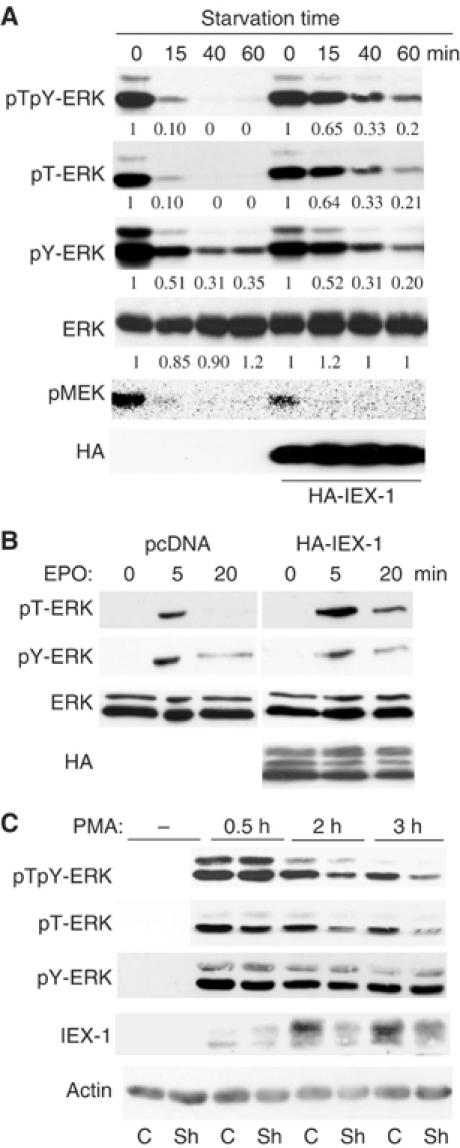
IEX-1 inhibits ERK dephosphorylation on threonine. (A, B) IEX-1 expression extends the pT-ERK signal. CHO-EpoR cells were transfected with either empty vector or HA-IEX-1. The cells were either stimulated with FCS for 5 min and then starved for various times (A) or stimulated for various times with 10 U/ml Epo (B). Quantification of the pERK levels in cells expressing IEX-1 or control, normalized to ERK levels, is expressed relative to the levels of phosphorylation at their zero time point. (C) IEX-1 downregulation reduces pT-ERK signal duration. HeLa cells expressing shRNA for IEX-1 (Sh) or GFP (C) were starved of FCS overnight and stimulated with PMA (20 ng/ml). ERK phosphorylation and IEX-1 expression were examined at various times following stimulation.
IEX-1 is an early-response gene product that is induced by various growth factors. Thus, pT-ERK phosphorylation should be more sustained in growth factor-treated cells where IEX-1 is expressed. To test this, we generated HeLa cells expressing small hairpin RNA (shRNA) for IEX-1. As a control, we used either an irrelevant shRNA (GFP) or an shRNA for IEX-1 that we previously found unable to downregulate IEX-1 expression. PMA-induced IEX-1 protein was greatly reduced in cells expressing IEX-1-shRNA, as compared to control (Figure 1C). Both the pTpY-ERK and pT-ERK signals were also decreased in these cells, at late but not early time points of PMA stimulation, that is, when endogenous IEX-1 is expressed. By contrast, the pY-ERK signal, which again disappeared at a slower rate, was not affected, confirming that IEX-1 does not act on MEK-mediated ERK phosphorylation. Similar effects were observed in HEK-293 and UT7 cells in response to PMA and TPO, respectively (data not shown). Thus, induction of IEX-1 plays an important role, in response to different growth factors and in different cell contexts, to sustain ERK signal by reducing ERK dephosphorylation specifically on Thr.
B56-containing PP2A dephosphorylate ERK and their action is inhibited by IEX-1
PP2A has been previously implicated in the dephosphorylation of ERK (Alessi et al, 1995). To confirm this role, we used okadaic acid (OA) under conditions shown to inhibit specifically PP2A in intact cells (Favre et al, 1997). Since MEK is also a substrate of PP2A (Gause et al, 1993; Sontag et al, 1993), CHO cells were stimulated and then treated with the MEK inhibitor U0126 alone or in the presence of OA. OA completely prevented the U0126-induced downregulation in pERK levels (Figure 2A). This effect was similar to that observed upon expression of IEX-1, and OA could not further increase ERK phosphorylation in IEX-1-expressing cells. This supports a role for PP2A as the major S/T phosphatase involved in the rapid ERK inactivation occurring when activation is stopped and suggests that IEX-1 may inhibit a PP2A activity.
Figure 2.
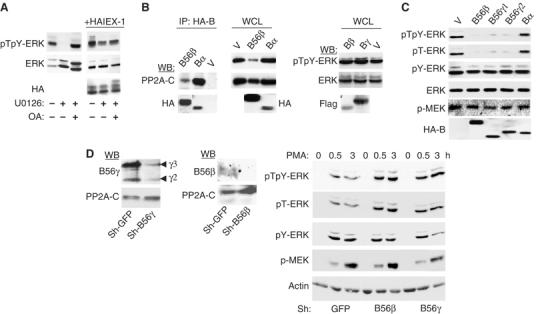
B56 subunits containing PP2A dephosphorylate ERK. (A) The PP2A inhibitor OA prevents ERK dephosphorylation. CHO cells were treated for 30 min at 37°C with 10 μM U0126 or vehicle alone, in the presence or absence of 0.1 μM OA and analyzed for ERK phosphorylation. (B, C) The B56- but not the B-family of PP2A dephosphorylates ERK. NIH-3T3 (B) or CHO (C) cells were transfected with empty vector (V) or the various PP2A-B subunits (4 × HA-B56β, HA-Bα, Flag-Bβ, Flag-Bγ, HA-B56γ). Anti-HA immunoprecipitates were blotted with anti-PP2A-C (B, left panel). ERK phosphorylation was analyzed in the whole-cell lysates (WCL). (D) Downregulation of B56-PP2A leads to increased ERK phosphorylation. Left panel: Expression of B56γ and B56β subunits in HeLa cells expressing the corresponding shRNA. Right panel: Cells expressing B56γ, B56β or control (GFP) shRNAs were starved of FCS overnight and stimulated with 10 ng/ml PMA for various times.
