

Abstract
Islet-brain 1 (IB1 or JIP-1) is a scaffold protein that interacts with components of the c-Jun N-terminal kinase (JNK) signal-transduction pathway. IB1 is expressed at high levels in neurons and in pancreatic β-cells, where it controls expression of several insulin-secretory components and secretion. IB1 has been shown to homodimerize, but neither the molecular mechanisms nor the function of dimerization have yet been characterized. Here, we show that IB1 homodimerizes through a novel and unique set of Src homology 3 (SH3)–SH3 interactions. X-ray crystallography studies show that the dimer interface covers a region usually engaged in PxxP-mediated ligand recognition, even though the IB1 SH3 domain lacks this motif. The highly stable IB1 homodimer can be significantly destabilized in vitro by three individual point mutations directed against key residues involved in dimerization. Each mutation reduces IB1-dependent basal JNK activity in 293T cells. Impaired dimerization also results in a reduction in glucose transporter type 2 expression and in glucose-dependent insulin secretion in pancreatic β-cells. Taken together, these results indicate that IB1 homodimerization through its SH3 domain has pleiotropic effects including regulation of the insulin secretion process.
Keywords: GLUT2, insulin secretion, JIP-1, MAPK pathway, scaffold protein
Introduction
Islet-brain 1 (IB1), also known as c-Jun N-terminal kinase (JNK)-interacting protein 1 (JIP1), is a scaffold protein that participates in the organization of the JNK signaling pathway. IB1 is highly expressed in neurons (Yasuda et al, 1999) and in pancreatic β-cells (Bonny et al, 1998), where it has been implicated in cell survival (Bonny et al, 2000), in regulating expression of the glucose transporter type 2 (GLUT2) (Bonny et al, 1998) and in glucose-induced insulin secretion (Waeber et al, 2000). IB1 is a highly conserved protein of 707, 711 and 715 residues in mouse, human and rat, respectively, which contains several protein–protein interaction domains, including a JNK-binding domain (JBD), a Src homology 3 domain (SH3) and a phosphotyrosine interaction domain (PID) (Figure 1).
Figure 1.
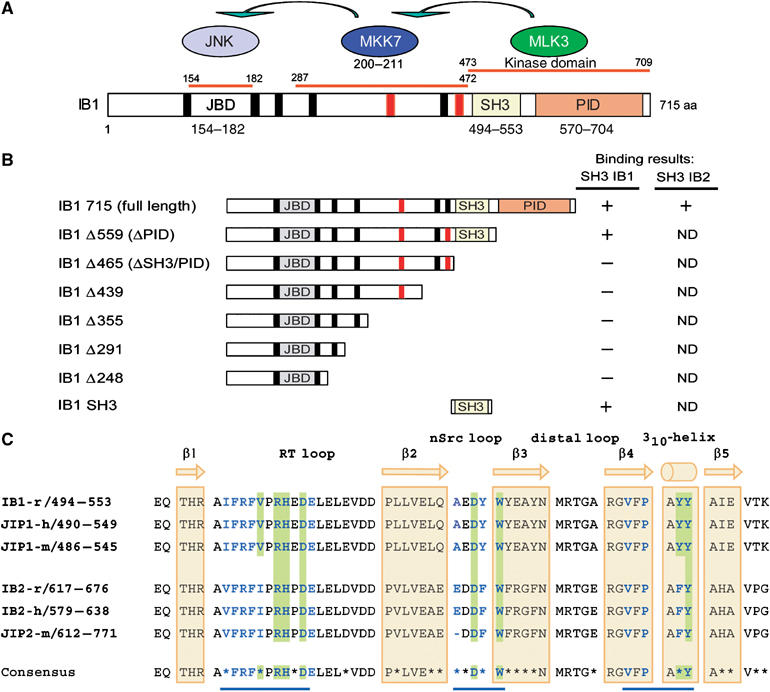
Protein interaction domains of IB1. (A) IB1 is characterized by a JBD (gray box, residues 154–182), SH3 (yellow box, residues 494–553) and a PID (orange box, residues 570–704). Residues are numbered according to the full-length rat IB1 sequence (GenBank, AF108959). The seven PxxP motifs are shown in black and red. Motifs marked in red are conserved in rat, human and mouse IB1 and IB2. MKK7 and MLK3, two of the known partners of IB1, bind to regions 287–472 and 473–709 of IB1, respectively. (B) Schematic representations of full-length and C-terminal deletion mutants of IB1. Numbering corresponds to the last amino-acid expressed in the various constructs. Binding results described in Figure 2 are summarized for all constructs. ND: not determined. (C) Sequence alignment of rat, mouse and human IB1/JIP1 and IB2/JIP2 SH3 domains. The sequence of rat IB1 (GenBank, AF108959), human JIP1 (Ensembl, ENSP00000241014), mouse JIP1 (Ensembl, ENSMUSP00000050773), rat IB2 (Ensembl, ENSRNOP00000050155), human IB2 (GenBank, AF218778) and mouse JIP2 (Ensembl, ENSMUSP00000023291) are included. The IB1 SH3 region is identical in all three species. Residues that participate to IB1 dimerization as well as those expected to do so in IB2 are shown in bold blue. Residues at the dimer interface involved in inter-protomer salt bridges or hydrogen bonds are shaded in green. Nonconserved amino acids between IB1 and IB2 are indicated with a star in the consensus sequence. The positions of the β strands 1–5 and the 310-helix are indicated in yellow.
Proteins that have been shown to associate with IB1 include the MAP kinase kinase kinase MLK3 (Whitmarsh et al, 1998; Mooney and Whitmarsh, 2004), the MAP kinase kinase MKK7 (Yasuda et al, 1999; Kelkar et al, 2000; Whitmarsh et al, 2001; Mooney and Whitmarsh, 2004), the MAPK phosphatase-7 (MKP7) (Willoughby et al, 2003), as well as several other ligands including kinesin light chain (Verhey et al, 2001). All these factors, except MLK3 and MKK7, have been shown to interact with the PID domain or the extreme C-terminal part (kinesin light chain) of IB1. However, although SH3 domains are important elements of signal transduction, no protein that interacts specifically with the SH3 domain of IB1 has been identified so far.
SH3 domains are central in the assembly of macromolecular complexes involved in many intracellular signaling pathways, where they mediate different functional roles. For example, the SH3 domains of members of the tyrosine kinase family (Src, c-Abl, Bcr-Abl) and of the serine/threonine kinase MLK3 autoinhibit their kinase activities (Nguyen and Lim, 1997; Brasher et al, 2001; Zhang and Gallo, 2001; Smith et al, 2003). SH3 domains are usually characterized by their preferred recognition of and association with canonical PxxP motifs (Kay et al, 2000). Recently, it has been shown that several SH3 domains recognize non-PxxP sequences. This is the case for the SH3 domains of Eps8 (recognizing the sequence PxxDY; Mongiovi et al, 1999), Gads (RxxK; Harkiolaki et al, 2003) or Fus1 (Arg-Ser-rich sequences; Tong et al, 2002) that interact with sequences only distantly or not related to the PxxP motif. In all these cases, a single linear sequence is sufficient for binding to the SH3 domain.
IB1 and another member of the family, IB2, have previously been shown to engage in oligomerization through their respective C-terminal regions that include the SH3 and PID domains (Yasuda et al, 1999; Nihalani et al, 2001). Here, we show that homo- and heterodimerization of IB1 and IB2 occur through a direct SH3–SH3 domain interaction. We report crystal structures of the domains, providing detailed insights into the first example of a direct SH3 domain homodimerization. Based on this information, we introduced individual point mutations directed against key residues involved in dimerization. Each of these mutations significantly destabilizes dimerization, thus allowing us to study the functional consequences of IB1 homodimerization.
Results
IB1 oligomerization is mediated by its own SH3 domain
The contribution of the SH3 domain to IB1 oligomerization was investigated by in vitro pull-down experiments using chimeric GST-SH3 protein constructs. The highly homologous SH3 domain of IB2 was produced as well, together with two unrelated SH3 domains that were used as specificity controls: the SH3 domain of the GTPase-activated protein (GAP) and the C-terminal SH3 domain of the growth factor receptor-bound protein 2 (Grb2).
