

Abstract
Mice lacking epidermal growth factor receptor (EGFR) develop a neurodegeneration of unknown etiology affecting exclusively the frontal cortex and olfactory bulbs. Here, we show that EGFR signaling controls cortical degeneration by regulating cortical astrocyte apoptosis. Whereas EGFR−/− midbrain astrocytes are unaffected, mutant cortical astrocytes display increased apoptosis mediated by an Akt-caspase-dependent mechanism and are unable to support neuronal survival. The expression of many neurotrophic factors is unaltered in EGFR−/− cortical astrocytes suggesting that neuronal loss occurs as a consequence of increased astrocyte apoptosis rather than impaired secretion of trophic factors. Neuron-specific expression of activated Ras can compensate for the deficiency of EGFR−/− cortical astrocytes and prevent neuronal death. These results identify two functionally distinct astrocyte populations, which differentially depend on EGFR signaling for their survival and also for their ability to support neuronal survival. These spatial differences in astrocyte composition provide a mechanism for the region-specific neurodegeneration in EGFR−/− mice.
Keywords: cortical astrocytes, epidermal growth factor receptor, knockout mice, midbrain astrocytes, neurodegeneration
Introduction
The epidermal growth factor receptor (EGFR/erbB1) is one of four members of the ErbB family of tyrosine kinase receptors and is activated by ligand-dependent homo- and heterodimerization (Olayioye et al, 2000; Yarden and Sliwkowski, 2001; Schlessinger, 2002). EGFR-mediated signaling can stimulate cell proliferation, differentiation and inhibit apoptosis (Yarden and Sliwkowski, 2001). EGFR mutations have been shown to occur at high frequency in tumors of epithelial and glial origin (Olayioye et al, 2000). The same cell types are also affected in mice lacking the EGFR (Miettinen et al, 1995; Sibilia and Wagner, 1995; Threadgill et al, 1995; Kornblum et al, 1998; Sibilia et al, 1998), suggesting that EGFR signaling is important in normal astrocyte and epithelial cell physiology. Recently, we demonstrated that transgenic mice expressing an active form of Son-of-Sevenless in epidermis develop skin papillomas in an EGFR-dependent manner. In these mice the EGFR provides an essential survival signal in epithelial tumor cells (Sibilia et al, 2000).
Several studies suggest that EGFR and its ligands are implicated in important developmental processes in the brain. EGF has been shown to act as a potent mitogen for neural stem cells (Weiss et al, 1996). However, EGFR−/− neurospheres are not impaired in their ability to self-renew in response to FGF and their overall differentiation into neuronal as well as glial lineages does not seem to be affected in vitro (Tropepe et al, 1999). Before embryonic day 14 (E14), EGFR expression in the brain is low and concentrated at the apical surface of cells lining the lateral ventricle, but increases in a small proportion of cells during later gestation (Caric et al, 2001). EGFR expression was shown to regulate the competence of neural progenitors to interpret LIF as an astrocyte-inducing signal (Viti et al, 2003). In addition, upregulation of EGFR affects cell migration, since telencephalic cells can respond chemotactically to EGFR ligands (Caric et al, 2001) and infusion of EGF into the lateral ventricle diverts cells from the rostral migratory stream to the cortex (Craig et al, 1996). Overexpression of EGFR in rat embryonic striatal and cortical progenitors also promotes their premature departure from the ventricular zone and their differentiation into astrocytes, indicating that EGFR signaling plays an important role during astrocyte development (Burrows et al, 1997, 2000). Recent findings show that asymmetric distribution of the EGFR in neural progenitors during mitosis generates cells with different fates. Interestingly, the resulting EGFRhigh daughter cells are committed along the radial glia/astrocyte lineage (Sun et al, 2005).