To determine whether a particular PP2A heterotrimer is involved in ERK dephosphorylation, we examined the effect of increased expression of various families of B-regulatory proteins on ERK phosphorylation. Transiently expressed B subunits can form functional trimeric PP2A (McCright et al, 1996). Indeed, both B (Bα) and B56 (B56β) family members co-precipitated with the endogenous C subunit when expressed in NIH-3T3 cells (Figure 2B, left panel). However, whereas expression of B56β greatly decreased endogenous ERK phosphorylation, Bα had no effect. Likewise, neither Bβ nor Bγ altered pERK levels (Figure 2B). The decrease in ERK phosphorylation induced by B56β is not specific for this B56 member, as it was also observed with B56γ1 and γ2 (Figure 2C). Interestingly, expression of B56 subunits decreased both pTpY-ERK and pT-ERK, whereas it did not change the levels of pY-ERK or pMEK. This indicates that B56-PP2A does not inhibit the phosphorylation of ERK by MEK but rather controls ERK dephosphorylation.
To confirm these results, we used shRNA to knockdown the expression of B56 subunits. HeLa cells expressing shRNAs that target all B56γ isoforms (Chen et al, 2004, 2005) or B56β showed increased and prolonged pT-ERK levels in response to PMA, as compared to control cells (Figure 2D). Consistent with the specific decrease in pT-ERK induced by expression of B56γ or β subunits, downregulation of neither of these subunits affected pY-ERK or pMEK levels. Thus, downregulation of either endogenous B56γ or B56β proteins is sufficient to alter significantly and specifically the pT-ERK signal, confirming the direct involvement of these enzymes in ERK dephosphorylation.
IEX-1 and B56-containing PP2A act in an opposite fashion on ERK phosphorylation, suggesting that IEX-1 may prevent their action. Indeed, overexpression of IEX-1 reversed the downregulation of the pT-ERK signal induced by B56β (Figure 3A) and B56γ (data not shown). This effect is specific for B56, since IEX-1 expression did not restore ERK phosphorylation in cells expressing MKP1 or MKP3 (Figure 3B). Conversely, B56β inhibited IEX-1-mediated extension of ERK Thr phosphorylation upon serum starvation, whereas neither Bα nor Bβ could do so (Figure 3C, right panel). B56γ1 subunits also competed in a dose-dependent fashion with IEX-1's ability to promote ERK activation (Figure 3D).
Figure 3.
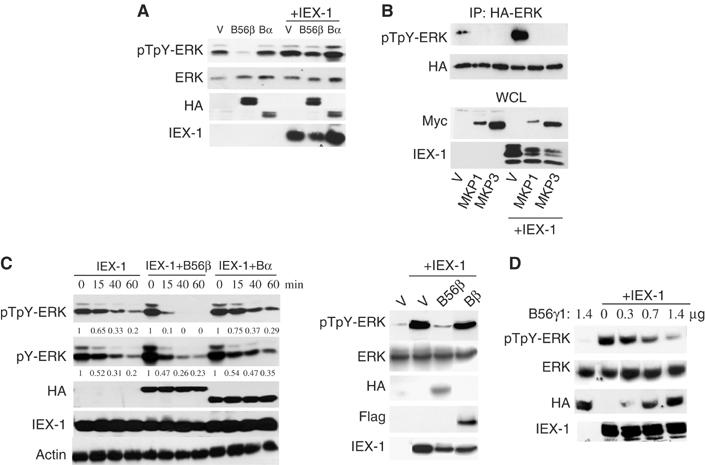
Competition between IEX-1 and B56-containing PP2A in ERK signaling. (A, B) IEX-1 reverses B56β- but not MKP-mediated ERK dephosphorylation. CHO cells were transfected with 1 μg of the various HA-B subunits (A) or with 0.7 μg HA-ERK and 0.7 μg Myc-MKP1/3 (B) in the presence of 1 μg of pcDNA (V) or His-IEX-1. ERK phosphorylation was detected 24 h following transfection in WCL (A) or anti-HA precipitates (B). (C, D) B56 but not B subunits compete with IEX-1 effect on ERK activation. (C) CHO-EpoR cells were transfected with 0.5 μg His-IEX-1 together with 0.5 μg of HA- or Flag-tagged B subunits. The cells were stimulated and starved as depicted in legend to Figure 1A. Quantification of pERK levels is shown relative to the levels of phosphorylation at zero time point; after normalization to actin levels. (D) ERK activation was assessed in CHO cells transfected with 0.4 μg of IEX-1 and the indicated amounts of HA-B56γ1 or empty vector.
IEX-1 interacts with B56 subunits and with pERK independently
To get insights into the mechanism by which IEX-1 affects B56 activity on ERK, we analyzed the interactions between B subunits and IEX-1. Figure 4A shows that IEX-1 co-precipitated with both B56β and γ1 subunits but not with Bα, in cells co-transfected with B subunits and IEX-1. Conversely, IEX-1 was found in the HA-B56β (Figure 4B) and B56γ (see below Figure 8C), but not in HA-Bα precipitates. Since antibodies to both B subunits and IEX-1 are poorly precipitating, the interaction between the two endogenous proteins could not be tested. However, anti-HA antibodies also co-precipitated specifically PMA-induced endogenous IEX-1 protein from HeLa stably expressing more limited amounts of HA-B56β (Figure 4C). Conversely, transfected IEX-1 was found to interact with the endogenous B56-PP2A holoenzymes (see below). This shows that the B56/IEX-1 interaction occurs also without massive overexpression of the two proteins, as it can be reached during transient transfections.
Figure 4.
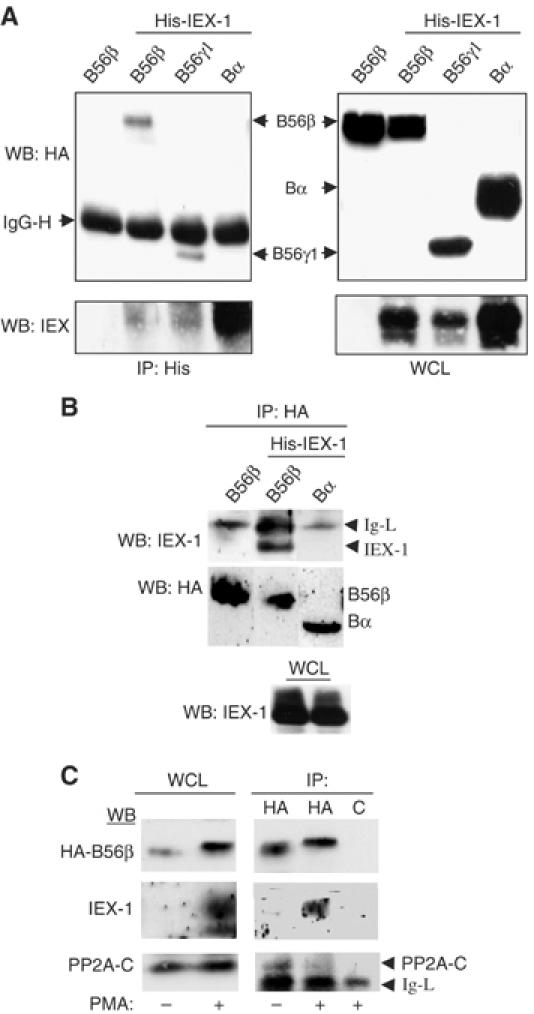
IEX-1 interacts with B56 but not with B subunits. (A, B) CHO cells were transfected with His-IEX-1 together with the indicated HA-B subunits. After 24 h, the lysates were immunoprecipitated with anti-His (A) or anti-HA (B) antibodies. (C) HeLa cells stably expressing HA-B56β were treated or not for 2 h with PMA to induce IEX-1 expression and the lysates were precipitated with either anti-HA or control (c) antibodies. The presence of B proteins and IEX-1 in the immunoprecipitates and WCL was detected by Western blotting.