35S-radiolabeled full-length IB1 was found to bind to the SH3 domains of IB1 and to a weaker extent to that of IB2, but neither to GST alone nor to the GAP or Grb2 SH3 domains (Figure 2A). MLK3 and MKK7, as potential partners binding to the large C-terminal part of IB1 (Kelkar et al, 2000), were assayed in the same pull-down experiments (Figure 2B). None of these proteins was able to bind to the IB1 SH3 domain. As expected, MKK4, which is an activator of JNK that does not bind to IB1 (Whitmarsh et al, 1998; Yasuda et al, 1999), was found not to interact with IB1.
Figure 2.

Pull-down experiments. (A) Involvement of the SH3 domain of IB1 in dimerization. Autoradiogram of separated 35S-labeled (*) full-length IB1 pull-down products observed using the GST-fused SH3 domains of IB1, IB2, Grb2 and GAP or GST alone (upper panel). IB1 specifically recognizes the SH3 domain of IB1, but not the SH3 domains of GAP or Grb2. As expected, we observe crossbinding with the SH3 domain of IB2. (Lower panel) Sample loading control, Coomassie-stained gel. (B) Interaction of the IB1 SH3 domain with known IB1 partners. SDS–PAGE autoradiogram analysis of pull-down products obtained using GST-fused IB1 SH3 and 35S-labeled (*) MLK3, MKK7 and MKK4 (upper panel). The SH3 domain of IB1 binds neither to MLK3 and MKK7 nor to the negative control MKK4. (Lower panel) Sample loading control, Coomassie-stained gel. (C) Mapping of IB1 sequences responsible for SH3 binding. Pull-down experiments were performed with GST-IB1 SH3 and a series of 35S-labeled (*) truncated C-terminal fragments (see Figure 1B). Autoradiograms (upper panels) and Coomassie staining (lower panels) of the SDS–PAGE gels are shown. Binding is lost when the C-terminal region that includes the SH3 domain of IB1 is deleted (ΔSH3/PID). (D) Stability of the IB1 SH3 dimer. Pull-down experiments were performed with GST-IB1 SH3 and 35S-labeled (*) SH3 proteins. The washed samples were incubated at increasing temperatures (4–67°C) and analyzed by SDS–PAGE, followed by autoradiography (top panel) of the Coomassie-stained gels (bottom panel). The complex is extremely stable, with a small fraction of dimers dissociating at 52°C and above. (E) Stability of the full-length IB1 dimer. Immunoprecipitation (IP) of Flag-IB1 was performed after transfection of 293T cells with Flag or Flag-IB1 together with GFP-IB1 constructs. The washed samples were incubated at increasing temperatures and analyzed by SDS–PAGE. Cosedimented GFP-IB1 was detected by GFP immunoblotting. The amount of immunoprecipitated proteins was verified with anti-Flag antibodies. The dimer is extremely stable, with binding still detectable at 80°C.
IB1 self-association occurs via homophilic SH3–SH3 interactions
To define the IB1 sequence(s) recognized by its own SH3 domain and to determine the potential role of PxxP motifs, 35S-radiolabeled full-length or C-terminal-truncated IB1 proteins (Figure 1B) was used in a pull-down assay using GST-IB1 SH3 as a bait. As shown in Figure 2C, full-length IB1, as well as PID-truncated IB1 (ΔPID), was able to bind to GST-IB1 SH3 (middle panel) but not to GST alone (left panel). In contrast, the ΔSH3/PID and shorter constructs failed to bind to GST-IB1 SH3. These results show that homophilic interactions occur between the SH3 domains of each IB1 molecule. The homophilic dimerization of IB1 via its SH3 domains was further substantiated by pull-down experiments using the GST-IB1 SH3 construct and 35S-radiolabeled SH3. As shown in Figure 2D, the SH3 domain of IB1 was found to bind to its own SH3, but not to GST alone. In this pull-down assay, the thermal stability of the SH3–SH3 complex was investigated. The SH3 dimer appeared extremely stable, with only a small fraction of dimers dissociating at 52°C and above. Gel filtration and dynamic light-scattering experiments confirmed a dimeric state in solution (see Supplementary data). The stability of the full-length IB1 dimer was further investigated by immunoprecipitation experiments in 293T cells co-transfected with either Flag or Flag-tagged IB1 (Flag-IB1) and green fluorescent protein (GFP)-tagged IB1 (GFP-IB1). As shown in Figure 2E, IB1 was found to homodimerize. The dimer was extremely stable, with binding still detectable at 80°C.
Structural insights to IB1 homodimerization
A total of three crystal forms of the IB1 SH3 domain were included in this study, see Table I. The structure from the type I crystals is shown in Figure 3A. The structures of the SH3 domain and of the SH3 homodimer obtained from the different crystal forms are identical and the type I structure is used as a reference throughout this paper. The molecule comprises a total of 62 residues, of which the two N-terminal amino acids are reminiscences of the expression vector. The fold is as expected from many previous structural investigations (Mayer, 2001). The structure is composed of five β-strands arranged into two antiparallel sheets. The first sheet is formed by strands 1 and 5 and the second sheet by strands 2, 3 and 4. β-Strands 1 and 2 are separated by the long RT-Src loop, strands 2 and 3 by the N-Src loop and strands 3 and 4 by the distal loop. The three residues connecting β-strands 4 and 5 form a 310-helix. Taken together, the two β-sheets form a barrel.
Table 1.
Data reduction, phasing and refinement statistics
| Type I, IB1 SH3-S, SeMet | Type II, IB1 SH3-S, SeMet | Type III, IB1 SH3-L | |
|---|---|---|---|
| Data collection | |||
| X-ray source, wavelength (Å) | BL711/MaxLab, 0.967 | X11/EMBL-Hamburg, 0.811 | ID29/ESRF, 0.979 |
| Space group | P42212 | P21212 | P212121 |
| Cell dimensions (Å) | a=b=114.5, c=48.4 | a=75.6, b=81.6, c=89.7 | a=45.9, b=57.0, c=145.5 |
| Monomer per asymmetric unit | 4 | 8 | 4 |
| Resolution range (Å) | 25–2.05 (2.16–2.05)a | 20–1.75 (1.84–1.75) | 30–3.0 (3.19–3.00) |
| Unique reflections | 37 925 (5354) | 54 422 (7198) | 8075 (782) |
| Average multiplicity | 8.0 (8.2) | 4.5 (3.7) | 4.6 (4.2) |
| Completeness (%) | 98.3 (97.4) | 96.2 (88.5) | 99.2 (100.0) |
| 〈I/σI〉 | 7.7 (2.3) | 20.8 (2.6) | 12.1 (2.9) |
| Rmergeb (%) | 9.6 (34.0) | 5.6 (32.4) | 10.2 (46.1) |
| Phasing | |||
| Heavy atom sites | 4 | ||
| Resolution range (Å) | 40–2.65 | ||
| Figure of merit (acentric/centric) | 0.34/0.11 | ||
| Phasing power (anomalous)c | 1.17 | ||
| Rcullisd (iso/ano) | 0.79 | ||
| Refinement | |||
| Rwork/Rfreee (%) | 18.4/22.8 | 20.4/23.5 | 22.4/25.1 |
| Number of atoms | |||
| Protein | 2100 | 4181 | 511 |
| Water | 249 | 597 | 0 |
| Hetero | 33 | 95 | 0 |
| Average B-factor (Å2) | |||
| Protein | 19 | 21 | 51 |
| Water | 29 | 34 | — |
| Hetero | 48 | 56 | — |
| R.m.s. deviation | |||
| Bond lengths (Å) | 0.006 | 0.005 | 0.007 |
| Angles (deg) | 1.4 | 1.4 | 1.4 |
| Ramachandran plotsf | |||
| Most favored regions (%) | 88.2 | 89.9 | 84.9 |
| Additional allowed regions (%) | 11.4 | 9.8 | 15.1 |
| aValues in parentheses refer to the highest resolution bin. | |||
| bRmerge=∑hkl(∑i(∣Ihkl, i−〈Ihkl〉∣))/∑hkl, i〈Ihkl〉, where Ihkl, i is the intensity of an individual measurement of the reflection with Miller indicies h, k and l, and 〈Ihkl〉 is the mean intensity of that reflection. | |||
| cPhasing power=∑hklFH, hkl/∑hkl∣FPH, obs, hkl−FPH, calc, hkl∣. | |||
| dRcullis=∑hkl∣∣FPH, hkl±FP, hkl∣−FH, calc, hkl∣/∑hkl∣FPH, hkl−FP, hkl∣, where FPH is the structure factor of the heavy atom derivative, FP is the structure factor of the native protein and FH, calc is the calculated structure factor for the heavy atom. | |||
| eRwork=∑hkl(∣∣Fobs, hkl∣−∣Fcalc, hkl∣∣)/∣Fobs, hkl∣, where ∣Fobs, hkl∣ and ∣Fcalc, hkl∣ are the observed and calculated structure factor amplitudes. Rfree is equivalent to the Rwork, but calculated with 5.7% (Type I), 2.1% (Type II) and 7.4% (Type III) of the reflections omitted from the refinement process. | |||
| fValues from PROCHECK (CCP4, 1994). | |||
Figure 3.
