

Abstract
In Escherichia coli, the Y-family DNA polymerases Pol IV (DinB) and Pol V (UmuD2′C) enhance cell survival upon DNA damage by bypassing replication-blocking DNA lesions. We report a unique function for these polymerases when DNA replication fork progression is arrested not by exogenous DNA damage, but with hydroxyurea (HU), thereby inhibiting ribonucleotide reductase, and bringing about damage-independent DNA replication stalling. Remarkably, the umuC122∷Tn5 allele of umuC, dinB, and certain forms of umuD gene products endow E. coli with the ability to withstand HU treatment (HUR). The catalytic activities of the UmuC122 and DinB proteins are both required for HUR. Moreover, the lethality brought about by such stalled replication forks in the wild-type derivatives appears to proceed through the toxin/antitoxin pairs mazEF and relBE. This novel function reveals a role for Y-family polymerases in enhancing cell survival under conditions of nucleotide starvation, in addition to their established functions in response to DNA damage.
Keywords: DinB, hydroxyurea, mazEF, UmuC, UmuD
Introduction
In both eukaryotes and prokaryotes (Boye et al, 1996; Bell and Dutta, 2002), initiation of DNA replication is exquisitely regulated, and sophisticated systems have evolved to contend with the potentially lethal consequences of inhibition of replication fork progression (Elledge, 1996). Depletion of deoxyribonucleotide triphosphate (dNTP) pools leads to arrest of cell division in eukaryotes (Tercero et al, 2003) and prokaryotes (Foti et al, 2005) until DNA replication is properly restored. Mutations in components of such checkpoints result in genomic instability and elevated mutation frequencies that may lead to cancer in higher organisms (Hartwell and Kastan, 1994). Responses to arrest of fork progression include induction of DNA damage tolerance pathways. Although the rationale for such a response is clear when stalling is brought about by exogenous DNA damage, it is more enigmatic (Kai and Wang, 2003a, 2003b) when replication fork progression is inhibited in a DNA damage-independent manner.
Y-family polymerases possess properties that are advantageous for the resolution of replication forks stalled by DNA damage as they have the ability to insert nucleotides opposite DNA lesions that block replicative DNA polymerases, a process termed translesion synthesis (TLS) (Friedberg et al, 2002). TLS often ensues with comparatively low fidelity, meaning that bypass of DNA damage takes place at a potentially mutagenic cost (Goodman, 2002). Notable exceptions exist, however, such as eukaryotic Pol η bypassing cyclobutane pyrimidine dimers (Washington et al, 2001).
The Y-family DNA polymerases are encoded in Escherichia coli by the dinB and umuDC genes, which are both regulated by the LexA transcriptional repressor as part of the SOS response to DNA damage (Sutton et al, 2000). Initially, full-length UmuD is expressed from the umuDC operon. The UmuD homodimer interacts with UmuC to effect a DNA damage checkpoint function (Opperman et al, 1999), and cold sensitivity due to overproduction of umuDC (Marsh and Walker, 1985) appears to result from an exaggeration of this function (Opperman et al, 1999; Sutton and Walker, 2001b). UmuD thereafter undergoes removal of its first 24 amino acids, dependent on the RecA nucleoprotein filament (Burckhardt et al, 1988; Shinagawa et al, 1988), to form UmuD′. The UmuD′ homodimer (UmuD2′) is a positive effector of UmuC, the catalytic subunit of Pol V (Nohmi et al, 1988). Transcription of the dinB gene is weakly repressed by LexA, so that basal levels of DinB are high compared to those of UmuC (Woodgate and Ennis, 1991; Kim et al, 2001). Indeed, upon SOS induction Pol IV is the most abundant DNA polymerase in the cell (Kim et al, 2001). Among Y-family polymerases, the DinB subfamily is strikingly conserved, and it is the only branch present in all domains of life (Ohmori et al, 2001).
Hydroxyurea (HU) has been widely used to investigate responses to DNA damage-independent replication arrest (Lopes et al, 2001; Sogo et al, 2002). HU inhibits class I ribonucleotide reductases (RNR), such as that of aerobically grown E. coli (Stubbe, 2003), by scavenging a stable di-iron tyrosyl radical that is essential for catalysis. RNRs catalyze the conversion of ribonucleotides into deoxyribonucleotides—the rate-limiting step in DNA biosynthesis in most organisms (Stubbe, 2003). Levels of intracellular dNTPs are thought to decline upon HU treatment such that DNA replication is arrested through substrate starvation (Sneeden and Loeb, 2004).
We report that the E. coli Y-family polymerases Pol IV and Pol V play a role upon DNA damage-independent replication stalling. Strains bearing novel umuC alleles are unexpectedly HUR, challenging the notion that replication inhibition by HU arises solely from dNTP starvation. Genetic analyses demonstrate that the dinB and umuD gene products also participate in the DNA damage-independent response to inhibition of replication fork progression. Together, these data suggest combined action of the UmuC derivatives together with the dinB and umuD gene products at these stalled replication forks. Moreover, we also find that the lethality of such replication fork arrest in wild-type derivatives is alleviated independently by mutation of the mazEF and relBE toxin/antitoxin pairs, suggesting that the action of these Y-family polymerases may prevent mazEF- or relBE-mediated lethality under conditions of nucleotide starvation.
Results
E. coli carrying the umuC122∷Tn5 allele are unexpectedly resistant to HU
We were interested in whether the umuC+ gene product might be part of the cellular response when replication fork progression is inhibited in a DNA damage-independent manner by dNTP depletion. We therefore examined a set of strains carrying null alleles of umuC for their sensitivity to killing by HU (Figure 1A). A strain in which the umuDC operon has been deleted is as sensitive to killing by HU as its umuD+C+ parent. Intriguingly, a strain carrying a precise ΔumuC deletion that leaves the umuD+ gene intact displays a modest level of resistance to killing (Figure 1A). Perhaps, either or both of the umuD+ gene products might contribute to HUR in the absence of UmuC (see below).
Figure 1.
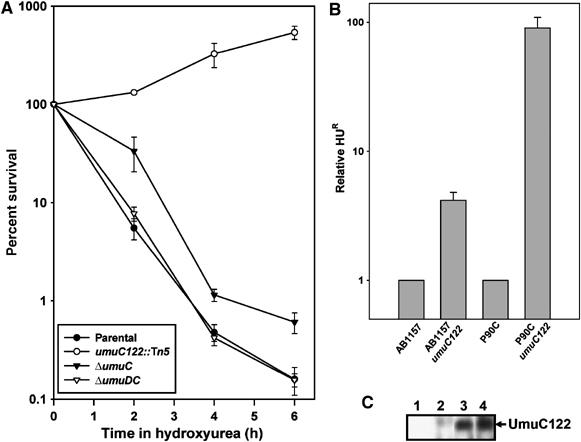
Bacterial cells bearing the umuC122 allele are HUR. (A) Survival time course in hydroxyurea reveals HUR of a strain bearing the umuC122 allele (open circles). In comparison, a strain bearing a ΔumuC allele (closed triangles) is slightly HUR, whereas both parental (closed circles) and ΔumuDC strains (open triangles) are sensitive to the reagent. CFUs were determined by serial dilution. Error bars represent the standard deviation determined from at least five samples. (B) Comparison of survival in HU of both AB1157 and P90C backgrounds. Error bars represent the standard deviation determined from at least five samples. (C) The truncated UmuC122 protein is expressed in vivo as determined by immunoblotting. Lane 1 shows a cell-free extract from a ΔumuDC strain with vector only (pGB2), lane 2 shows the same strain but instead bearing the plasmid pDC, and lanes 3 and 4 show two independent isolates of the same strain bearing pDC122. Plasmid-borne copies were used to facilitate detection of UmuC in the absence of SOS induction.