Figure 8.

IEX-1 induces ERK-dependent dissociation of B56/A/C trimers. (A–C) IEX-1 displaces B56 from the PP2A-C subunit. CHO cells were transfected with 1 μg of HA-B56β (A, B) or HA-B56γ1 (C) and either 2 μg of pcDNA, His-IEX-1-Wt or His-IEX-1-ΔBD. The anti-HA (A, C) or anti-PP2A-C (B) precipitates and WCL were blotted with the indicated antibodies. (D) ERK-mediated phosphorylation of B56γ1 decreases its interaction with PP2A-A/C. Sepharose-bound GST-B56γ1 was phosphorylated (+ERK2-PP) or not in vitro with ERK2, and incubated for 2 h at 4°C with CHO cell lysates. The presence of endogenous PP2A-A and C subunits in the precipitates was assessed. Autoradiography indicates the phosphorylation of GST-B56γ1. (E, F) IEX-1 interacts with the PP2A-C subunit when ERK is not activated. CHO cells transfected with HA-B56β (D) or HA-IEX-1 (E) were starved or not (FCS) of serum for 4 h. The lysates were either precipitated with Sepharose bound-GST fusion proteins (D) or anti-HA antibodies.
IEX-1 binds to active pERK through a DEF-type docking domain (Garcia et al, 2002). IEX-1 mutated at this site (IEX-ΔBD) had lost the ability to bind to pERK but remained fully able to precipitate the B56β subunit in GST-pull-down (Figure 5A) or immunoprecipitation (Figure 5B) assays. This shows that the B56 subunits and pERK bind to different sites on IEX-1. While no pERK was detected in the anti-HA immunoprecipitates in cells transfected with HA-B56 alone, B56 was found associated with both IEX-1 and endogenous pERK in cells coexpressing IEX-1 (Figure 5C). This indicates that B56 binds to pERK through IEX-1, forming a pERK/IEX-1/B56 ternary complex in vivo.
Figure 5.
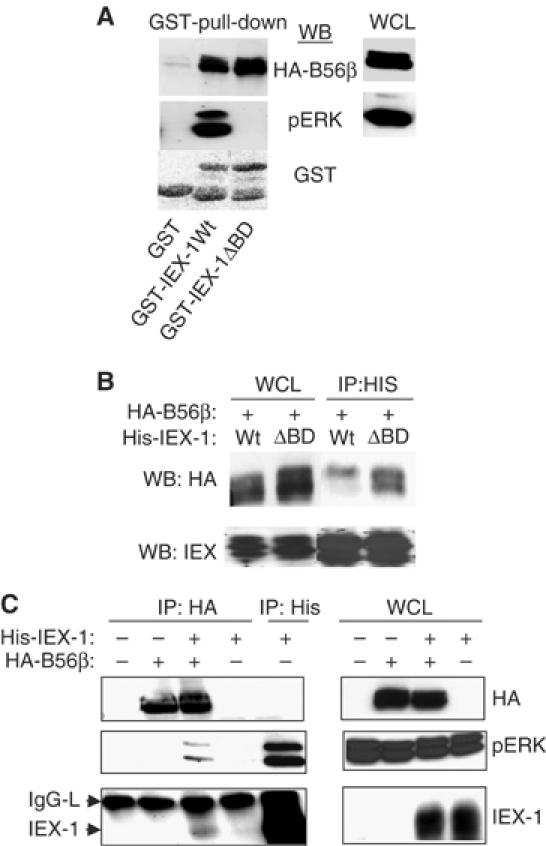
IEX-1 forms a ternary complex with B56 and perk. (A) Lysates from CHO cells transfected with HA-B56β were reacted with Sepharose-bound-GST, GST-IEX-1-Wt or GST-IEX-1-ΔBD for 1 h at 4°C and washed. The presence of pERK and B56β was analyzed. (B, C) CHO cells were transfected with His-IEX-1 (Wt or ΔBD) HA-B56β subunit alone or together. The indicated antibodies were used to blot anti-His (B, C) and anti-HA (C) immunoprecipitates and WCL.
IEX-1 increases ERK-dependent phosphorylation of B56 regulatory subunits
The ability of IEX-1 to enhance ERK activation is strictly dependent on IEX-1 binding to pERK, as the IEX-1-ΔBD mutant has lost this function (Garcia et al, 2002). The results above indicate that it also correlates with its binding to the B56 subunits. We therefore wished to determine the role played by pERK in the capacity of IEX-1 to reverse B56 effects.
HA-B56β was detected as a doublet in lysates from serum-grown cells (Figure 6A). The slower migrating band disappeared in cells treated with U0126 and, conversely, increased in cells expressing constitutively active MEK, indicating that it results from an ERK-dependent phosphorylation event. Interestingly, we noticed that although the slower migrating phosphorylated species was the far less abundant species detected in the whole-cell lysates, it was the only one found associated to IEX-1 in IEX-1 immunoprecipitates (Figure 5B). However, when the lysates from cells expressing B56β were incubated with Sepharose-bound GST-IEX-1 at 4°C, the two bands of B56 were found in the precipitate in a ratio similar to that found in total lysates (Figure 5A). Thus, IEX-1 seems to bind equally well to the phosphorylated and the unphosphorylated B56 species in vitro but, in vivo, IEX-1-bound B56β is shifted to the slower migrating form. This suggests that IEX-1 may favor phosphorylation of B56 by ERK in the pERK/IEX-1/B56 complex. In agreement with this, expression of IEX-1 induced a shift in mobility of both B56β and B56γ1 subunits (Figure 6B). To further assess the role of IEX-1-bound pERK in this event, we analyzed the effect of the IEX-1-ΔBD mutant on B56 migration. As shown in Figure 6B, no change in B56β migration was observed upon expression of IEX-1-ΔBD. In addition, contrasting with what we observed with IEX-1-Wt, the ratio between the phosphorylated versus the nonphosphorylated B56β species co-precipitated with IEX-1-ΔBD and in the whole-cell lysates are similar (Figure 5B). Thus, IEX-1-induced B56 mobility shift in vivo is dependent on the formation of a pERK/IEX-1/B56 complex, strongly suggesting that it results from ERK-mediated phosphorylation.