Analysis of mice deficient for the EGFR family members revealed important functions of these receptors during development of the nervous system. Mice lacking erbB2, erbB3, erbB4 or the ligand heregulin are embryonic lethal and display heart defects and abnormal development of the central and peripheral nervous system (Gassmann et al, 1995; Lee et al, 1995; Meyer and Birchmeier, 1995; Riethmacher et al, 1997). Mice lacking erbB2 or erbB3 almost completely lack Schwann cells and motor and sensory neurons undergo cell death in later stages of development (Erickson et al, 1997; Riethmacher et al, 1997; Lin et al, 2000; Crone et al, 2003). Therefore, it appears that erbB2 and erbB3 exert important functions in the development and maintenance of the peripheral nervous system in both neurons as well as glial cells.
In contrast, EGFR seems to be important in the central nervous system. Mice lacking the EGFR die between midgestation and postnatal day 20 depending on the genetic background. Mutant mice exhibit epithelial (Miettinen et al, 1995; Sibilia and Wagner, 1995; Threadgill et al, 1995; Sibilia et al, 2003) and bone defects (Sibilia et al, 2003; Wang et al, 2004) and develop a progressive neurodegeneration starting after birth in the forebrain and affecting particularly the frontal cortex and olfactory bulbs. This degeneration is characterized by massive apoptotic cell death affecting both neurons and glial cells (Kornblum et al, 1998; Sibilia et al, 1998). Interestingly, other parts of the brain such as midbrain do not show any signs of degeneration. A migratory disorder is detected in the white matter of the hippocampus with nests of ectopic neurons, which are also undergoing apoptosis (Sibilia et al, 1998, 2003). Cerebral cortices from EGFR−/− mice contain lower numbers of astrocytes and their expansion is severely compromised also in vitro (Sibilia et al, 1998). These results suggest that EGFR signaling is involved in the proliferation and/or differentiation of astrocytes and the survival and migration of postmitotic neurons (Kornblum et al, 1998; Sibilia et al, 1998). However, it is not clear whether the massive cortical degeneration is caused by an intrinsic defect of the neurons, a defect of the astrocytes, which cannot support the survival of neurons or a combination of both.
Here, we investigated whether the astrocyte defects observed in EGFR−/− mice are sufficient to induce the neurodegeneration and whether the restriction of the neurodegeneration to the cortex is due to regional differences in astrocyte composition. In addition, we also asked whether triggering neuroprotective stimuli through the expression of RasV12 in postmitotic neurons is able to rescue the neurodegeneration of EGFR−/− mice.
Results
EGFR signaling is dispensable in neurons but differently required in cortical and midbrain astrocytes
Since the neurodegeneration in EGFR−/− mice is confined to the cortex, it is possible that this restriction reflects regional differences in neural cell composition and that mainly cortical cells are affected by the lack of EGFR. Comparison of neurons isolated from the cortex of EGFR−/− and wild-type mice did not reveal morphological differences when cultured in serum-free medium (Neurobasal supplemented with B27) that promotes the survival of neurons in the absence of astrocytes (Brewer et al, 1993). Both populations extended long axons after several days in culture indicating that neurons are not primarily affected in EGFR−/− cortex (Figure 1A and B). Midbrain and cortical mutant and wild-type astrocytes also appeared morphologically similar and glial fibrillary acidic protein (GFAP) staining revealed that differentiated astrocytes can be generated ex vivo from both brain regions regardless of the genotype (Figure 1C–F). However, midbrain mutant astrocytes did not show a growth defect in vitro as observed for EGFR−/− cortical astrocytes (Figure 1G) (Sibilia et al, 1998). After 50 days in culture, midbrain astrocyte numbers were similar to controls whereas the cumulative cell number of EGFR−/− cortical astrocytes was only 25% of controls (Figure 1G). These results show that EGFR is dispensable in neurons but differently required in midbrain versus cortical astrocytes.
Figure 1.