We also tested umuC122∷Tn5 (umuC122), which is known to behave as a umuC null allele with respect to induced mutagenesis caused by UV radiation and many chemicals (Elledge and Walker, 1983; Sargentini and Smith, 1984; Christensen et al, 1988; Bates et al, 1991). We found that strains carrying umuC122 are at least 100-fold more resistant to killing by HU than their umuC+ parents and can in fact multiply during HU treatment (Figure 1A). We observed this HUR phenotype in all strain backgrounds tested, including AB1157 (Figure 1B; Bachmann, 1987). These observations indicate that umuC122 is a gain-of-function umuC allele with regard to cell survival after HU treatment. This is plausible as the Tn5 insertion results in a missense mutation followed immediately by a termination codon giving rise to a predicted 32 kDa UmuC protein lacking its last 102 residues (Koch et al, 1992). The truncation occurs downstream of the conserved polymerase domain common to Y-family DNA polymerases (Boudsocq et al, 2002). Immunoblotting confirmed that the umuC122 allele indeed encodes a UmuC derivative of this molecular weight (Figure 1C). We observed that the truncated UmuC122 protein appears to be expressed at higher levels than wild-type UmuC (data not shown), although this may be because one of the synthetic peptides used to raise antibodies against UmuC lies immediately at the C-terminus of the UmuC122 protein, perhaps resulting in a more accessible epitope relative to full-length UmuC. The UmuC122 protein may also lack one or more C-terminal motifs that would normally target the protein for Lon-mediated proteolytic degradation (Frank et al, 1996). Overexpression of UmuC did not confer statistically significant HUR (data not shown).
The umuC122 allele alleviates the lethal effects of class I RNR inhibition by HU
The observation of an HUR phenotype as a consequence of a umuC mutation was unanticipated as most previously reported HUR mutants affect RNR (Sneeden and Loeb, 2004). By immunoblotting, we showed that the levels of the small and large subunits of RNR are not affected during HU treatment in strains bearing the umuC122 allele (Figure 2A). Also, we found that the protective effect of umuC122 is observed with other RNR inhibitors such as guanazole (Figure 2B). We sought evidence that the umuC122 mutation helps cells recover from the lethal consequences of HU-mediated RNR inhibition instead of acting by some other mechanism. Therefore, we took advantage of the fact that anaerobically grown E. coli utilize an HU-insensitive class III RNR rather than the HU-sensitive class I RNR used during aerobic growth (Fontecave et al, 1989). As shown in Figure 2C, we found that the anaerobically grown HU-treated umuC+ and umuC122 strains were both insensitive to HU. These observations indicate that the umuC122 mutation alleviates the lethality caused by HU inhibition of the class I RNR in E. coli through a mechanism that does not involve alteration of RNR protein levels.
Figure 2.
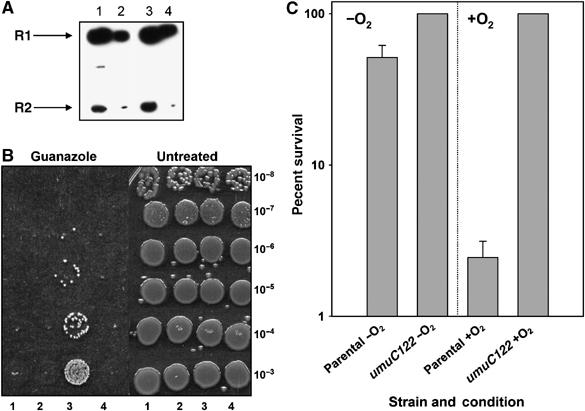
HUR proceeds through RNR inhibition. (A) Immunoblot of large and small subunits of RNR shows no difference in levels between wild-type (lanes 1 and 2) and umuC122 (lanes 3 and 4) strains during HU treatment. Lanes 1 and 3 contain twice as much total protein as lanes 2 and 4 (3.25 μg of total protein). (B) umuC122 also alleviates cell death during challenge with other RNR inhibitors. The left panel shows results of treatment with 100 mM guanazole, whereas untreated results are shown on the right. Lane 1 shows the parental P90C strain, lane 2 shows the ΔumuDC strain, lane 3 shows umuC122 and lane 4 shows the ΔumuC strain. (C) Class I RNR is sensitive to HU, whereas class III RNR, used exclusively in anaerobic growth, is indifferent to the reagent. The parental (P90C) and umuC122 strains were treated with HU for 6 h with (+O2) and without (−O2) oxygen. CFUs reported are the average of four samples and error bars represent the standard deviation as determined from these samples.
Resistance to HU requires the catalytic activity of the truncated UmuC122 protein
To facilitate further analysis of the genetic requirements for umuC122-mediated HUR, we tested whether a plasmid-borne umuDumuC122 (pDC122), expressed in a ΔumuDC derivative, conferred HUR. This was indeed the case (Figure 3A). To determine whether this HUR requires the catalytic activity of UmuC122, we used the umuC104 allele (D101N) (Figure 3D) (Koch et al, 1992), which alters a conserved catalytic residue common to all Y-family polymerases (Boudsocq et al, 2002). The addition of pDC104 had little effect on resistance to killing by HU (Figure 3A). However, introduction of the D101N mutation into pDC122 eliminated HUR (Figure 3A), indicating that the UmuC122 protein must be catalytically active to observe this phenotype.
Figure 3.

umuC requirements for observation of HUR. (A) In a ΔumuDC strain, addition of the plasmid-borne umuC alleles pDC122 and pDC125 confer HUR. pDC122 carries the umuD+ gene, but umuC has a stop codon at residue 322, thus reconstructing the truncated allele present on the chromosome by virtue of the Tn5 insertion. pDC104 encodes UmuC(D101N), rendering UmuC catalytically inactive, whereas pDC122C104 encodes UmuC122(D101N). pDC125 encodes UmuC(A39V), an allele that separates the UV-induced mutagenesis and cold-sensitivity phenotypes of umuC. CFUs were determined by serial dilution, and treatment was carried out with Sp (for plasmid maintenance) and 50 mM HU. Values reported are the average of three experiments and error bars represent the standard deviation obtained from those values. (B) The umuD gene products are also required for HUR. The resistance conferred by a plasmid-borne umuC122 allele depends upon the umuD gene products. pD(S60A)C122 is as pDC122 but encodes a UmuD protein with a mutation (S60A) rendering the protein unable to undergo autoproteolysis to become UmuD′. The plasmid pD′C122 is as pDC122 but encodes only UmuD′ instead of the full-length protein. Reported values are the average of three experiments and error bars represent the standard deviation as determined from those experiments. (C) The umuD gene products are also required for the HUR conferred by umuC125. Plasmids and data analysis are as in (B). (D) A structural representation of the UmuC active site reveals the proximity of A39 to residues essential for catalysis (D6, D101). The template is shown in red, and the primer in green. Model is courtesy of Dr D Barksy (LLNL, Livermore, CA).