Figure 6.
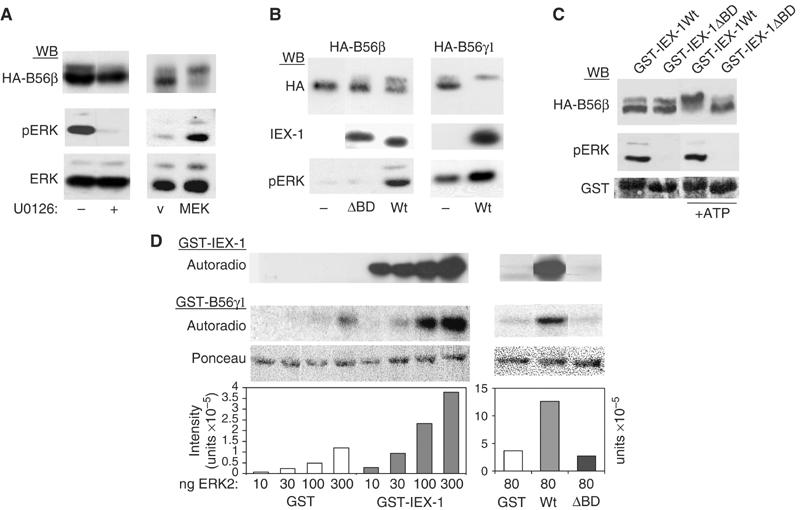
IEX-1 increases the phosphorylation of B56 subunits by ERK. (A, B) ERK-dependent phosphorylation of B56 subunits. CHO cells were transfected with HA-B56β or B56γ1, alone or together with either constitutively active MEK (A) or IEX-1 (B). The shift in B56 mobility and IEX-1 and pERK expressions were analyzed on 7 and 10% acrylamide SDS gels, respectively. (A) U0126 (10 μM) was added 40 min before cell lysis. (C) IEX-1-associated ERK induces B56β mobility shift. Lysates from CHO cells transfected with HA-B56β were reacted with Sepharose-bound GST-IEX-1-Wt or GST-IEX-1-ΔBD. After six washes, the beads were incubated at 30°C for 20 min in kinase buffer containing or not ATP before Western blotting with the indicated antibodies. (D) IEX-1 increases ERK activity towards the B56γ1 subunit in vitro. Then, 1 μg GST-B56γ1 was subjected to in vitro kinase assays with the indicated amounts of recombinant ERK2, in the presence of 1 μg of either GST, GST-IEX-1-Wt or GST-IEX-1-ΔBD. Autoradiographies of GST-B56 and GST-IEX-1 are shown. The plots represent quantification of the radioactivity incorporated into GST-B56γ. Ponceau staining shows equal loading of GST-B56γ1.
We then tested whether IEX-1-associated ERK is sufficient to induce a shift in mobility of B56 in the complex. For that, cells were transfected with B56β and the pERK/IEX-1/B56 complex was precipitated with Sepharose-bound GST-IEX-1 and incubated at 30°C in a kinase buffer containing or not ATP. As shown in Figure 6C, HA-B56β bound to GST-IEX-1 was completely shifted to the slower migrating form upon addition of ATP. No shift was observed when the reaction was performed with GST-IEX-1-ΔBD. Since no pERK was found in these complexes, this result shows that the retarded band of B56β is resulting from phosphorylation by IEX-1-associated ERK.
We then assess whether IEX-1 could augment ERK catalytic activity towards B56 in the complex. Thus, various levels of active recombinant ERK2 were preincubated with either purified GST or GST-IEX-1 proteins and then used to phosphorylate GST-B56γ1 in the presence of radio-labelled ATP. Contrasting with GST-IEX-1, which was readily phosphorylated at all ERK2 concentrations tested, GST-B56γ1 phosphorylation occurred only at the highest dose (Figure 6D). This phosphorylation was, however, greatly increased in the presence of GST-IEX-1. The ERK docking domain of IEX-1 is required for this effect as shown by the inability of GST-IEX-1-ΔBD to augment B56γ1 phosphorylation. Thus, only IEX-1-bound ERK has an increased activity towards B56γ1. Altogether, these results show that, by binding to both pERK and B56, IEX-1 enhances the ability of ERK to phosphorylate the B56 proteins in vivo and in vitro.
The similar action of IEX-1 on β and γ B56 members suggests that ERK may phosphorylate a common site in these proteins. Sequence analysis reveals two conserved ERK consensus Ser/Pro phosphorylation sites (positions 136 and 327 in B56γ1). To determine which site was phosphorylated in the B56/IEX-1/pERK complex, GST-B56γ1-S136A and B56γ1-S327A were used in an in vitro kinase assay in the presence or absence of IEX-1, as above. As shown in Figure 7A, the Ser327A mutation almost completely abolished both basal and IEX-1-increased B56γ1 phosphorylation, whereas the phosphorylation of the B56γ-S136A mutant remained similar to that of B56-Wt. This indicates that S327 is the major ERK phosphorylation site in B56γ1. In vivo, B56γ1-S327A, but not B56γ1-S136A, was much more efficient than B56γ1-Wt at reversing IEX-1 effect on ERK phosphorylation (Figure 7B). This was particularly striking at low levels of expression of the B56 subunits where the B56γ1-S327A mutant inhibited completely ERK activation even in the presence of IEX-1, while under these conditions neither B56-Wt or the S136A mutant had an effect (Figure 7B). Thus, IEX-1 cannot inhibit the phosphatase activity of a B56 species that is unresponsive to ERK phosphorylation. Altogether, these results indicate that ERK-mediated phosphorylation of the B56γ1, subunit at Ser327 negatively controls its activity on ERK and that IEX-1-induced increase in phosphorylation at this conserved S/P site is required for its ability to prevent ERK inactivation by B56 family of PP2A.
Figure 7.