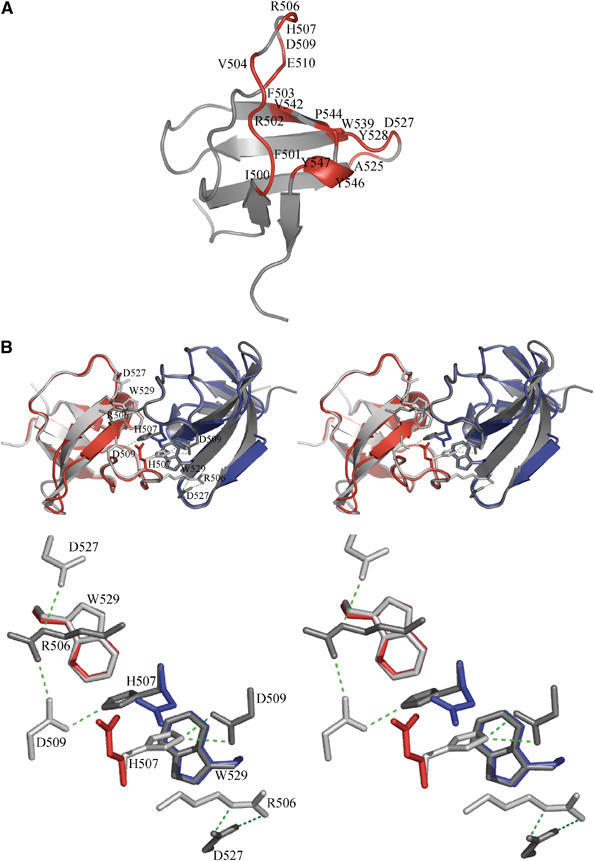
Structure of the IB1 SH3 domain. (A) Cartoon representation of the IB1 SH3 domain (in gray). The structure is composed of five β-strands arranged into two antiparallel sheets. The first sheet is formed by strands 1 and 5 and the second sheet by the strands 2, 3 and 4. The IB1 SH3 domain forms a dimer. Residues contacting the other protomer (Table II) are colored in red and labeled. (B) Cartoon representations of the experimentally determined IB1 SH3 homodimer (gray) and of the artificial C-terminal Grb2 SH3 dimer (in red and blue) generated by superposition onto the IB1 SH3 backbone (shown in stereo). The two IB1 SH3 protomers in the dimer are in a cis arrangement and it is likely that the two PID domains are in close proximity to each other. The superimposed Grb2 dimer illustrates that in this structure two opposing glutamate residues (red and blue) are incompatible with IB1-like homodimerization (see enlargement).
As expected from the pull-down experiments, the IB1 SH3 forms homodimers (see Figure 3B). The area of the dimer interface is 860 Å2 (per monomer) as determined with the protein–protein interaction server (Jones and Thornton, 1996). This interface is composed of three regions: residues 500–510, 525–529 and 542–547, and consequently the epitope must be characterized as discontinuous (Figure 4A). In the type I crystals, two dimers are found (formed by molecules A/C and B/D). The 17 residues in monomer A that are contacting monomer C are indicated in Figure 3A. The interactions are summarized in Table II. The interface is predominantly hydrophobic (70% of the amino acids). A total of 10 direct hydrogen bonds and one salt bridge (Arg506-Asp527) are found at the interface and in addition 10 bridging water molecules are observed. This provides the basis for a very stable dimer structure.
Figure 4.
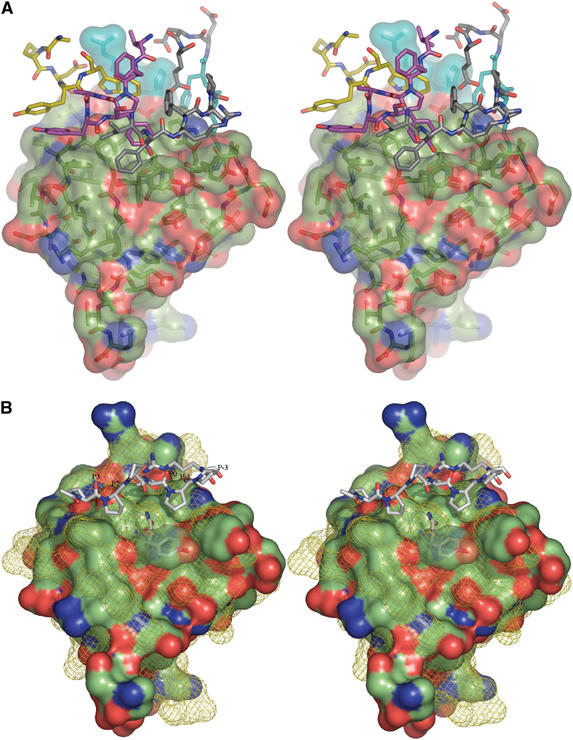
Stereographic representation of the IB1 SH3 domain. (A) The solvent-accessible surface of the IB1 SH3 domain. Contact residues from the other molecule of the homodimer colored after atom type are shown as sticks. Carbon atoms from residues 500 to 510 are colored in gray, those from residues 525 to 529 are in yellow and the 542 to 547 region is shown in magenta. Residues 506 and 507 from both monomers are colored in cyan. (B) The solvent-accessible surfaces of the IB1 and SEM-5 SH3 domains. The surface of the IB1 SH3 domain is colored after atom type. The superimposed solvent-accessible surface of SEM-5 (Lim et al, 1994) is shown as a yellow mesh, along with the bound mSos-derived peptide PPPVPPR in sticks representation. The canonical PPII binding sites are labeled (Yu et al, 1994).
Table 2.
Residues defining the IB1-SH3 homodimer interface using the protein–protein interaction server
| Residue | Interface (Å2) | % Interface | H-bonds |
|---|---|---|---|
| Ile500 | 2.61 | 0.30 | 0 |
| Phe501 | 62.80 | 7.31 | 0 |
| Arg502 | 49.41 | 5.75 | 0 |
| Phe503 | 26.48 | 3.08 | 0 |
| Val504 | 56.08 | 6.53 | 1 |
| Arg506 | 136.22 | 15.86 | 2 |
| His507 | 82.98 | 9.66 | 1 |
| Asp509 | 21.96 | 2.56 | 1 |
| Glu510 | 6.23 | 0.73 | 0 |
| Ala525 | 14.01 | 1.63 | 0 |
| Asp527 | 56.90 | 6.62 | 2 |
| Tyr528 | 49.81 | 5.80 | 0 |
| Trp529 | 90.77 | 10.57 | 1 |
| Val542 | 22.10 | 2.57 | 0 |
| Pro544 | 36.11 | 4.20 | 0 |
| Tyr546 | 82.22 | 9.57 | 1 |
| Tyr547 | 62.32 | 7.25 | 1 |
| Interface contributions from individual residues are given both as the interacting area (Å2) and as percentages of the total dimer surface. The numbers of hydrogen bonds from each residue are also reported. | |||
Although the IB1 SH3 dimers show almost perfect two-fold symmetry, the hydrogen-bonding pattern involves side chains from the three residues Arg506, His507 and Asp509 in one of the monomers. The Asp residue shows a strong interaction with both the His and Arg side chains of the other molecule. The symmetrically equivalent residue of the partner Asp509* preserves strong binding to the neighboring His507, but does not engage in the hydrogen-bonding network to Arg506 (Figure 3B).