A unique umuC missense allele also confers resistance to HU
We also tested the response to HU of umuC125, a umuC allele bearing an A39V mutation, which does not affect the ability of UmuC to function in UV mutagenesis, but eliminates the cold sensitivity observed when it is overexpressed together with UmuD (Marsh et al, 1991; Sutton and Walker, 2001b). We found that ΔumuDC cells containing pDC125, the plasmid-borne version of umuC125, are also resistant to HU, although not to as high a level as observed with pDC122 (Figure 3A). This observation indicates that HUR is not a unique property of the umuC122 allele, but can be mimicked, at least in part, by a simple missense mutation affecting the N-terminus of UmuC.
The umuD+ gene is required for HU resistance
The data presented in Figure 1A suggest that the umuD+ gene product(s) might contribute to HUR in the absence of UmuC. Furthermore, the umuD+ gene products influence the biological function of UmuC (Nohmi et al, 1988; Woodgate et al, 1989). We therefore assessed whether either form of the umuD+ gene product is required for the high level of HUR we observed in umuC122 bearing strains. The umuD(S60A) mutation (Koch et al, 1992; McLenigan et al, 1998) eliminates the serine that serves as the nucleophile in RecA-mediated UmuD autocleavage, so that only full-length UmuD is produced [pD(S60A)C]. Alternatively, the DNA encoding the first 24 amino acids in the N-terminus of UmuD can be deleted so that UmuD′ is synthesized directly (pD′C) (Nohmi et al, 1988).
As shown in Figure 3B, ΔumuDC cells with a plasmid carrying umuD(S60A)umuC122 [pD(S60A)C122] exhibited a lower level of HUR than the corresponding cells bearing the umuC122 plasmid (pDC122), but nevertheless were substantially HUR. Similarly, ΔumuDC cells bearing pD′C122 exhibited a lower level of HUR than the corresponding pDC122 bearing strain, but were still HUR. These results suggest that the full degree of HUR displayed by a umuD+umuC122 strain requires both forms of the umuD+ gene product. The two forms might act sequentially, first the UmuD2 homodimer and then the UmuD2′ homodimer. If so, it would appear that the component of HUR requiring UmuD2 is more substantial than the component requiring UmuD2′. Another possibility is that a component of the HUR requires the action of the UmuD·UmuD′ heterodimer, which is known to be the most stable form in vitro (Battista et al, 1990).
We performed similar experiments with the umuC125 plasmid-borne allele (Figure 3C, note y-axis scale) in which we examined HUR when umuD(S60A) and umuD′ were combined with umuC125. Interestingly, in contrast to the situation with umuC122, the strain bearing pD(S60A)C125 displayed comparable HUR relative to the strain bearing pDC125, whereas the strain bearing pD′C125 showed substantially less HUR. These data, combined with the fact that the level of HUR of a umuC125 strain is less than that of a umuC122 strain (Figure 3A), suggest that the UmuC125 protein is less proficient at the UmuD′-dependent component of HUR than the UmuC122 protein.
The dinB+ gene is required for HUR
The results presented to this point indicate that the high-level resistance of certain umuC mutants to killing by HU also requires certain forms of the UmuD protein. Involvement of DinB in HUR would be consistent with reports that DinB cooperates with UmuC in TLS past certain lesions (Napolitano et al, 2000; Sommer et al, 2003). Furthermore, under both induced and uninduced conditions, the intracellular levels of the umuD gene products are much higher than the estimated intracellular concentrations of UmuC, but are approximately equal to those of DinB (Woodgate and Ennis, 1991; Kim et al, 2001). Therefore, we constructed a strain with a precise deletion of the dinB+ gene in umuC+ and umuC122 backgrounds. In a umuC+ strain, loss of dinB+ results in a slight sensitivity to HU (Figure 4A). However, introduction of the ΔdinB mutation into the strain carrying the umuC122 allele eliminates the high level of HUR observed in this strain (Figure 4A). Thus, the dinB+ gene product is essential for the HUR exhibited in umuC122 strains.
Figure 4.

The dinB gene and its catalytic activity are necessary to avert HU lethality. (A) The HUR of a umuC122 dinB+ strain (open circles) is eliminated by deletion of the dinB gene (closed triangles). In contrast, deletion of the dinB gene has only a mild effect on the parental strain (open triangles and closed circles). (B) Reconstruction of the dinB+ locus on the chromosome restores HUR to the umuC122 ΔdinB strain. However, transduction of the dinB003 allele, which encodes a catalytically inactive DinB(D103N), does not restore HUR, indicating that the catalytic activity of DinB is required. Treatment was for 6 h with 100 mM HU in rich medium. umuC122 dinB+ refers to the reconstructed wild-type gene with a linked cat gene upstream the dinB promoter. Reported values are the average of three experiments and error bars represent one standard deviation.
We asked whether HUR could be restored in a umuC122ΔdinB mutant by introducing plasmids carrying the dinB+ gene. We were unable to complement HUR in trans with low- or high-copy number plasmids bearing dinB+. However, by transducing the wild-type copy of the dinB+ gene into the umuC122ΔdinB mutant, the HUR phenotype was restored (Figure 4B). The possibility that the restoration is due to a closely linked locus rather than to dinB+ is inconsistent with the data presented in the following section. These observations suggest that the level of DinB expression or a cis-regulatory element is critical for the ability of dinB+ to contribute to the HUR of a umuC122 strain. Perhaps, DinB cannot contribute to HUR if its levels do not correlate with those of the products of the umuD+ gene.
The catalytic activity of DinB is required for HU resistance
To test whether DinB must be catalytically active to contribute to HUR, we introduced the dinB003 mutation into the chromosome of a umuC122 strain. This mutation (D103N) alters a conserved aspartic acid residue required for phosphodiester bond formation (Wagner and Nohmi, 2000). The large loss of HUR we observed (Figure 4B) suggests that DinB is indeed acting as a DNA polymerase as it contributes to HUR. Thus, it appears that HUR results from the combined action of two DNA polymerases, DinB and a mutant form of UmuC, acting together with UmuD and UmuD′.
DNA synthesis is slowed in both parental and umuC122 strains during HU challenge
To explain the observation that both Y-family polymerases are required for HUR, we asked whether HUR was simply due to an extensive alteration in the rate of DNA replication. We measured DNA synthesis by examining the ability of thymidilate synthase-negative (thyA−) derivatives of wild-type and umuC122 strains to incorporate thymidine (3H-Thy) in 10 min pulses during HU treatment. We found that the amount of DNA synthesis is reduced during HU treatment in both wild-type and umuC122 strains compared to untreated controls (Figures 5A and B). Any minor changes that we observe in the ability to incorporate 3H-Thy into the DNA do not appear to account for the striking difference in viability, that is, competence to develop colonies, between the HU-treated wild-type and umuC122 strains. This remarkable and unexpected result led us to examine the cells microscopically during HU treatment (see below).
Figure 5.