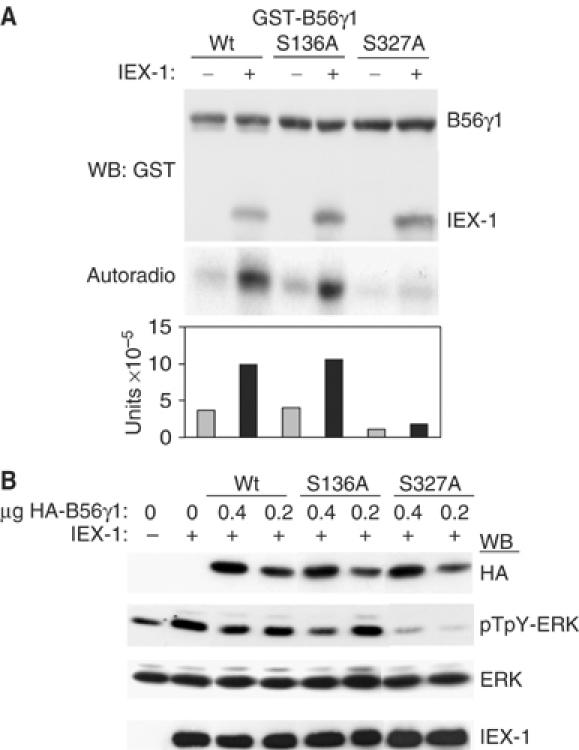
ERK-mediated phosphorylation of B56γ1 at Ser327 leads to inhibition of its activity towards ERK. (A) Identification of the ERK phosphorylation site in B56γ1. GST-B56γ1 Wt, S136A or S327A were subjected to in vitro kinase assays with 100 ng ERK, in the presence or absence of GST-IEX-1, as in legend to Figure 6D. The plot shows the radioactivity incorporated in B56γ1, normalized to the levels of the GST detected in the anti-GST blot. (B) Effect of B56γ1 mutants on ERK activation. CHO cells were tansfected with 0.4 μg of IEX-1 together with various amounts of the indicated B56γ1 forms and assayed for ERK phosphorylation.
IEX-1 induces the dissociation of B56 from the PP2A catalytic subunit in an ERK-dependent fashion
We next examined by which mechanism ERK-mediated phosphorylation of the B56 subunit leads to inhibition of the phosphatase activity. Active PP2A holoenzymes are formed by association of B subunits with A/C dimers. Therefore, we analyzed the association of B56 with PP2A-C in the presence of IEX-1. The levels of endogenous PP2A-C subunit co-precipitated with HA-B56β (Figures 4C and 8A) or B56γ1 (Figure 8C) were greatly reduced in cells expressing IEX-1. Antibodies to PP2A-C also precipitated much greater amounts of B56β in the absence than in the presence of IEX-1 (Figure 8B). IEX-1 was found in anti-B56 but not in anti-PP2A-C precipitates. Thus, the associations of B56 with C and with IEX-1 seem mutually exclusive, suggesting that IEX-1, by binding to the B56 subunit, induces the dissociation of the holoenzyme. The amount of PP2A-A detected in the anti-B56 immunoprecipitates in our hands was too small to analyze the displacement of this subunit by IEX-1. However, since PP2A-A and C form a dimeric core in vivo, our results suggest that IEX-1 triggers the dissociation of B56 from the AC dimer, providing a possible mechanism by which IEX-1 may inhibit B56-containing PP2A enzymes.
Expression of the IEX-1-ΔBD mutant did not change the B56γ1/C interaction, despite its efficient interaction with B56 (Figure 8C). This indicates that IEX-1-increased ERK-mediated phosphorylation of B56 is involved in the dissociation of PP2A-C from B56. Indeed, GST-B56γ1 phosphorylated in vitro by ERK2 has a decreased ability to bind both PP2A-C and A subunits (decrease by 61±8%, n=3, as compared to the nonphosphorylated protein, Figure 8D). In addition, we found that GST-IEX-1 could precipitate more endogenous PP2A-C from lysates of serum-starved than from serum-grown B56β-transfected cells (Figure 8E). More importantly, together with pERK, small amounts of endogenous PP2A-C were detected in IEX-1 precipitates from serum-starved cells but not from cells cultured in the presence of serum (Figure 8F). In this experiment, no B56 protein was transfected, showing that IEX-1 can associate not only with overexpressed B56 subunits but also with endogenous B56-PP2A holoenzymes, providing that there is no ERK activation.
Altogether, these results show that IEX-1 favors the dissociation of B56-PP2A by binding to B56-containing holoenzymes and allowing phosphorylation of the B56 subunit by the associated pERK.
Discussion
The importance of PP2A in the regulation of ERK signaling has long been recognized (Gause et al, 1993; Sontag et al, 1993; Alessi et al, 1995; Ory et al, 2003; Wang et al, 2003). However, neither the nature of the holoenzyme involved nor the mechanism controlling this activity has yet been elucidated. This is most probably due to the fact that PP2A acts on several kinases and has both positive and negative effects in this pathway, making difficult the analysis of its action on ERK itself. Here, by starting from the observation that the ERK substrate IEX-1 could inhibit specifically the dephosphorylation of ERK on Thr, we identified B56 regulatory subunit-containing PP2As as regulators of ERK dephosphorylation and underscored a mechanism by which their activity is controlled during mitogen stimulation. Indeed, a key finding of this study is that IEX-1-mediated inhibition of B56-PP2A activity is ERK-dependent and involves a new mode of ERK action by substrate transphosphorylation: by docking both active ERK and the B56 subunit, IEX-1 forms a ternary complex in which the ability of ERK to phosphorylate B56 is greatly increased. Phosphorylation occurs at a Ser-Pro site conserved in all B56 members; it triggers dissociation of B56 from the C subunit, thereby leading to inhibition of the phosphatase activity towards ERK. The importance of this regulation in ERK signaling is illustrated by the altered length of ERK activation occurring either by preventing IEX-1 induction or by downregulating B56 subunits expression.
Expression of B56-, but not of B-, family subunits decreased phosphorylation of ERK on Thr without affecting pY-ERK or pMEK levels. This indicates that B56 acts on ERK directly. Likewise, the activation of the other MAPKs, JNK and p38, remained unchanged in B56-expressing cells (data not shown). Thus, although ERKs are certainly not the only kinases targeted by B56 enzymes, as far as MAPK are concerned, they may have a restricted specificity on ERK1/2. There is a high homology between the different B56 members (McCright et al, 1996). ERK dephosphorylation is not linked to a specific B56 subunit since the same effect was observed with B56β, and the three γ isoforms. Likewise, downregulation of either B56γ or B56β expression increased basal ERK activation and prolonged ERK signal during stimulation, showing that these B56 members have redundant effects on ERK. How ERKs are dephosphorylated by B56-containing PP2As is not yet clear. B56 subunits might target the holoenzyme to ERK by interacting directly with the kinase. However, no ERK was detected in B56 immunoprecipitates in the absence of IEX-1. This could be due to the fact that the interaction is indirect or to the formation of unstable complexes. The reciprocal regulation of PP2A and ERK activities observed here makes this possibility likely.