To better characterize the surface of the interface, the SH3 structure of SEM-5, the Caenorhabditis elegans homologue for Grb2 and the IB1 SH3 structure have been superimposed (Figure 4B). This maps out the canonical polyproline helix type II (PPII) binding sites (P−3, P−1, P0, P+2, P+3) (Yu et al, 1994) on the surface of the IB1 SH3 domain. It is remarkable that all the SH3 residues of IB1 involved in dimerization (Figure 3A) are also those that are usually involved in contacts to PPII ligands, see for example, Harkiolaki et al (2003).
Figure 4A shows how IB1 SH3 (molecule A) residues interact with the PPII binding pockets of molecule C. The Tyr546 of the molecule A (TyrA546) occupies the P+3 recognition site of molecule C, stacking partly with PheC501 and interacting via further hydrophobic contacts with TyrC547. Residue 546 is usually a tyrosine in other SH3 domains (Larson and Davidson, 2000), and often hydrogen bonds with its hydroxyl group to the backbone carbonyl of the residue that is located in the P+2 site. There is no residue in the P+2 site in the IB1 SH3 homodimer, but TyrC546(OH) hydrogen bonds to a water molecule (Wat15). Wat15 is further hydrogen bonding with the carbonyl of TyrA528 and Wat29, resulting in a stabilization of the dimer. ProA544 occupies the P0 site and is located in a strongly hydrophobic environment containing residues PheA503, TrpA529, PheA543, TyrA547, PheC503, ProC544 and TyrC547. Besides the surrounding hydrophobic contacts, TrpA529 and TyrA547 are further stabilized by hydrogen bonds from TrpA529(NE1) to the ValC504 carbonyl oxygen and between TyrA547(OH) and the TyrC546 amide nitrogen, respectively. PheA503 occupies the P−1 site and is involved in hydrophobic contacts with residues TrpC529 and ProC544. The P−3 site is typically occupied by a positively charged residue like arginine as is the case in the SEM-5 structure where side-chain atoms engage in hydrogen bonding to Glu172 and a water molecule. The SEM-5 residue Glu172 corresponds to GluC510 in the IB1 SH3 structure. GluC510 makes strong interactions with the ArgC506 main chain amide and the HisC507 side chain of the same molecule, occluding the role of arginine as a favored P−3 residue. Instead, binding energy is provided at the P−3 site by the perfectly stacking residues HisC507 and HisA507 as well as through hydrogen bonding between the residues AspA509 and HisC507.
Arg506 mutations destabilize IB1 dimerization
The crystal structures suggested that residue Arg506 is important for dimerization. Therefore, we examined two IB1 SH3 mutants in which Arg506 was replaced by alanine (IB1 SH3 R506A) or glutamate (IB1 SH3 R506E). 35S-radiolabeled wild–type (wt) or mutant IB1 SH3 proteins (SH3 wt, SH3 R506A and SH3 R506E) was used in a pull-down assay using GST-IB1 SH3 as a bait (Figure 5A). In contrast to wt IB1 SH3, both SH3 mutants lost their capacity to interact with the wt SH3 domain.
Figure 5.
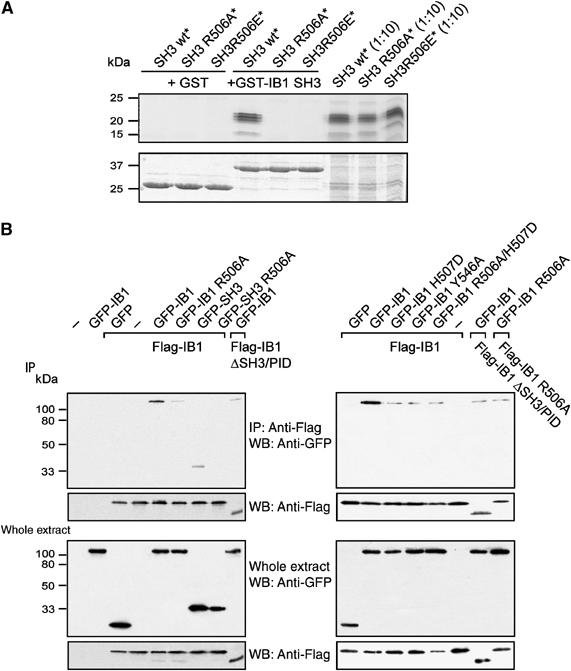
Mutations R506A, R506E, H507D, Y546A and R506A/H507D destabilize IB1 dimerization. (A) Pull-down experiments were performed with GST-SH3 and 35S-labeled (*) wt or mutant SH3 IB1 constructs (SH3 wt, SH3 R506A or SH3 R506E). Aliquots of 35S-labeled proteins were analyzed by SDS–PAGE and autoradiography (top panels) of the Coomassie-stained gel (bottom panels). (B) Flag-tagged IB1 was immunoprecipitated from extracts of 293T cells co-transfected with Flag-IB1 constructs (Flag-IB1 or Flag-IB1 ΔSH3/PID) and full-length wt or mutant GFP-IB1 constructs (GFP-IB1 wt, GFP-IB1 R506A, GFP-IB1 H507D, GFP-IB1 Y546A or GFP-IB1 R506A/H507D). Similar experiments were performed from cells co-transfected with Flag-IB1 and wt or mutant GFP-SH3 fusions (GFP-SH3, GFP-SH3 R506A). Co-immunoprecipitated proteins and immunoprecipitation efficiency were assessed with anti-GFP and anti-Flag antibodies, respectively. Transfection efficiencies were verified by anti-Flag and anti-GFP antibodies. IP: immunoprecipitation; WB: western blot.
To provide further evidence for the Arg506 involvement in IB1 dimerization, we performed immunoprecipitation in 293T cells co-transfected with Flag-tagged IB1 constructs (Flag-IB1, Flag-IB1 ΔSH3/PID) and wt or mutant GFP-tagged IB1 (GFP-IB1 wt, GFP-IB1 R506A). Likewise, cells were co-transfected with either wt or mutant GFP-tagged SH3 constructs (GFP-SH3 wt, GFP-SH3 R506A). Immunoprecipitations were carried out using an anti-Flag resin and cosedimented proteins were detected by an anti-GFP antibody. As shown in Figure 5B (left panel), mutation of Arg506 in both the full-length IB1 and the SH3 domain alone led to a massive destabilization of the dimer. Nonetheless, neither Arg506 replacement nor deletion of the SH3-PID domains, is sufficient to block dimerization completely in the full-length protein, suggesting that additional N-terminal regions are likely to contribute to dimerization. As the crystal structure suggested that residues His507 and Tyr546 are also involved in IB1 dimerization, we performed similar immunoprecipitation experiments with two additional single mutants (GFP-IB1 H507D and GFP-IB1 Y546A) and a double mutant (GFP-IB1 R506A/H507D). As shown in Figure 5B (right panel), all these mutations led to a massive destabilization of the dimer, similar to the one observed with either the deletion of the SH3-PID domains or the Arg506 mutation in both Flag and GFP constructs. Gel filtration and limited trypsinolysis experiments confirm that mutations at the IB1 SH3–SH3 interface hinder dimerization (see Supplementary data).