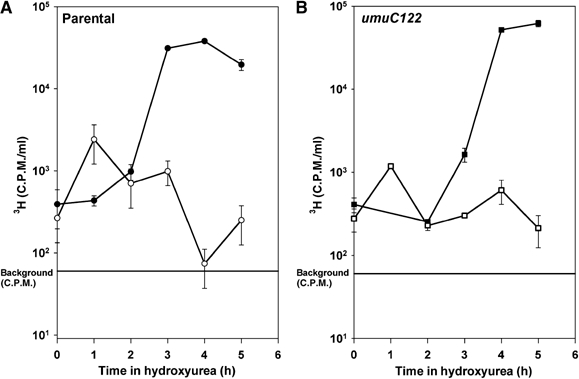
DNA synthesis is slowed in both wild-type and umuC122 strains. Thymidine-requiring derivatives of both strains were used for the experiments shown. 3H-Thy was added at 1 μCi/ml for 10 min at each time point shown, after which cells were immediately precipitated with 10% TCA. For both the wild-type shown in (A) (circles) and umuC122 shown in (B) (squares) strains, bulk DNA replication is slowed during hydroxyurea treatment. The straight line represents the background c.p.m. Error bars represent the standard deviation of three samples.
A strain bearing a mazEF or relBE mutation is also resistant to HU
Although wild-type and umuC122 strains display comparable levels of bulk DNA synthesis during HU treatment, only in the umuC122 mutant is this activity beneficial for survival. It seemed possible that the wild-type strain loses viability not directly due to stalled replication forks that arise during HU treatment, but instead due to events that occur downstream of such stalled forks. Examination by microscopy of an HU-treated parental culture revealed drastically fewer cells (>90% reduction at 5 h) than in the umuC122 strain, most likely due to cell lysis. Hence, we considered the phenomenon of thymineless death, which is also thought to be the product of stalled replication forks formed by substrate starvation (Ahmad et al, 1998). In E. coli strain MC4100, thymineless death is mediated at least in part by the mazEF genes (Sat et al, 2003), which encode a toxin–antitoxin pair. We speculated that HUR and thymineless death may proceed through similar mechanisms.
Therefore, we examined the sensitivity to HU of an MC4100 derivative harboring a deletion of the mazEF genes (Aizenman et al, 1996). Not only does deletion of these genes protect cells from the lethal consequences of HU challenge (Figure 6A), but the mechanism of HUR is also likely to be related to that of the umuC122 strain. Microscopical examination during HU treatment indicates that umuC122 and mazEF strains appear quite similar at the single-cell level (Figure 6B). No morphological difference is visible among the strains 1 h into HU treatment (panels A–D), but each HUS parental strain had to be concentrated an additional five-fold to analyze comparable numbers of cells relative to its HUR derivative. Finally, at 5 h (panels E–H), we observed similar responses in both HUS parental strains (concentrated 15-fold relative to their HUR derivatives). In comparison, the umuC122 and mazEF strains show extreme elongation and no dead cells, suggesting that HUR may arise through a similar mechanism in both strains. Therefore, it is plausible that the HUR phenotype of the umuC122 mutant may be due to a failure to transduce a signal in a mazEF-dependent pathway leading to cell death and lysis (Aizenman et al, 1996).
Figure 6.
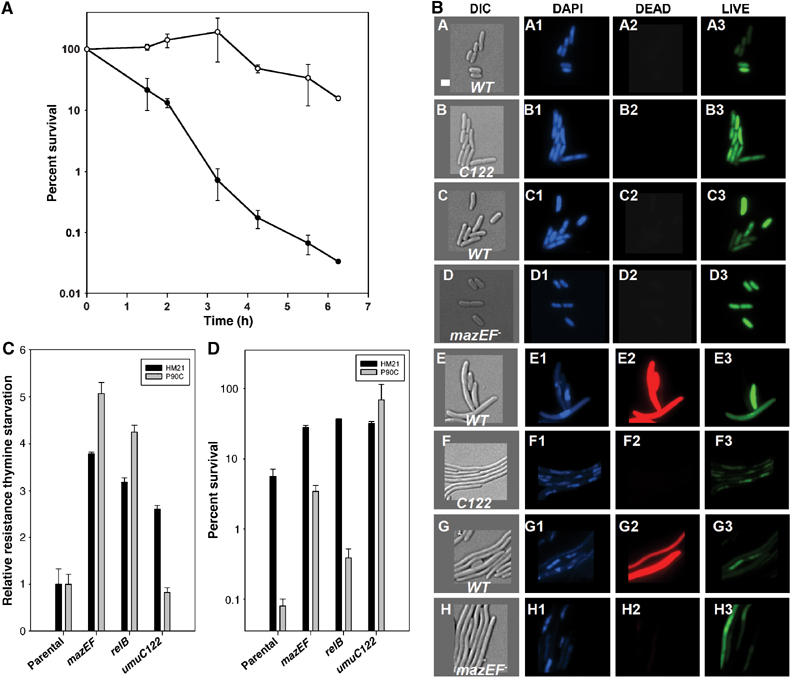
Survival phenotypes under dNTP starvation. (A) Survival time course in 100 mM HU of the parental MC4100 derivative (closed circles) and the mazEF mutant strain (open circles) in LB. Error bars shown represent the standard deviation of two samples. (B) Strains bearing the indicated alleles and wild-type control backgrounds were treated with 100 mM HU to determine cell morphology under HU treatment. Micrographs are presented for treated cells only because untreated samples of each strain showed indistinguishable morphologies over 5 h without HU. Panels A–D show representative images of cells after 1 h of HU treatment. (A, B) P90C wild-type and umuC122 control DIC image, (C, D) MC4100 wild-type and mazEF− control DIC image. Images labeled 1 show DAPI staining, images labeled 2 show DEAD staining and images labeled 3 show LIVE staining. Panels E–H are corresponding representative images of cells following treatment with HU for 5 h. Images were colorized using OpenLab software (Improvision) and were sized in Canvas (Deneba Systems). The white bar in (A) represents 2 μm. Exposure times for the images were as follows: DIC, 0.03 s; DAPI, 0.13 s; LIVE, 0.01 s; and DEAD, 0.13 s. The LIVE/DEAD stain was used according to the manufacturer's recommendations (Molecular Probes). (C) Mutation in the relBE gene products protects (denotes relB in this figure) cells from thymine starvation. Error bars represent the standard deviation of three samples. (D) Deletion of the relBE genes also promotes HUR. Treatment with Tp in HM21 (4 μg/ml) and P90C (7 μg/ml) strains was performed in appropriately supplemented M9 minimal medium. CFUs were determined after 16 h incubation. HU challenge (100 mM) was carried out in LB appropriately supplemented medium. Error bars shown represent the standard deviation of three samples.
We then tested whether a different TA pair may protect cells from the lethality caused by thymine starvation or HU challenge. Thus, we constructed P90C derivatives that harbored deletions of either the mazEF or relBE genes (Gerdes et al, 2005). We also transduced the umuC122 allele into the E. coli strain HM21, the donor of the mazEF and relBE deletion alleles. We tested the mazEF and relBE strains in both backgrounds for HUR and response to thymine starvation using trimethoprim (Tp) to inhibit thyA. We found that the relBE deletion protects cells from inhibition of fork progression upon thymine starvation similarly to mazEF (Figure 6C), and that both strains showed comparable responses upon HU challenge (Figure 6D). Moreover, we found the umuC122 allele confers resistance to both Tp (Figure 6C) and HU (Figure 6D) in the HM21 strain background, although this HUR is of a lower magnitude than that observed in the P90C strain. In contrast, the umuC122 allele does not confer resistance to Tp in the P90C background. This results suggest that there may be communication between pathways that couple HU- and Tp-induced stalled replication forks to cell death, and that a factor(s) involved in such communication is/are absent in the P90C strain, which bears a ∼105 kb deletion on its chromosome. Moreover, both pathways appear to utilize the relBE and mazEF TA pairs as their ultimate executioners.