Recently, conflicting data have been reported regarding the effect of B and/or B56 holoenzymes on ERK signaling. A negative regulatory role of B but not of B56 PP2A has been observed in Drosophila Schneider S2 cells (Silverstein et al, 2002). However, this effect might be accounted for by inactivation of Raf or MEK rather than of ERK. Species differences in the nature of the PP2A holoenzymes acting at different steps may also exist. Indeed, while PP2A acts as a negative regulator of Raf in Drosophila, it plays the opposite role in mammalian cells by dephosphorylating inhibitory residues on Raf-1 (Ory et al, 2003). A study just demonstrated the involvement of PP2A containing Bα and Bδ members in Raf-1 activation in HEK cells. No effect of these enzymes was found on ERK (Adams et al, 2005). Contrasting with both these results and ours, a recent report shows that knockdown of Bα or Bδ increases ERK activation in PC6-3 cells, while that of B56 has no effect (Van Kanegan et al, 2005). These discrepancies might be explained by experimental differences. Particularly, Van Kanegan et al did not monitor ERK phosphorylation directly but by measuring activation of its downstream target Elk1 which might be itself a substrate for PP2A. On the other hand, activation was analyzed in the presence of constitutively active MEK or Ras. This strong activation signal could blur a positive effect of B56 subunit knockdown on ERK. Alternatively, cell-type differences in the use of the different holoenzymes might exist.
The diversity of the PP2A regulatory subunits and their ability to target different substrates suggest specific regulations of the individual holoenzymes. The opposite effects of IEX-1 and B56β and γ subunits on ERK phosphorylation, either when overexpressed or downregulated, show that IEX-1 is a specific inhibitor of the action of these enzymes on ERK. This conclusion is also supported by the ability of B56β and γ, but not B, subunits to compete with IEX-1 for ERK phosphorylation specifically on Thr. The mechanism by which IEX-1 inhibits B56-containing PP2A is complex, involving the formation of a ternary complex between B56, IEX-1 and active ERK, the phosphorylation of B56 by ERK in this complex and the displacement of B56 from the A/C core enzyme. The fact that phosphorylation occurs on a Ser residue which is conserved in all B56 members, strongly suggests that IEX-1 is a general inhibitor of the B56 family of PP2A. Although other protein inhibitors of PP2A has been isolated (Li et al, 1995), IEX-1 is, to our knowledge, the first nonviral protein identified which inhibits a specific B subunit-containing PP2A. PP2A complexes composed of B56α, B56β and B56ɛ localize to the cytoplasm, whereas those composed of B56δ and B56γ are concentrated in the nucleus (McCright et al, 1996). By inhibiting different B56 members, IEX-1 will protect ERK from dephosphorylation in both compartments. This is consistent with the fact that IEX-1 is found in both the nucleus and the cytoplasm and that its capacity to control ERK activation is independent of pERK delocalization (Garcia et al, 2002).
A previous study had shown that the B56 subunits are phosphoproteins (McCright et al, 1996). Several kinases, including PKR, PKA and Chk2 (Usui et al, 1998; Xu and Williams, 2000; Dozier et al, 2004), have been reported to phosphorylate B56 members, leading to activation or change in the specificity of these enzymes. Our results with the MEK inhibitor indicate that, at least in serum growing cells, ERK is probably responsible for most of the B56 electrophoretic shift observed. This is also supported by the fact that only the wild-type IEX-1 but not the ΔBD mutant can greatly increase the appearance of the slower migrating B56 forms. ERK-mediated phosphorylation inhibits B56γ1-PP2A activity on ERK as shown by the increased capacity of the B56γ1-S327A mutant to induce ERK dephosphorylation. A previous report has shown that B56 subunits form trimeric holoenzymes independently of their phosphorylation states (McCright et al, 1996). By contrast, we found that ERK-phosphorylated GST-B56γ1 showed a decreased binding to A and C subunits and that IEX-1-induced B56/C dissociation is ERK-dependent. The reasons for these differences are not clear. However, it is possible that, in addition to increasing the phosphorylation of B56 by ERK, IEX-1 may help in the dissociation of the B56/A/C trimers.
ERKs can be targeted to their substrates through two conserved docking sites: the D-domain, a stretch of positively charged amino acids; or the DEF motif, defined by the conserved FXF consensus sequence (Jacobs et al, 1999). This docking system suggests a competition between substrates for phosphorylation by ERK. We show here that this does not seem to be always the rule. Indeed, we found that IEX-1, while itself heavily phosphorylated by ERK, greatly enhances the phosphorylation of B56 subunits, not only in vivo but also in vitro when only the three recombinant proteins are present. Thus, increased B56 phosphorylation is not resulting from unmasking ERK phosphorylation sites on B56 which would have been hidden by the A/C core enzyme. Rather, this demonstrates that, in the presence of IEX-1, ERK has an increased ability to phosphorylate PP2A B56 subunits.
Docking domains affect the velocity of ERK phosphorylation and the residues that are phosphorylated. Their presence determines whether a protein is a good ERK substrate or not (Jacobs et al, 1999). In the case of IEX-1, association of ERK to the DEF motif is absolutely required for its phosphorylation in vivo as well as in vitro (Figure 6; Garcia et al, 2002). The model of recognition of the DEF motif by ERK2 predicts that, because of steric hindrance, ERK bound to a DEF site should be able to phosphorylate only Ser/Thr located within 10 residues from this domain (Lee et al, 2004). Sequence analysis of B56 members revealed no D-domain. Although a DEF domain is present in the N-terminus part of B56γ1, its position is not within distance compatible with phosphorylation at Ser327. This probably explains why B56 subunits are poor ERK substrates. IEX-1, by interacting with both B56 and active ERK, may put this site and the kinase in a favorable conformation, providing the missing ERK-docking site on B56 subunits required for making them reasonably good ERK substrates. The inability of the IEX-1-ΔBD to enhance B56 phosphorylation supports this hypothesis. Whether IEX-1 increases ERK phosphorylation of other associated proteins and whether this mechanism of transphosphorylation between ERK substrates is specific for the IEX-1 DEF domain, is currently unknown.