Dimerization of IB1 increases basal JNK activity
To investigate the role of IB1 dimerization on JNK activity, we performed transfection studies using increasing amounts of IB1, and then measured JNK activity in a sensitive assay using GST-c-Jun (residues 1–79) as a JNK-specific substrate. As proposed previously (Elion, 1998), JNK activity displays a bell-shaped IB1 concentration dependence up to a 2.6-fold increase ±0.4 (s.e.m.) (as shown from eight experiments; P<0.001 for IB1 wt compared with each mutant, see Supplementary data for statistical details) at the optimal concentration (0.1 μg of IB1 DNA; Figure 6). In contrast, none of the R506A (n=7), H507D (n=3), Y546A (n=3) and R506A/H507D (n=3) IB1 mutants promote JNK activity (P>0.05 for all mutants compared to each other); rather, we consistently observed a trend towards inhibition, which reaches statistical significance at the highest IB1 concentration (P<0.0001, for all mutants at 2.0 μg versus no IB1 mutant DNA).
Figure 6.
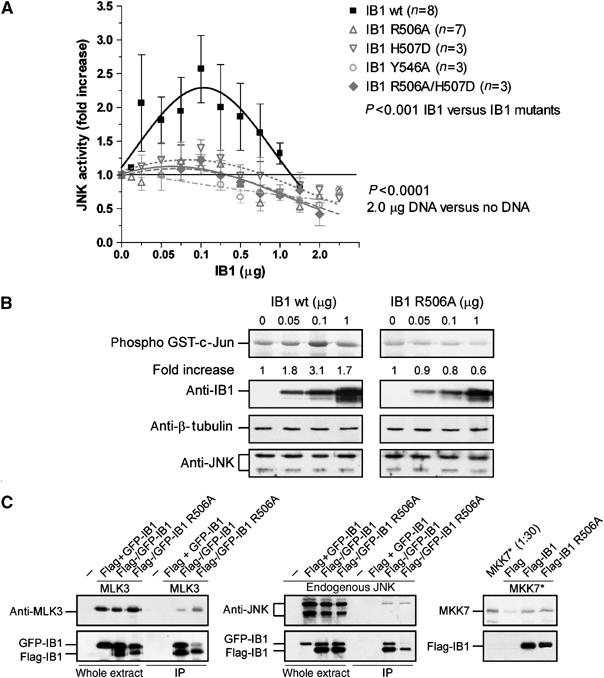
IB1 dimerization increases JNK basal activity. (A) The 293T cells were transiently transfected with increasing levels of wt (IB1 wt) or mutant IB1 (IB1 R506A, IB1 H507D, IB1 Y546A or IB1 R506A/H507D). JNK activity was measured in cell extracts using GST-c-Jun as substrate. The maximal fold increase of basal JNK activity is reached in cells transfected with 0.1 μg of wt IB1 DNA. (B) Transfection efficiencies and protein quantification were verified by Western blotting with anti-IB1, anti-β-tubulin and anti-JNK antibodies as indicated. (C) Relative binding of JNK, MKK7 and MLK3 to wt or mutant IB1. Flag-tagged IB1 was immunoprecipitated from extracts of 293T cells co-transfected with Flag- and GFP-IB1 constructs (Flag-IB1 wt, Flag-IB1 R506A or Flag and GFP-IB1 wt or GFP-IB1 R506A) in the presence or absence of MLK3-pCMVsport6 construct. Co-immunoprecipitated MLK3 or endogenous JNK was assessed with anti-MLK3 and anti-JNK antibodies, respectively. Pull-down experiments were performed with immunoprecipitated Flag, wt or mutant Flag-IB1 and 35S-labeled (*) MKK7. Binding of MKK7 was detected by autoradiography.
The Arg506Ala IB1 mutation does not affect binding to JNK, MKK7 or MLK3
The observed increase in IB1-dependent JNK basal activity might be mediated by a difference in binding affinity of either one of the three members of the MAPK module binding to IB1. We performed co-immunoprecipitation experiments in 293T cells co-transfected with either wt Flag- and GFP-tagged IB1 or mutant Flag- and GFP-tagged IB1 (Flag-IB1 wt, GFP-IB1 wt or Flag-IB1 R506A, GFP-IB1 R506A) in the absence or presence of MLK3. Immunoprecipitations were carried out using an anti-Flag resin and cosedimented proteins were detected by the appropriate antibodies. In addition, 35S-radiolabeled MKK7 was used in a pull-down assay with either wt or mutant Flag-tagged IB1 (Flag-IB1 wt, Flag-IB1 R506A) bound to an anti-Flag resin. All three kinases, JNK, MKK7 and MLK3, appear to bind to the IB1 mutants to a similar extent as to the wt protein (Figure 6C).
Functional role of IB1 dimerization in GLUT2 expression and in insulin secretion
IB1 has been shown to participate in the regulation of important cellular events in neurons and pancreatic β-cells, including apoptosis, expression of the GLUT2 and glucose-induced insulin secretion (Bonny et al, 1998; Waeber et al, 2000). We first investigated whether IB1 dimerization could affect apoptosis of pancreatic β-cells. To this end, we attempted to decrease formation of the IB1 dimers by overexpressing either the wt or the mutated IB1 SH3 domains linked to GFP. As shown in Figure 7A, only the overexpression of the wt IB1 SH3 domain (GFP-SH3 wt), but not the mutant one (GFP-SH3 R506A), is able to destabilize homodimerization in a dose-dependent manner. The wt or the mutated IB1 SH3 domains fused to GFP was then overexpressed in INS-1E cells, and the morphology of the nuclei of green fluorescent cells was evaluated using a combination of Hoechst and propidium iodide staining (Bonny et al, 2001). These treatments did not affect IL-1β-stimulated apoptosis.
Figure 7.
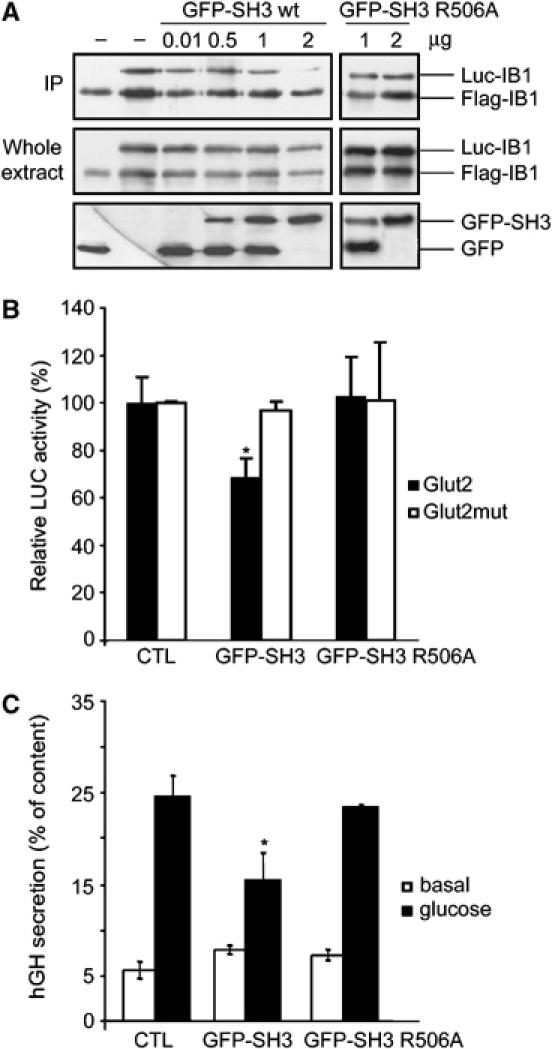
Functional role of the IB1 dimer. (A) Overexpression of the IB1 SH3 domain results in destabilization of the IB1 dimer. INS-1E cells were co-transfected with Flag-IB1, Luc-IB1 and increasing amount of GFP-SH3 wt and mutant constructs. Flag-tagged IB1 was immunoprecipitated from INS-1E extracts. Co-immunoprecipitated Luc-IB1 and immunoprecipitation efficiency were assessed with anti-IB1 antibodies. Transfection efficiencies were verified by anti-IB1 and anti-GFP antibodies. IP: immunoprecipitation. (B) INS-1E cells were transiently co-transfected with a luciferase-encoding plasmid driven by the Glut2 promoter (Glut2) or by a mutant form (Glut2mut), with an empty plasmid (control) or wt or mutant GFP-tagged SH3 expression vectors (GFP-SH3 wt, GFP-SH3 R506A). The relative luciferase activities are reported. Overexpression of wt SH3 leads to a significant decrease in the expression of GLUT2, whereas the mutated domain did not have any effect. (C) Dimerization of IB1 is required for glucose-induced insulin secretion. INS-1E cells were transiently co-transfected with an hGH-encoding plasmid and with an empty plasmid (control) or a wt or mutant GFP-tagged SH3 expression vectors (GFP-SH3 wt, GFP-SH3 R506A). Cells were later incubated under either basal or stimulatory conditions. The total amount of hGH present in the cells and the fraction released in the medium were measured by ELISA. Overexpression of wt SH3 leads to a significant decrease in stimulated secretion, whereas the mutated domain had no effect.