HU-treated strains bearing the truncated UmuC protein have a high mutation frequency
Our findings raise the possibility that the four proteins we have identified as being critical for HUR—DinB, a UmuC derivative, UmuD, and UmuD′—enhance cell survival under conditions of low dNTP concentrations. They may even take over much of DNA replication, thereby helping cells to replicate even in the presence of HU (Figure 1A). If DNA replication upon HU challenge is DinB- and UmuC dependent, one would expect such DNA synthesis on undamaged DNA to be less accurate than that carried out by the DNA Pol III holoenzyme. Therefore, we tested whether the mutation frequency to rifampicin resistance is changed before or after HU treatment in a umuC122 strain. We determined that untreated strains encoding UmuC122 protein have a spontaneous mutation frequency of 4±2 × 10−7, identical to the mutation frequency of the untreated umuC+ parental strain (4±3 × 10−7). However, after HU treatment, the mutation frequency of the umuC122 strain increases ca. 100-fold to 7±3 × 10−5, whereas the mutation frequency of the umuC+ parental strain remains at ca. 10−7. These data suggest that it may be possible to explain the HUR phenotype of strains bearing the umuC122 allele by a model in which one or both of the Y-family polymerases are responsible for a significantly greater proportion of DNA replication during HU treatment than under normal conditions.
Discussion
We examined the effect of inhibiting replication fork progression in a DNA damage-independent manner with HU in strains bearing different alleles of the umuC gene and found that cells bearing a carboxy-terminal truncation allele umuC122∷Tn5 (Elledge and Walker, 1983) are strikingly resistant to HU treatment (Figure 1A). Moreover, an unusual point mutation in UmuC (umuC125 allele, A39V) (Marsh et al, 1991) displays a similar phenotype (Figures 3A and D). We have shown that umuC122 is a gain-of-function allele that mediates HUR and encodes a gene product that could, in principle, perform DNA polymerization as its polymerase domain is intact (Boudsocq et al, 2002). DNA polymerase activity in such a mutant protein is not unprecedented as truncations of the carboxy-terminal domain of human Y-family polymerase η are TLS proficient in vitro (Broughton et al, 2002). XP-V patients (Masutani et al, 1999) bearing these C-terminal truncations tend to have more tumors than those carrying other Pol η alleles (Broughton et al, 2002). Indeed, we show that cells expressing a catalytically inactive UmuC122 protein are sensitive to HU (Figure 3A). We have also shown that the DinB protein (Figure 4A), and its catalytic activity (Figure 4B), is needed to observe the phenotype. In addition, we have learned that certain umuD gene products are required for the HUR phenotype (Figures 3B and C).
Analysis of HU-treated cultures by microscopy (Figure 6B) revealed not only that the HU-treated wild-type cells die, but that many also disappear over the course of treatment, presumably through cell lysis. These data challenged our expectation that stalled replication forks would simply arrest cell division and prevent colony formation. We had not anticipated that they would bring about cell lysis in and of themselves.
We have shown that cells treated with HU are affected in a process downstream of RNR inhibition (Figures 2B and C). The current model for replication stalling elicited by dNTP depletion is that substrate starvation brings about fork arrest and concomitant cell death (Roy et al, 2004). However, HU-treated Saccharomyces cerevisiae cells have been shown to exhibit both normal replication forks that can still sustain very slow DNA synthesis, as well as stalled replication forks (Sogo et al, 2002; Lopes et al, 2003). Moreover, HU-treated S. cerevisiae show a reduction in levels, but not an absence, of dNTPs (Koc et al, 2004). Hence, the dNTP starvation model may be too simplistic to account for all these observations.
Therefore, we considered whether the HUR mediated by these gain-of-function alleles of umuC is due to an abrogation in a pathway that would normally lead to cell death under conditions of dNTP starvation. We found that E. coli strains bearing a deletion of such a function (mazEF∷Kan) (Aizenman et al, 1996) are also HUR (Figures 6A and B). We also found that deletion of relBE protects cells from both thymine starvation and HU challenge (Figures 6C and D). It is likely that the function of the mazEF and relBE gene products is to slow metabolism, thereby enabling stasis and resumption of balanced growth (Pedersen et al, 2002; Gerdes et al, 2005). However, when challenged with dNTP starvation, cells are unable to recover from this stasis and eventually perish. Based on these data, HU-induced death of E. coli may be brought about not by stalled replication forks directly, but rather through a series of downstream processes involving the TA pairs mazEF and relBE. The UmuC variants, acting in combination with the dinB and umuD gene products, may mitigate such mazEF- or relBE-induced death, either directly or indirectly. Further studies will be needed to establish whether and to what extent replication fork collapse is required to signal such lethal pathways, as well as other factors that might be involved. It will be interesting to look for a function that would bestow TpR in the P90C umuC122 derivative (Figure 6D). This strain harbors a large deletion (Δ(lac-pro), ca. 105 kb) compared to the HM21 background, where the umuC122 derivative is TpR (Figure 6C).
In E. coli, intracellular dNTP pools are at least 10-fold lower (10 μM) in the presence of HU than in untreated cells (100 μM) (Sinha and Snustad, 1972; Mathews and Sinha, 1982). One explanation for HU-induced stalled replication forks is that the replicative DNA polymerase cannot catalyze efficient DNA synthesis as its Km for dNTPs (3–40 μM for DNA Pol III) (Kornberg and Baker, 1991) is higher than the concentrations of dNTPs present in the HU-treated cells. In comparison, the Km for dNTPs of Pol IV (0.12 μM for His-DinB with the β processivity clamp) and Pol V (0.08 μM with RecA versus 1200 μM without) are much lower (Tang et al, 2000; Wagner et al, 2000). Therefore, it appears the E. coli Y-family DNA polymerases have the potential to operate efficiently at low dNTP concentrations, conditions at which DNA Pol III would operate poorly. Furthermore, such capabilities seem to be dramatically regulated through protein–protein interactions.
All these data are consistent with the notion that DinB, UmuC, and the umuD gene products are recruited to stalled replication forks upon HU treatment. We propose that the UmuC derivatives alter the highly dynamic process of polymerase switching, so that Y-family polymerases are defective in the switch back to the replicative polymerase. Ordinarily, UmuC, UmuD, and DinB would be part of a transient complex relieving arrested replication forks, regardless of how they arise. Both Y-family polymerases would work together to enhance cell survival, perhaps with DinB extending primers that are misaligned on their templates (Wagner et al, 1999) and UmuC continuing replication before hand off of the primer terminus to the replicative DNA polymerase. Such polymerase switching is regulated by numerous factors in E. coli including the umuD gene products (Sutton and Walker, 2001a). In contrast, the UmuC variants would be recruited to HU-induced stalled forks and would be proficient to catalyze DNA synthesis, but would be unable to sense the signal to hand off the primer terminus to the replicative DNA polymerase. Hence, these UmuC derivatives would retain access to the replication fork unlike the wild-type protein. The unexpected finding (Figure 5A) that wild-type cells still carry out DNA replication upon HU challenge may be explained by a futile cycling of Y-family polymerase recruitment and subsequent handoff to the replicative DNA polymerase which cannot function effectively at the low dNTP levels of the cell. Furthermore, although umuC122 is nonmutable in vivo with respect to UV, its gene product may be able to catalyze DNA polymerization on undamaged templates. Under normal circumstances, such prolonged access to the fork would be detrimental, but during the unique stress of HU treatment (low dNTPs), it is advantageous for survival, albeit at a mutagenic penalty.