PP2A phosphatases are constitutively active. Inhibition of MEK activation leads to a rapid arrest in ERK signal due to a decreased phosphorylation on Thr. Likewise, the pT-ERK levels decay more rapidly than pY-ERK levels during mitogen stimulation. This confirms previous studies showing that dephosphorylation of ERK on Thr is rate limiting (Alessi et al, 1995) and suggests that inhibition of PP2A activity is an important step in the control of ERK signaling. Our study provides a mechanism by which this can be done. Although B56 are poor ERK substrates, we observed a small ERK-dependent shift in mobility for both β and γ subunits upon serum stimulation. In addition, recombinant phosphorylated B56 subunits have a decreased ability to associate with A and C subunits in vitro. This suggests a model (Figure 9) in which, at early time points following mitogen stimulation, the levels of ERK activation probably overcome that of B56-PP2A activity and/or are sufficient to induce its phosphorylation and inhibition, allowing ERK signaling to proceed. At later times however, the ratio between ERK and B56-PP2A activities is shifted in favor of the phosphatase, leading eventually to extinction of the signal. When IEX-1 is induced, phosphorylation of B56 subunits is greatly enhanced at low levels of active ERK, leading to sustained inhibition of PP2A and prolongation of ERK Thr phosphorylation. Prevention of IEX-1 induction by using shRNA reduced the duration of ERK activation (Figure 1; Garcia et al, 2002). This indicates that the levels of endogenous IEX-1 protein induced are sufficient to counteract B56-PP2A actions on ERK. The high ERK activity observed in IEX-1-expressing cells even at low doses of growth factors or under apoptotic conditions (Garcia et al, 2002), that is, when ERK upstream activation is low, also supports this model.
Figure 9.

A model for the control of ERK signal by PP2A and IEX-1. (A) Early time points of growth factor stimulation. The high ERK activation by upstream kinases overcomes the activity of B56-PP2A and the signal proceeds. (B, C) Late time points of growth factor stimulation. Upstream activation of ERK is low and B56-PP2A activity overcomes that of ERK, leading signal arrest. In cells where IEX-1 is induced (C), inhibition of B56-PP2A leads to sustained ERK signal (see text for details).
IEX-1 expression is high in many tumor cells (Wu, 2003), and forcing its expression can lead to tumor development (Zhang et al, 2003). Its ability to inhibit the activity of B56-containing PP2A most probably accounts for these effects. Recent studies have shown that B56-PP2A contribute to cancer development. Indeed, decreased B56γ expression or activity are involved in SV40 small t tumoral activities (Chen et al, 2004) and melanoma metastasis progression (Ito et al, 2000). In addition, two cancer-associated PP2A-A mutants are specifically defective in B56 subunit binding (Ruediger et al, 2001). Although the tumor suppressor function of B56 enzymes may be related to direct interactions with the tumor suppressor APC (Seeling et al, 1999), their ability to negatively regulate ERK phosphorylation demonstrated here is also likely to play an important role in this process. The elucidation of the signaling pathways regulated by B56-PP2A and the mechanisms affecting their activity will help the discovery of new drugs to block tumor progression.
Materials and methods
Plasmids and antibodies
Plasmids encoding IEX-1 were described (Garcia et al, 2002). Expression vectors for 4xHA-B56β, HA-Bα, HA-B56γ1, HA-B56γ2, GST-B56γ1, Flag-Bβ, Flag-Bγ, Myc-MKP1 and Myc-MKP3 were gifts of Drs D Virshup (University of Utah), E Ogris (Institute of Medical Biochemistry, Vienna), N Nojima (Osaka university), E Cohen (University of Montreal) and S Keyse (Ninewells Hospital and Medical School, Dundee), respectively. pFC-MEK218E/S222E was from Stratagene. B56γ1 phosphorylation mutants were generated using the QuickChange site-directed mutagenesis kit (Stratagene). Recombinant active His6-ERK2 was purified as described (Garcia et al, 2002). The antibodies used in this study were anti-phospho-specific antibodies recognizing pY-ERK (SC-7976, Santa Cruz), pT-ERK (Promega), pTpY-ERK and pMEK (Cell Signaling Technology); Anti-ERK1 and anti-B56β (Santa Cruz), anti-PP2A-C (clone ID6, Upstate Biotechnology), anti-His (Covance); anti-HA (Roche); rabbit anti-pan B56γ was a generous gift of Dr Mumby (University of Texas); anti-IEX-1 antiserum was previously described (Garcia et al, 2002).
RNA interference
The target sequences for IEX-1 (nucleotides 250–270, accession number AF083421) and GFP were described previously (Garcia et al, 2002). Vectors that drive the expression of shRNA were generated by cloning the above nucleotides, followed by a 6 bp loop and the corresponding antisense sequence and by five thymidines, in a pcDNA vector in which the CMV promoter was replaced by the H1 promoter (pcDNA-H1). Two B56γ shRNA were used: nucleotide sequence 238–258 (Chen et al, 2005) was cloned in the pcDNA-H1; pMKO-1 expressing ShRNA B56γ nucleotide sequence 423–442 was a gift of Dr W Hahn (Dana Farber Cancer Institute, Boston (Chen et al, 2004)). pSM2c vectors expressing B56β shRNA (NM006244, nucleotides 975–994 and 1716–1735) were from Open Biosystems.
Cell culture, transfections and transfection-based assays
CHO expressing the Epo receptor (CHO-EpoR) cells (Garcia et al, 2002) were cultured in Dulbecco's MEM nut Mix F-12 (HAM) medium containing 7% fetal calf serum (FCS). NIH-3T3 and HeLa cells were maintained in Dulbecco's MEM supplemented with 10% FCS. To generate stable clones of HeLa cells, 3 × 105 cells were transfected with 2 μg of vectors expressing IEX-1-shRNA, B56shRNA or 4 × HA-B56β and 48 h later, the cells were selected either in G418 (1 mg/ml), puromycin (0.5 μg/ml) or hygromycin (200 μg/ml), respectively. Transient transfections were performed with lipofectAMINE plus reagent (Life Technologies). At 24 h post transfection, the cells were incubated for various times in the presence or absence of U0126 (Promega) and/or OA (Calbiochem). To follow the kinetics of ERK inactivation, the cells were washed three times in PBS and incubated for various times at 37°C in serum-free medium. In some experiments, 24 h post transfection, cells were deprived of serum overnight, and stimulated with Epo (Boeringher Mannheim), PMA or (Sigma) before lysis.
Immunoprecipitation and Western blotting
Transfected cells were harvested 24 h after transfection and lysed in Tris 50 mM pH 7.5 buffer containing 137 mM NaCl, 0.5% NP-40, 10% glycerol, 1 mM EDTA, 1 mM orthovanadate, 20 mM β-glycerophosphate, 20 mM NaF, 1 mM sodium pyrophosphate and a protease inhibitor cocktail (Roche). Clear lysates were obtained by centrifugation. Immunoprecipitation, gel electrophoresis and immunoblotting were performed as described (Garcia et al, 2002).