To determine whether IB1 dimerization is implicated in the regulation of GLUT2 expression, INS-1E cells were transiently co-transfected with either wt or mutant SH3 constructs (GFP-SH3 wt, GFP-SH3 R506A) and with constructs encoding the luciferase reporter gene driven by the wt (Glut2) or a mutant form (Glut2mut) of the GLUT2 promoter. At 24 h after the transfection, luciferase activities were measured. The destabilization of the IB1 dimer results in a 29.5±8.1% (s.e.m.) reduction of the expression of GLUT2 (Figure 7B).
To study the functional relevance of IB1 dimerization on insulin secretion, INS-1E cells were similarly transiently co-transfected with either wt or mutant SH3 constructs (GFP-SH3 wt, GFP-SH3 R506A) and with a plasmid encoding the human growth hormone (hGH). hGH is targeted to secretory granules and is coreleased with insulin during exocytosis (Iezzi et al, 1999). At 3 days after transfection, cells were incubated under resting conditions or under conditions that stimulate insulin release. Overexpression of wt SH3 results in a significant decrease in stimulated secretion (42±13% s.e.m.), whereas the mutated domain did not affect secretion (Figure 7C). These findings demonstrate that IB1 dimerization through the SH3 domain is required for an appropriate function of β-cells such as expression of GLUT2 and glucose-induced insulin secretion, but is not relevant for apoptosis under the conditions investigated.
Discussion
IB1 dimerizes through a novel SH3–SH3 interaction
We have shown in this study that IB1 homodimerizes through a novel type of SH3–SH3 domain interaction. The pull-down and the co-immunoprecipitation experiments show that IB1 dimerizes through its own SH3 domain. Gel filtration experiments, dynamic light-scattering and structure determinations based on X-ray crystallography support this observation. Taken together, our results reveal that the SH3 domain of IB1 dimerizes both in solution and in crystals and that other regions of IB1 have a minor contribution to dimerization.
The majority of the structures of complexes between SH3 domains and ligands deposited in the RCSB Protein Data Bank show interactions between the SH3 domain and proline-rich peptides or proteins containing proline-rich segments, such as the structure of the complex between SEM-5 and the mSos-derived peptide PPPVPPRRR (Lim et al, 1994). Although other structures exist where SH3 domains bind one another (Nishida et al, 2001; Delbruck et al, 2002; Harkiolaki et al, 2003), the homodimerization of the IB1 SH3 domain is unique as it involves the canonical PPII recognition sites without actually having the PxxP motif. A total of seven PxxP motifs are found in the sequence of IB1 (Figure 1A), two of which are conserved in mouse, rat and human. Since neither the SH3 domain of MLK3 nor other IB1-associated components (DLK, MKK7 or JNK) have been implicated in specific recognition of these IB1 PxxP motifs, they may either have been introduced by chance or harbor interactions sites with hitherto unknown partners.
We have investigated the possibility of IB1 SH3-like homodimerization in other SH3 domains in order to examine whether this novel use of SH3 domains is unique to IB1-like systems. Analysis of 396 human SH3 domains retrieved from the SMART database (Letunic et al, 2004) shows that the IB1 SH3 domain is not phylogenetically exceptional. Based on the IB1 SH3 dimer, we have also modeled a Grb2-C dimer (Figure 3B). Here, dimerization immediately appeared very unlikely since the two opposing glutamate residues (Glu171 of Grb2), which occupy the positions of the two stacking histidine residues in the IB1 SH3 domain, would introduce repulsion. Along these lines, we have scanned the human SH3 sequences for conservation of the residue pairs His507-Asp509 and Arg506-Asp527 involved in strong IB1 SH3 dimer interactions. This identified only the different IB1-related variants, and it revealed that this particular dimerization scheme is unique to the IB1 system. The SH3 domain of Lck contains residues that are equivalent of the IB1 SH3 His507-Asp509 pair. Biochemical studies pointed towards the Lck SH3–SH2 region as responsible for a physiologically relevant dimerization of full-length Lck (Eck et al, 1994; Lee-Fruman et al, 1996). However, the Lck SH3–SH2 crystal structure indicates that it does not involve SH3–SH3 homodimerization, but a classical SH3–polyproline interaction (Eck et al, 1994).
Although the SH3 surfaces of IB1 and SEM-5 look similar (Figure 4B), a notable difference is found at position 546, a tyrosine in IB1 and an asparagine in SEM-5. We have shown that Tyr546 is important for IB1 homodimerization. Since Tyr546 points away from the PPII binding sites, this disrupts the cavity at the IB1 SH3 surface. As in SEM-5 and in most other SH3 domains (Larson and Davidson, 2000), this residue is an aspargine, and it contributes one of the three important hydrogen bonds to the backbone carbonyls of the polyproline helix. It seems unlikely that the IB1 SH3 domain can make conformational changes that would allow it to provide a hydrogen-bond donor at this position. Consequently, the affinity of the IB1 SH3 domain towards a canonical PxxP motif alone is expected to be reduced. However, this does not rule out the possibility that IB1 could bind a PxxP-containing epitope, but rather that additional binding energy must be provided by specific residues outside this region. Our results suggest that if an exogenous PxxP ligand were to compete for binding at the IB1 SH3 dimer interface, it requires that interactions are preserved in the specificity-determining region around residues Arg506 and His507 (Figures 3B and 4A).
Functional consequences of IB1 dimerization
IB1 dimerization appears to result in an increase in IB1-mediated JNK activity. These data add a further level of complexity to the regulation of JNK signaling components by IB1. IB1 has been initially characterized both as a cytoplasmic inhibitor and as an activator of the JNK signaling pathway. Evidence pointing to a negative role of IB1 are numerous: first, IB1 has been shown to both sequester JNK into the cytoplasm and to prevent its access to c-Jun by a direct competitive mechanism (Dickens et al, 1997; Bonny et al, 2001). Accordingly, in pancreatic β-cells and in urothelium, it was demonstrated that IB1 decreases JNK-mediated c-Jun phosphorylation, and concomitant apoptosis (Bonny et al, 2000; Tawadros et al, 2002). Peptides derived from IB1 further inhibit c-Jun phosphorylation and apoptosis in neuronal cells (Borsello et al, 2003). Second, MLK3 is maintained in a monomeric, autoinhibited, unphosphorylated and catalytically inactive form when bound to IB1 (Nihalani et al, 2001). It has also been suggested that MKK7 binds to the leucine zipper of MLK3, which in turn prevents homodimerization and activation of MLK3 (Mooney and Whitmarsh, 2004). Third, when JNK binds to IB1, the rearrangements that follow induce a distortion in the ATP binding site of JNK, providing another mechanism of allosteric inhibition of JNK kinase activity (Heo et al, 2004). Fourth, IB1 can selectively bind the phosphatase MKP7, which leads to the dephosphorylation and inactivation of JNK bound to IB1, and consequently further reduces the phosphorylation of c-Jun (Willoughby et al, 2003).