Why does this apparent failure to hand off to the replicative polymerase in the umuC mutants prevent HU-induced death? Although it is possible that UmuC communicates directly with either or both of the mazEF and relBE gene products, thereby signaling cell death in response to stalled replication forks, it is perhaps more likely that the prolonged action of the UmuC derivatives at the replication fork prevents the generation of an intermediate that would lead to the mazEF- and relBE-dependent process of cell death and lysis. We suggest a factor that responds to one of these intermediates that is specific to thymineless death is missing in the P90C strain, explaining why the umuC122 derivative behaves as the wild type upon Tp challenge. The carboxy-terminus of UmuC harbors interaction sites for both UmuD2 and UmuD2′ (Jonczyk and Nowicka, 1996; Sutton and Walker, 2001b), which are absent in the UmuC122 protein. Perhaps, the lack of this domain alters the ability of the UmuC122 protein to return the primer terminus to the replicative DNA polymerase. Moreover, the data in Figures 3B and C highlight the role of UmuD cleavage in HUR. Alternatively, the truncated UmuC122 protein may remain at the replication fork due to altered interaction with the β-subunit of Pol III as deletion of its C-terminus may modify the accessibility of its β-binding motif (residues 357–361) (Becherel et al, 2002). It is clear that umuC122 and ΔumuC are both loss of function alleles for UV- and chemical-induced mutagenesis in exponentially growing cells. However, phenomena tested using umuC122 should be reevaluated. In comparison, the A39V mutation in the UmuC125 protein is in close proximity to the active site (ca. 6 Å; Figure 3D). The phenotype conferred by the umuC125 allele may be due to either disruption of regulatory protein–protein interactions with similar consequences to the umuC122 mutation or to alteration of the biochemical properties of the protein, such as a reduction in koff for the primer/template, Km for dNTP substrates, or both. In either case, the consequence is prolonged access to the replication fork under conditions of nucleotide starvation, resulting in survival during HU challenge.
If these polymerases replicate DNA in the presence of HU, mutability should be markedly higher in the mutant strains relative to the wild type. Indeed, the umuC122 bearing strain displays a 100-fold higher mutation frequency upon HU treatment than its untreated counterpart or the wild-type strain. Intriguingly, before the discovery of Y-family polymerases, it has been reported that imbalances in dNTP pools increase mutagenesis, perhaps by decreasing the fidelity of DNA synthesis (Sargent and Mathews, 1987; Ji and Mathews, 1991; Mun and Mathews, 1991; Zhang et al, 1996). This reduction in fidelity could perhaps now be attributed to the recruitment of such Y-family polymerases to the replication forks under conditions of nucleotide imbalance.
Materials and methods
Strains and plasmids
We used different E. coli K12 strains and their isogenic derivatives (Table I): P90C (Cairns and Foster, 1991), AB1157 (Bachmann, 1987), and HM21 (Moyed and Bertrand, 1983). A precise deletion of dinB was constructed using the method described by Wanner et al (Datsenko and Wanner, 2000) with primers FW2 (5′acgcgttaaatgctgaatctttacgcatttctcaaacc3′) and RW2 (5′gtgatattgaccgatttttcagcgagaattcgatgcat3′). The deletion was transduced by P1 (Miller, 1974) into the appropriate strains from BW25113 (Datsenko and Wanner, 2000). P1 transduction was also used to transfer the umuC122 allele (Elledge and Walker, 1983), a deletion of the umuDC operon (Woodgate, 1992), and a precise deletion of umuC. Wild-type and umuC122 thyA− derivatives were constructed by P1 transduction from the strain EGSC#6827. The dinB003 allele (Wagner et al, 1999) was constructed on the chromosome of BW25113 using the plasmid-borne allele as a template. The umuDC-containing plasmids are derivatives of pGB2 (Sutton and Walker, 2001b). The noncleavable UmuD(S60A) allele (Nohmi et al, 1988) was introduced by site-directed mutagenesis using a Quickchange kit (Stratagene, La Jolla, CA) with the following oligonucleotide (5′gcaagtggtgatgctatgattgatggtgg3′) and its reverse complement. The umuC122 allele was reconstructed in the same plasmid system using the primer (5′ccactcaggacagcagggattgaatagatagttaaacgcgatctctggatgc3′) and its reverse complement.
Table 1.
Strains and plasmids used in this study
| Description | Reference | |
|---|---|---|
| Bacterial strains | ||
| P90C | Δ(lac-pro)XIII thi ara | Cairns and Foster (1991) |
| P90C umuC122::Tn5 | As P90C, but with Tn5 insertion in the umuC gene | This work |
| P90C ΔdinB | As P90C, but bearing a precise deletion of the dinB gene and replacement by cat | This work |
| P90C ΔumuC | As P90C, but bearing a deletion of the umuC gene | S Lovett |
| P90C ΔumuDC | As P90C, but bearing a deletion of the umuDC genes and replacement by cat | This work |
| P90C umuC122ΔdinB | As P90C umuC122, but bearing a precise deletion of dinB gene and replacement by cat | This work |
| P90C umuC122dinB003 | As P90C umuC122, but with dinB003 encoding DinB D103N | This work |
| P90C umuC122dinB+ | As P90C umuC122, but with cat upstream of dinB | This work |
| P90C ΔumuDCΔdinB | As P90CΔdinB, but bearing a deletion in the umuDC genes | This work |
| P90C thyA | As P90C, but with a deletion in the thyA gene linked to Tn10 | This work |
| P90C umuC122::Tn5 thyA | As P90C umuC122::Tn5, but with a deletion in the thyA gene linked to Tn10 | This work |
| P90C ΔrelBE | As P90C, but with a deletion of the relBE genes and replacement by a KanR marker | This work |
| P90C ΔmazEF | As P90C, but with a deletion of the mazEF genes and replacement by a KanR marker | This work |
| AB1157 | F− thr-1 leuB6 proA2 his4 thi1 argE3 lacY1 galK2 rpsL supE44 ara-14 xyl-15 mtl-1, txs-33 | Walker Lab Stock |
| AB1157 umuC122::Tn5 | As AB1157, but bearing a Tn5 insertion in the umuC gene | This work, Walker Lab Stock |
| HM21 | F+ dapA zde-264::Tn10 | K Lewis |
| HM21 ΔrelBE | As HM21, but bearing a deletion of the relBE genes and replacement by a KanR marker | K Lewis |
| HM21 ΔmazEF | As HM21, but bearing a deletion of the mazEF genes and replacement by a KanR marker | K Lewis |
| HM21 umuC122::Tn5 | As HM21, but carrying a Tn5 insertion in umuC gene | This work |
| MC4100 relA+ | araD139 Δ(argF-lac)205 flb-5301 pstF25 rpsL150 deoC1 | H Engelberg-Kulka |
| MC4100 relA+ ΔmazEF | As MC4100, but bearing a deletion of the mazEF genes and replacement by a KanR marker | H Engelberg-Kulka |
| Plasmids | ||
| pGB2 | pSC101 derivative, bearing an SpR marker | Walker Lab Stock |
| pDC | As pGB2 bearing the umuDC genes | Walker Lab Stock |
| pDC125 | As pDC, but the umuC gene carries a A39V mutation | Walker Lab Stock |
| pD′C125 | As pDC125, but the umuD gene encodes only the 24 aa shorter protein UmuD′ | This work |
| pDC122 | As pDC, but carrying a truncation in the umuC gene | This work |
| pD′C122 | As pDC122, but the umuD gene encodes only for the 24 aa shorter protein UmuD′ | This work |