GST phosphorylation and pull-down assays
Purified GST-B56γ1 (1 μg) was incubated with various amounts of active ERK2 in the presence or in the absence 1 μg of purified GST-IEX-1-Wt or IEX-1-ΔBD, in 50 μl kinase buffer (Hepes 10 mM, pH 7.5, 10 mM MgCl2 and 1 mM DTT) containing 50 μM ATP and 3 μCi of [γ32P]ATP, for 20 min at 30°C. The products were resolved on polyacrylamide gels and transferred to nitrocellulose membranes. Radioactive bands were visualized by autoradiography and quantified by phosphoimaging. To test if phosphorylation of B56 affects its capacity to bind to the A/C core enzyme, in vitro phosphorylated or untreated GST-B56γ1 bound to Sepharose was incubated with lysates from CHO cells for 2 h at 4°C. After six washes in lysis buffer, samples were analyzed by Western blotting with anti-PP2A-C and anti-PP2A-A antibodies. Pull-down with GST-IEX-1 Wt or mutant proteins were performed as described (Garcia et al, 2002). In some experiments, after six washes, the beads were incubated for 20 min at 30°C in 50 μl of kinase buffer containing 200 μM ATP. After three washes, the samples were analyzed by Western blotting.
Acknowledgments
We are grateful to Drs D Virshup, E Ogris, N Nojima, W Hahn, E Cohen, S Keyse and M Mumby, for the gifts of plasmids and antibodies. This work was supported by the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique and by grants from the Ligue Contre le Cancer, comité Ile de France, and the Association pour la Recherche contre le Cancer.
References
- Adams DG, Coffee RL Jr, Zhang H, Pelech S, Strack S, Wadzinski BE (2005) Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J Biol Chem 280: 42644–42654 [DOI] [PubMed] [Google Scholar]
- Alessi DR, Gomez N, Moorhead G, Lewis T, Keyse SM, Cohen P (1995) Inactivation of p42 MAP kinase by protein phosphatase 2A and a protein tyrosine phosphatase, but not CL100, in various cell lines. Curr Biol 5: 283–295 [DOI] [PubMed] [Google Scholar]
- Brondello JM, Pouyssegur J, McKenzie FR (1999) Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 286: 2514–2517 [DOI] [PubMed] [Google Scholar]
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S (1998) Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 280: 1262–1265 [DOI] [PubMed] [Google Scholar]
- Chen J, St-Germain JR, Li Q (2005) B56 regulatory subunit of protein phosphatase 2A mediates valproic acid-induced p300 degradation. Mol Cell Biol 25: 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC (2004) Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5: 127–136 [DOI] [PubMed] [Google Scholar]
- Dozier C, Bonyadi M, Baricault L, Tonasso L, Darbon JM (2004) Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B' regulatory subunit. Biol Cell 96: 509–517 [DOI] [PubMed] [Google Scholar]
- Favre B, Turowski P, Hemmings BA (1997) Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J Biol Chem 272: 13856–13863 [DOI] [PubMed] [Google Scholar]
- Garcia J, Ye Y, Arranz V, Letourneux C, Pezeron G, Porteu F (2002) IEX-1: a new ERK substrate involved in both ERK survival activity and ERK activation. EMBO J 21: 5151–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gause KC, Homma MK, Licciardi KA, Seger R, Ahn NG, Peterson MJ, Krebs EG, Meier KE (1993) Effects of phorbol ester on mitogen-activated protein kinase kinase activity in wild-type and phorbol ester-resistant EL4 thymoma cells. J Biol Chem 268: 16124–16129 [PubMed] [Google Scholar]
- Ito A, Kataoka TR, Watanabe M, Nishiyama K, Mazaki Y, Sabe H, Kitamura Y, Nojima H (2000) A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. EMBO J 19: 562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13: 163–175 [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J (2001) Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353: 417–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyse SM (2000) Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12: 186–192 [DOI] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell 14: 43–55 [DOI] [PubMed] [Google Scholar]
- Li M, Guo H, Damuni Z (1995) Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34: 1988–1996 [DOI] [PubMed] [Google Scholar]
- Lin YW, Chuang SM, Yang JL (2003) ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin–proteasome pathway. J Biol Chem 278: 21534–21541 [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185 [DOI] [PubMed] [Google Scholar]
- McCright B, Rivers AM, Audlin S, Virshup DM (1996) The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem 271: 22081–22089 [DOI] [PubMed] [Google Scholar]
- Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4: 556–564 [DOI] [PubMed] [Google Scholar]
- Ogris E, Gibson DM, Pallas DC (1997) Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene 15: 911–917 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Li H, Jensen MR, Zhang T, Taya Y, Thorgeirsson SS, Prives C (2002) Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell 9: 761–771 [DOI] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK (2003) Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 13: 1356–1364 [DOI] [PubMed] [Google Scholar]
- Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM (1990) Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell 60: 167–176 [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22: 153–183 [DOI] [PubMed] [Google Scholar]
- Pulido R, Zuniga A, Ullrich A (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J 17: 7337–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger R, Fields K, Walter G (1999) Binding specificity of protein phosphatase 2A core enzyme for regulatory B subunits and T antigens. J Virol 73: 839–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger R, Pham HT, Walter G (2001) Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene 20: 10–15 [DOI] [PubMed] [Google Scholar]
- Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM (1999) Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science 283: 2089–2091 [DOI] [PubMed] [Google Scholar]
- Silverstein AM, Barrow CA, Davis AJ, Mumby MC (2002) Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci USA 99: 4221–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M (1993) The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75: 887–897 [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC (1995) A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol 128: 1131–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui H, Inoue R, Tanabe O, Nishito Y, Shimizu M, Hayashi H, Kagamiyama H, Takeda M (1998) Activation of protein phosphatase 2A by cAMP-dependent protein kinase-catalyzed phosphorylation of the 74-kDa B′′ (delta) regulatory subunit in vitro and identification of the phosphorylation sites. FEBS Lett 430: 312–316 [DOI] [PubMed] [Google Scholar]
- Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S (2005) Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem 280: 36029–36036 [DOI] [PubMed] [Google Scholar]
- Wang PY, Liu P, Weng J, Sontag E, Anderson RG (2003) A cholesterol-regulated PP2A/HePTP complex with dual specificity ERK1/2 phosphatase activity. EMBO J 22: 2658–2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MX (2003) Roles of the stress-induced gene IEX-1 in regulation of cell death and oncogenesis. Apoptosis 8: 11–18 [DOI] [PubMed] [Google Scholar]
- Xu Z, Williams BR (2000) The B56alpha regulatory subunit of protein phosphatase 2A is a target for regulation by double-stranded RNA-dependent protein kinase PKR. Mol Cell Biol 20: 5285–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Finegold MJ, Porteu F, Kanteti P, Wu MX (2003) Development of T-cell lymphomas in Emu-IEX-1 mice. Oncogene 22: 6845–6851 [DOI] [PubMed] [Google Scholar]