In contrast, experiments using transiently transfected cell lines have suggested that IB1 can enhance the activation of JNK by MLK3 or MKK7 (Whitmarsh et al, 1998) and even facilitate MLK-dependent signal transduction of the JNK module (Davis, 2000). IB1 has also been shown to enhance the JNK-mediated phosphorylation of APP and to link APP to the kinesin light chain (Inomata et al, 2003; Matsuda et al, 2003). IB1-deficient mice suffer from impaired JNK activation and increased neuronal cell death (Whitmarsh et al, 2001). Finally, the results reported here indicate that IB1 homodimerization increases JNK activity in the absence of exogenous stimulation. This might not be particularly surprising as dimerization would potentially double the local concentration of MKK7 to IB1-bound JNK, thus resulting in an elevated phosphorylation of JNK by its upstream kinase. However, the relative low level of JNK activation by IB1 might not be enough to induce stimulus-independent apoptosis.
One of the important functions of IB1 (Bonny et al, 1998; Waeber et al, 2000) is the control of the expression of GLUT2 and the regulation of the insulin-secretory process. Our data link IB1 dimerization to these important regulatory functions. The experiments with temperature-dependent pull-down assays and co-immunoprecipitation experiments, the dimerization observed in crystals and monitored in solution by gel filtration and dynamic light scattering, suggest that IB1 exists as a dimer in vivo. Further studies will be aimed at understanding whether the novel SH3–SH3 interaction described here is dynamically regulated, for example, in the presence of exogenous stimuli controlling the insulin-secretion process.
Materials and methods
DNA constructs and mutagenesis
All constructs used in this study were prepared by standard methods (Sambrook and Russell, 2001; see Supplementary data). In addition to a long rat IB1 SH3 variant (IB1 SH3-L, amino acids 489–559) (Dar et al, 2003), we prepared a construct optimized for crystallization (IB1 SH3-S, amino acids 494–553).
Radiolabeled proteins
35S-methionine-labeled IB1, truncated IB1, kinase domain of MLK3, MKK7α1 and MKK4, IB1 SH3 wt, IB1 SH3 R506A and IB1 SH3 R506E were prepared by in vitro transcription and translation using the TNT-coupled reticulocyte lysate system (Promega).
Pull-down assays
GST-fusion proteins bound to glutathione–agarose beads were incubated with 35S-labeled proteins and the beads were washed before SDS–PAGE analyses. In a similar way, immunoprecipitated Flag, Flag-IB1 wt or mutant was incubated with 35S-labeled MKK7α1. Proteins were analyzed by SDS–PAGE and autoradiography or immunoblotting with anti-IB1 antibodies (see Supplementary data).
Dissociation experiments
Pull-down experiments and co-immunoprecipitation were performed with GST-fused proteins and 35S-labeled SH3 wt constructs or Flag-tagged proteins and GFP-IB1. After the final washing step, the samples were incubated for half an hour at increasing temperatures (4–95°C). After centrifugation, the samples, 10% of the supernatant and aliquots of 35S-labeled SH3 wt, were resuspended in SDS buffer and boiled for 5 min. Proteins were analyzed by SDS–PAGE (15 or 8%) and autoradiography or immunoblotting with anti-GFP and anti-Flag antibodies.
Cell culture, transfection and JNK assays
Human embryonic HEK 293T cells and the insulin-secreting cell line INS-1E were cultured and transfected by standard methods and JNK activity was measured by an established assay (see Supplementary data).
Immunoprecipitations
Cells were transiently transfected with the appropriate expression vectors: the Flag-IB1 constructs as indicated together with the vectors expressing GFP-IB1 (GFP-IB1 wt, GFP-IB1 R506A, GFP-IB1 H507D, GFP-IB1 Y546A or GFP-IB1 R506A/H507D) or GFP-SH3 constructs (GFP-SH3 wt, GFP-SH3 R506A) and, if necessary, with MLK3-pCMVsport6 or Rluc-IB1 constructs. After 24 h, cells were lysed and 200 μg of crude extracts were incubated 60 min with anti-Flag agarose resin (Sigma) at 20°C in PBS solution. After five washes in the same buffer, Flag-IB1 and co-immunoprecipitated partners were subjected to SDS–PAGE and immunoblotting with anti-GFP, anti-Flag and anti-IB1 antibodies, or used in pull-down experiments with 35S-labeled MKK7α1.
Western blotting and antibodies
Standard procedures and relevant antibodies were used throughout (see Supplementary data).
Apoptotic counts
The insulin-secreting cell line INS-1E was transfected with GFP, GPF-fused SH3 wt or mutant (GFP-SH3 wt, GFP-SH3 R506A). Apoptotic cells were counted 48 h after the addition of IL-1β (10 ng/ml) by propidium iodide and Hoechst 33342 staining (Bonny et al, 2000). The number of apoptotic cells in experiments involving transfected GFP constructs was evaluated using an inverted fluorescence microscope (Axiovert 25; Zeiss). Apoptotic cells were discriminated from normal cells by the characteristic blebbing of the cytoplasm, which was easily determined from the fluorescence emitted by the GFP. A minimum of 1000 cells in duplicate was counted for each experiment.
Transcription experiments
INS-1E cells (1 × 105) plated in 24-well dishes were transiently co-transfected with constructs encoding the firefly luciferase reporter gene driven by a region of the rat Glut2 promoter (−338 to +49) (Glut2) in the pGL3 vector. The mutated Glut2 promoter (Glut2mut) and the empty vector driven by the minimal tk promoter (pGL3basic) constructs were used as controls in these experiments. At 24 h after the transfection, the cells were harvested with 50 μl of the passive lysis buffer (Promega). Luciferase activities (from Glut2 promoter and normalization with Renilla luciferase vector (pRL-CMV-renilla)) were measured with 50 μl of protein extract using the dual-luciferase reporter assay system (Promega). All experiments were repeated three times in triplicate.
Secretion experiments
INS-1E cells (3 × 105) plated in 24-well dishes were transiently co-transfected with a construct encoding the hGH (Nicholls, San Juan Capistrano, CA). After 3 days, the cells were washed and preincubated for 30 min in buffer (140 mM NaCl, 3.6 mM KCl, 0.5 mM NaH2PO4, 0.5 mM MgSO4, 1.5 mM CaCl2, 2 mM NaHCO3, 10 mM Hepes and 0.1% bovine serum albumin) containing 2 mM glucose (basal conditions). The medium was then removed and the cells incubated for 45 min in the same buffer (basal conditions) or in a buffer containing 20 mM glucose, 10 μM forskolin and 100 μM IBMX (stimulatory conditions). The total amount of hGH produced by transfected cells and the fraction released into the medium during the incubation period were determined by ELISA (Roche Diagnostics).
Crystallization and data collection
Preparation of protein for crystallization followed standard procedures (see Supplementary data). IB1 SH3-L was crystallized and data collected as reported previously (Dar et al, 2003). Crystals of the SeMet-IB1 SH3-S protein were obtained at 22°C by the hanging drop vapor diffusion method. Prism-shaped crystals (type I) with maximum dimensions of 0.03 × 0.03 × 0.1 mm3 appeared within 12 h after mixing 2 μl protein of a 1 mg/ml sample and 2 μl reservoir solution (100 mM Bicine, pH 9.0 and 3.2 M AmS). Larger IB1 SH3-S crystals (type II) were produced using a modified reservoir solution (100 mM Bicine, pH 9.0, 3.1 M AmS and 2% polyethylene glycol (PEG) 400). Diffraction data were collected at BL711, MaxLab, Lund, Sweden from a cryoprotected (20% trehalose, 100 mM Bicine and 3.3 M AmS) type I crystal. Type II data were collected at X11, DESY, EMBL, Hamburg, Germany. Type I and II data were integrated and scaled using MOSFLM and SCALA (CCP4, 1994), see Table I.