| pDC104 | As pDC, but carrying a D104N mutation in the umuC gene | Walker Lab Stock |
| pDC122/C104 | As pDC, but carrying a truncation in the umuC gene and a D104N mutation | This work |
| pD(S60A)C122 | As pDC, but carrying an S60A mutation in the umuD gene | This work |
| pD(S60A)C125 | As pDC125, but carrying a S60A mutation in the umuD gene | This work |
Strains were grown routinely in liquid or solid media (LB) or in minimal M9 medium with the addition of HU (30–100 mM), ampicillin (Amp; 100 μg/ml), spectinomycin (Sp; 60 μg/ml), chloramphenicol (Cm; 10–20 μg/ml), kanamycin (Kan; 50 μg/ml), rifampicin (Rif; 100 μg/ml), trimethoprim (Tp; 3–7 μg/ml), diaminopimelic acid (DAP; 30 μg/ml) and thymine (Thy; 50 μg/ml) whenever required. The dinB+ locus was reconstructed on the chromosome using the same approach as the dinB003 construction in the CmS derivative of umuC122ΔdinB mutant. The locus was transduced with P1 phage, and the presence of the full-length dinB+ gene was verified by PCR with the primers dinBF, 5′atgcgtaaaatcattcatgtgga3′ and dinBR, 5′tcataatcccagcaccagttgt3′.
HU treatment
Cultures were routinely treated in LB broth containing HU (Calbiochem) by diluting saturated cultures 1:1000. Treatment of ca. 106 bacteria/ml was for 6 h or as noted in the text or figure legends. Viability was checked throughout treatment. For anaerobic treatment with HU, cultures were treated as above for 6 h with 55 mM HU in an anaerobic chamber (Coy Laboratory Products) with a mixture of 5% carbon dioxide, 10% hydrogen, and 85% nitrogen. Samples for Western blotting were either TCA precipitated (20%) or concentrated 100-fold. The αUmuC antibody was used at a dilution of 1:20 000. The secondary antibody dilution and further detection were performed following the manufacturer's instructions (Pierce Biotechnology).
For the thymidine incorporation during HU treatment (100 mM), we used a 1:1 mixture of M9 medium (Miller, 1974) with 0.3% casein to LB with 10 μg/ml of thymidine. The 3H-Thy (Perkin-Elmer) incorporation was carried out in 10 min pulses, after which the sample was immediately TCA precipitated (10% final).
Acknowledgments
We would like to thank Sue Lovett (Brandeis) for providing us with the ΔumuC mutant strain, Hannah Engelberg-Kulka (Hebrew University) for the mazEF strains, Kim Lewis (Northeastern University) for the HM21 mazEF and relBE strains, and the E. coli genetic stock center for the #6827 strain. We also thank JoAnne Stubbe (MIT) for the antibodies to RNR subunits, Alan Grossman (MIT) for use of the microscope, and Michael Malamy (Tufts Medical School) for use of the anaerobic chamber. LAS was supported in part by a postdoctoral fellowship from NCI. This work was supported with the NIH Grant No. CA21615-27. GCW is an American Cancer Society Research Professor.
References
- Ahmad SI, Kirk SH, Eisenstark A (1998) Thymine metabolism and thymineless death in prokaryotes and eukaryotes. Annu Rev Microbiol 52: 591–625 [DOI] [PubMed] [Google Scholar]
- Aizenman E, Engelberg-Kulka H, Glaser G (1996) An Escherichia coli chromosomal ‘addiction module' regulated by guanosine [corrected] 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci USA 93: 6059–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann JB (1987) Escherichia coli and Salmonella typhimurium. Cellular and Molecular Biology. Washington, DC: American Society for Microbiology [Google Scholar]
- Bates H, Bridges BA, Woodgate R (1991) Mutagenic DNA repair in Escherichia coli, XX. Overproduction of UmuD′ protein results in suppression of the umuC36 mutation in excision defective bacteria. Mutat Res 250: 99–204 [DOI] [PubMed] [Google Scholar]
- Battista JR, Ohta T, Nohmi T, Sun W, Walker GC (1990) Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc Natl Acad Sci USA 87: 7190–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becherel OJ, Fuchs RP, Wagner J (2002) Pivotal role of the beta-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair (Amsterdam) 1: 703–708 [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Boudsocq F, Ling H, Yang W, Woodgate R (2002) Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair (Amsterdam) 1: 343–358 [DOI] [PubMed] [Google Scholar]
- Boye E, Stokke T, Kleckner N, Skarstad K (1996) Coordinating DNA replication initiation with cell growth: differential roles for DnaA and SeqA proteins. Proc Natl Acad Sci USA 93: 12206–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton BC, Cordonnier A, Kleijer WJ, Jaspers NG, Fawcett H, Raams A, Garritsen VH, Stary A, Avril MF, Boudsocq F, Masutani C, Hanaoka F, Fuchs RP, Sarasin A, Lehmann AR (2002) Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc Natl Acad Sci USA 99: 815–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burckhardt SE, Woodgate R, Scheuermann RH, Echols H (1988) UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc Natl Acad Sci USA 85: 1811–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns J, Foster PL (1991) Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128: 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JR, LeClerc JE, Tata PV, Christensen RB, Lawrence CW (1988) UmuC function is not essential for the production of all targeted lacI mutations induced by ultraviolet light. J Mol Biol 203: 635–641 [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274: 1664–1672 [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Walker GC (1983) Proteins required for ultraviolet light and chemical mutagenesis. Identification of the products of the umuC locus of Escherichia coli. J Mol Biol 164: 175–192 [DOI] [PubMed] [Google Scholar]
- Fontecave M, Eliasson R, Reichard P (1989) Oxygen-sensitive ribonucleoside triphosphate reductase is present in anaerobic Escherichia coli. Proc Natl Acad Sci USA 86: 2147–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti JJ, Schienda J, Sutera VA Jr, Lovett ST (2005) A bacterial G protein-mediated response to replication arrest. Mol Cell 17: 549–560 [DOI] [PubMed] [Google Scholar]
- Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R (1996) Regulation of SOS mutagenesis by proteolysis. Proc Natl Acad Sci USA 93: 10291–10296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Wagner R, Radman M (2002) Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science 296: 1627–1630 [DOI] [PubMed] [Google Scholar]
- Gerdes K, Christensen SK, Lobner-Olesen A (2005) Prokaryotic toxin–antitoxin stress response loci. Nat Rev Microbiol 3: 371–382 [DOI] [PubMed] [Google Scholar]
- Goodman MF (2002) Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem 71: 17–50 [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Kastan MB (1994) Cell cycle control and cancer. Science 266: 1821–1828 [DOI] [PubMed] [Google Scholar]
- Ji JP, Mathews CK (1991) Analysis of mutagenesis induced by a thermolabile T4 phage deoxycytidylate hydroxymethylase suggests localized deoxyribonucleotide pool imbalance. Mol Gen Genet 226: 257–264 [DOI] [PubMed] [Google Scholar]
- Jonczyk P, Nowicka A (1996) Specific in vivo protein–protein interactions between Escherichia coli SOS mutagenesis proteins. J Bacteriol 178: 2580–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Wang TS (2003a) Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev 17: 64–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Wang TS (2003b) Checkpoint responses to replication stalling: inducing tolerance and preventing mutagenesis. Mutat Res 532: 59–73 [DOI] [PubMed] [Google Scholar]
- Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T (2001) Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266: 207–215 [DOI] [PubMed] [Google Scholar]
- Koc A, Wheeler LJ, Mathews CK, Merrill GF (2004) Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem 279: 223–230 [DOI] [PubMed] [Google Scholar]
- Koch WH, Ennis DG, Levine AS, Woodgate R (1992) Escherichia coli umuDC mutants: DNA sequence alterations and UmuD cleavage. Mol Gen Genet 233: 443–448 [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker TA (1991) DNA Replication. New York: WH Freeman & Company [Google Scholar]
- Lopes M, Cotta-Ramusino C, Liberi G, Foiani M (2003) Branch migrating sister chromatid junctions form at replication origins through Rad51/Rad52-independent mechanisms. Mol Cell 12: 1499–1510 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- Marsh L, Nohmi T, Hinton S, Walker GC (1991) New mutations in cloned Escherichia coli umuDC genes: novel phenotypes of strains carrying a umuC125 plasmid. Mutat Res 250: 183–197 [DOI] [PubMed] [Google Scholar]
- Marsh L, Walker GC (1985) Cold sensitivity induced by overproduction of UmuDC in Escherichia coli. J Bacteriol 162: 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399: 700–704 [DOI] [PubMed] [Google Scholar]
- Mathews CK, Sinha NK (1982) Are DNA precursors concentrated at replication sites? Proc Natl Acad Sci USA 79: 302–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLenigan M, Peat TS, Frank EG, McDonald JP, Gonzalez M, Levine AS, Hendrickson WA, Woodgate R (1998) Novel Escherichia coli umuD′ mutants: structure–function insights into SOS mutagenesis. J Bacteriol 180: 4658–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH (1974) Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory [Google Scholar]
- Moyed HS, Bertrand KP (1983) hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murine synthesis. J Bacteriol 155: 768–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun BJ, Mathews CK (1991) Cell cycle-dependent variations in deoxyribonucleotide metabolism among Chinese hamster cell lines bearing the Thy− mutator phenotype. Mol Cell Biol 11: 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP (2000) All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J 19: 6259–6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohmi T, Battista JR, Dodson LA, Walker GC (1988) RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc Natl Acad Sci USA 85: 1816–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori H, Friedberg EC, Fuchs RP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R (2001) The Y-family of DNA polymerases. Mol Cell 8: 7–8 [DOI] [PubMed] [Google Scholar]
- Opperman T, Murli S, Smith BT, Walker GC (1999) A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc Natl Acad Sci USA 96: 9218–9223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Christensen SK, Gerdes K (2002) Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol Microbiol 45: 501–510 [DOI] [PubMed] [Google Scholar]
- Roy B, Guittet O, Beuneu C, Lemaire G, Lepoivre M (2004) Depletion of deoxyribonucleoside triphosphate pools in tumor cells by nitric oxide. Free Radic Biol Med 36: 507–516 [DOI] [PubMed] [Google Scholar]
- Sargent RG, Mathews CK (1987) Imbalanced deoxyribonucleoside triphosphate pools and spontaneous mutation rates determined during dCMP deaminase-defective bacteriophage T4 infections. J Biol Chem 262: 5546–5553 [PubMed] [Google Scholar]
- Sargentini NJ, Smith KC (1984) umuC-dependent and umuC-independent gamma- and UV-radiation mutagenesis in Escherichia coli. Mutat Res 128: 1–9 [DOI] [PubMed] [Google Scholar]
- Sat B, Reches M, Engelberg-Kulka H (2003) The Escherichia coli mazEF suicide module mediates thymineless death. J Bacteriol 185: 1803–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinagawa H, Iwasaki H, Kato T, Nakata A (1988) RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc Natl Acad Sci USA 85: 1806–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha NK, Snustad DP (1972) Mechanism of inhibition of deoxyribonucleic acid synthesis in Escherichia coli by hydroxyurea. J Bacteriol 112: 1321–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeden J, Loeb L (2004) Mutations in the R2 subunit of ribonucleotide reductase that confer resistance to hydroxyurea. J Biol Chem 279: 40723–40728 [DOI] [PubMed] [Google Scholar]
- Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- Sommer S, Becherel OJ, Coste G, Bailone A, Fuchs RP (2003) Altered translesion synthesis in E. coli Pol V mutants selected for increased recombination inhibition. DNA Repair (Amsterdam) 2: 1361–1369 [DOI] [PubMed] [Google Scholar]
- Stubbe J (2003) Di-iron-tyrosyl radical ribonucleotide reductases. Curr Opin Chem Biol 7: 183–188 [DOI] [PubMed] [Google Scholar]
- Sutton MD, Smith BT, Godoy VG, Walker GC (2000) The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu Rev Genet 34: 479–497 [DOI] [PubMed] [Google Scholar]
- Sutton MD, Walker GC (2001a) Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc Natl Acad Sci USA 98: 8342–8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MD, Walker GC (2001b) umuDC-mediated cold sensitivity is a manifestation of functions of the UmuD(2)C complex involved in a DNA damage checkpoint control. J Bacteriol 183: 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, Goodman MF (2000) Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 404: 1014–1018 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP (2000) The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep 1: 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T (1999) The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol Cell 4: 281–286 [DOI] [PubMed] [Google Scholar]
- Wagner J, Nohmi T (2000) Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity. J Bacteriol 182: 4587–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash L, Prakash S (2001) Accuracy of lesion bypass by yeast and human DNA polymerase eta. Proc Natl Acad Sci USA 98: 8355–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgate R (1992) Construction of a umuDC operon substitution mutation in Escherichia coli. Mutat Res 281: 221–225 [DOI] [PubMed] [Google Scholar]
- Woodgate R, Ennis DG (1991) Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol Gen Genet 229: 10–16 [DOI] [PubMed] [Google Scholar]
- Woodgate R, Rajagopalan M, Lu C, Echols H (1989) UmuC mutagenesis protein of Escherichia coli: purification and interaction with UmuD and UmuD′. Proc Natl Acad Sci USA 86: 7301–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lu Q, Inouye M, Mathews CK (1996) Effects of T4 phage infection and anaerobiosis upon nucleotide pools and mutagenesis in nucleoside diphosphokinase-defective Escherichia coli strains. J Bacteriol 178: 4115–4121 [DOI] [PMC free article] [PubMed] [Google Scholar]