Structure determination and refinement
The program SOLVE (Terwilliger and Berendzen, 1999) was used to locate the four expected heavy-atom positions based on the type I SeMet data. Phases were optimized using the program SHARP (Fortelle and Bricogne, 1997) and improved by density modification. Model building was performed using the Arp/Warp program (CCP4, 1994), completed manually in the program O (Jones et al, 1990) and refined using the CNS program (Brunger et al, 1998). Both the type II and type III crystal structures were solved by molecular replacement based on the derived type I model and refined using CNS (Brunger et al, 1998). Noncrystallographic symmetry (NCS) restraints were not imposed in the type I and II refinements, while strict NCS was imposed on the type III structure. In the type I crystal structure, a trehalose molecule and three sulfate ions were located at the surface of the protein. In the type II crystal structure, we have located three PEG polymers in addition to seven sulfate ions. The three structures have been deposited in the RCSB Protein Data Bank with entry codes 2FPD, 2FPE and 2FPF.
Modeling
The Grb2-C dimer (Figure 3B) was constructed by superpositions of monomers onto backbone atoms of a representative IB1 SH3 dimer structure. Geometric adjustment of the model was achieved by simple homology modeling using the program Modeler (Sali and Blundell, 1993). The program PyMOL (DeLano, 2002) was used to prepare figures.
Supplementary Material
Supplementary Figure S1
Supplementary Data and Table S1
Supplementary Data and References
Acknowledgments
We appreciate the support in data collection from the staff at the MaxLab (BL711), ESRF (ID29) and EMBL (X11, Hamburg) beamlines. This work was supported by DANSYNC, the Danish Medical Research Council, the Danish Natural Science Research Council, the European Community (Contract number HPRI-CT-1999-00017), the Swiss National Science Foundation (FN 3200BO-105595), the Swiss–French Diabetes Foundation and the Botnar Foundation. We thank Séverine Arcioni and Laurent Spack for critical comments and helpful discussions. We also thank Christelle Bielmann and Valérie Buchillier for their excellent technical assistance.
References
- Bonny C, Nicod P, Waeber G (1998) IB1, a JIP-1-related nuclear protein present in insulin-secreting cells. J Biol Chem 273: 1843–1846 [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50: 77–82 [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G (2000) IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem 275: 16466–16472 [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9: 1180–1186 [DOI] [PubMed] [Google Scholar]
- Brasher BB, Roumiantsev S, Van Etten RA (2001) Mutational analysis of the regulatory function of the c-Abl Src homology 3 domain. Oncogene 20: 7744–7752 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- CCP4, Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Dar I, Bonny C, Pedersen JT, Gajhede M, Kristensen O (2003) Crystallization and preliminary crystallographic characterization of an SH3 domain from the IB1 scaffold protein. Acta Crystallogr D 59: 2300–2302 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific [Google Scholar]
- Delbruck H, Ziegelin G, Lanka E, Heinemann U (2002) An Src homology 3-like domain is responsible for dimerization of the repressor protein KorB encoded by the promiscuous IncP plasmid RP4. J Biol Chem 277: 4191–4198 [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ (1997) A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 277: 693–696 [DOI] [PubMed] [Google Scholar]
- Eck MJ, Atwell SK, Shoelson SE, Harrison SC (1994) Structure of the regulatory domains of the Src-family tyrosine kinase Lck. Nature 368: 764–769 [DOI] [PubMed] [Google Scholar]
- Elion EA (1998) Routing MAP kinase cascades. Science 281: 1625–1626 [DOI] [PubMed] [Google Scholar]
- Fortelle EDL, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. In Methods in Enzymology, Macromolecular Crystallography Part A, Carter CW, Sweet RM (eds), Vol. 276, pp 472–494. San Diego, CA: Academic Press [DOI] [PubMed] [Google Scholar]
- Harkiolaki M, Lewitzky M, Gilbert RJ, Jones EY, Bourette RP, Mouchiroud G, Sondermann H, Moarefi I, Feller SM (2003) Structural basis for SH3 domain-mediated high-affinity binding between Mona/Gads and SLP-76. EMBO J 22: 2571–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH (2004) Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J 23: 2185–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iezzi M, Escher G, Meda P, Charollais A, Baldini G, Darchen F, Wollheim CB, Regazzi R (1999) Subcellular distribution and function of Rab3A, B, C, and D isoforms in insulin-secreting cells. Mol Endocrinol 13: 202–212 [DOI] [PubMed] [Google Scholar]
- Inomata H, Nakamura Y, Hayakawa A, Takata H, Suzuki T, Miyazawa K, Kitamura N (2003) A scaffold protein JIP-1b enhances amyloid precursor protein phosphorylation by JNK and Its association with kinesin light chain 1. J Biol Chem 278: 22946–22955 [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM (1996) Principles of protein–protein interactions. Proc Natl Acad Sci USA 93: 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Bergdoll M, Kjeldgarrd M (1990) O: a macromolecular modeling environment. In Crystallographic and Modeling Methods in Molecular Design, Bugg C, Ealick S (eds), pp 189–195. Berlin: Springer-Verlag Press [Google Scholar]
- Kay BK, Williamson MP, Sudol M (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 14: 231–241 [PubMed] [Google Scholar]
- Kelkar N, Gupta S, Dickens M, Davis RJ (2000) Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol Cell Biol 20: 1030–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson SM, Davidson AR (2000) The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci 9: 2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Fruman KK, Collins TL, Burakoff SJ (1996) Role of the Lck Src homology 2 and 3 domains in protein tyrosine phosphorylation. J Biol Chem 271: 25003–25010 [DOI] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P (2004) SMART 4.0: towards genomic data integration. Nucleic Acids Res 32: D142–D144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, Fox RO, Richards FM (1994) Stability and peptide binding affinity of an SH3 domain from the Caenorhabditis elegans signaling protein Sem-5. Protein Sci 3: 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda S, Matsuda Y, D'Adamio L (2003) Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J Biol Chem 278: 38601–38606 [DOI] [PubMed] [Google Scholar]
- Mayer BJ (2001) SH3 domains: complexity in moderation. J Cell Sci 114: 1253–1263 [DOI] [PubMed] [Google Scholar]
- Mongiovi AM, Romano PR, Panni S, Mendoza M, Wong WT, Musacchio A, Cesareni G, Di Fiore PP (1999) A novel peptide-SH3 interaction. EMBO J 18: 5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney LM, Whitmarsh AJ (2004) Docking interactions in the c-Jun N-terminal kinase pathway. J Biol Chem 279: 11843–11852 [DOI] [PubMed] [Google Scholar]
- Nguyen JT, Lim WA (1997) How Src exercises self-restraint. Nat Struct Biol 4: 256–260 [DOI] [PubMed] [Google Scholar]
- Nihalani D, Meyer D, Pajni S, Holzman LB (2001) Mixed lineage kinase-dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. EMBO J 20: 3447–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Nagata K, Hachimori Y, Horiuchi M, Ogura K, Mandiyan V, Schlessinger J, Inagaki F (2001) Novel recognition mode between Vav and Grb2 SH3 domains. EMBO J 20: 2995–3007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Smith KM, Yacobi R, Van Etten RA (2003) Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell 12: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawadros T, Formenton A, Dudler J, Thompson N, Nicod P, Leisinger HJ, Waeber G, Haefliger JA (2002) The scaffold protein IB1/JIP-1 controls the activation of JNK in rat stressed urothelium. J Cell Sci 115: 385–393 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista M, Ferracuti S, Nelson B, Paoluzi S, Quondam M, Zucconi A, Hogue CW, Fields S, Boone C, Cesareni G (2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295: 321–324 [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol 152: 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P (2000) The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nat Genet 24: 291–295 [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281: 1671–1674 [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, Rakic P, Davis RJ (2001) Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev 15: 2421–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby EA, Perkins GR, Collins MK, Whitmarsh AJ (2003) The JNK-interacting protein-1 scaffold protein targets MAPK phosphatase-7 to dephosphorylate JNK. J Biol Chem 278: 10731–10736 [DOI] [PubMed] [Google Scholar]
- Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ (1999) The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol 19: 7245–7254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL (1994) Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76: 933–945 [DOI] [PubMed] [Google Scholar]
- Zhang H, Gallo KA (2001) Autoinhibition of mixed lineage kinase 3 through its Src homology 3 domain. J Biol Chem 276: 45598–45603 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Data and Table S1
Supplementary Data and References