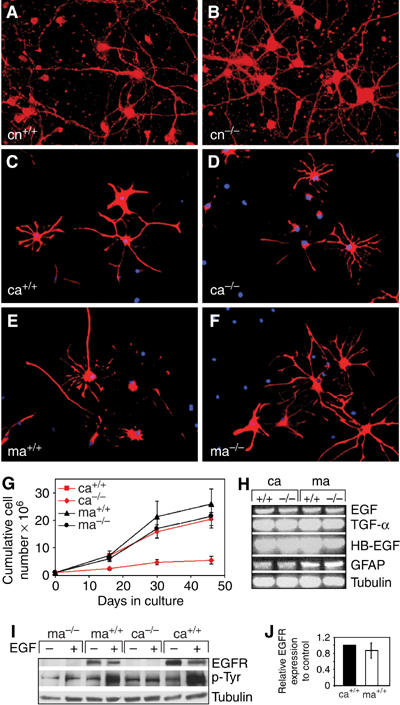
The EGFR is dispensable in neurons but differently required in astrocytes from various brain regions. (A, B) Cultured EGFR+/+ (cn+/+) and EGFR−/− (cn−/−) cortical neurons stained for the neuronal marker GAP-43 (red). (C–F) Immunofluorescence staining for GFAP (red) of cultured cortical EGFR+/+ (ca+/+) and EGFR−/− (ca−/−) and midbrain EGFR+/+ (ma+/+) and EGFR−/− (ma−/−) astrocytes. DAPI (blue) was used as a nuclear counterstain. (G) Cumulative cell numbers of EGFR+/+ and EGFR−/− cortical and midbrain astrocytes. Data represent mean±s.e.m. of four independent experiments. (H) RT–PCR analysis showing expression of GFAP and of the EGFR ligands EGF, TGFα and HB-EGF. Tubulin is used as loading control. (I) Western Blot analysis showing EGFR protein expression and tyrosine phosphorylation after stimulation with 50 ng/ml EGF. Tubulin is used as loading control. (J) Quantification of EGFR protein levels present in cortical and midbrain astrocytes relative to tubulin. Data represent mean±s.e.m. of three independent Western blot analyses normalized to control.
Analysis of EGFR expression in both astrocyte populations revealed that midbrain and cortical wild-type astrocytes expressed similar levels of EGFR protein, which was efficiently phosphorylated after EGF stimulation (Figure 1I and J). Moreover, RT–PCR analysis showed that the relative levels of EGFR ligands such as EGF, HB-EGF and TGFα expressed by wild-type and EGFR−/− cortical and midbrain astrocytes were comparable (Figure 1H). Similarly, the lack of EGFR did not affect the expression of GFAP (Figure 1H). These results show that, although cortical and midbrain astrocytes express comparable levels of EGFR and its ligands, midbrain astrocytes are not dependent on EGFR signaling for their expansion.
EGFR controls survival of cortical astrocytes
We next investigated the possible molecular mechanism responsible for the different requirement for EGFR signaling in cortical and midbrain astrocytes. Although expansion of EGFR−/− cortical astrocytes in vitro was severely impaired, the number of BrdU-positive cells was comparable among cortical and midbrain astrocytes regardless of their genotype (Figure 2A). FACS analysis of BrdU-labeled astrocytes also did not reveal any differences in cell cycle length between control and mutant cortical astrocytes (Figure 2B). In accordance with these findings, the expression of PCNA (Figure 2C) and several cell cycle regulators such as cyclins, cyclin-dependent kinases (CDKs) and their inhibitors p27, p16 (data not shown) was similar in all astrocyte populations. These results suggest that cell division was not primarily affected by the absence of EGFR. However, the number of apoptotic astrocytes was 3–4 times higher in EGFR−/− cortical cultures (Figure 2D). This was observed with two independent assays (Tunel and AnnexinV) and at different days after preparation (Figure 2D). Increased expression of genes directly involved in the execution of the apoptotic program could be detected in EGFR-deficient cortical astrocytes (Figure 2C and E). RNAse protection analysis revealed that the expression of caspase-1, -3, -8 and -12 was upregulated in EGFR−/− cortical astrocytes which was accompanied by a slight increase of active caspase-3 protein (Figure 2C and E). In contrast, the expression of AP-1 family members, which have also been implicated in neuronal apoptosis (Eferl and Wagner, 2003), was not significantly altered (data not shown). These results demonstrate that EGFR signaling is required for the survival but not for the proliferation of cortical astrocytes.
Figure 2.
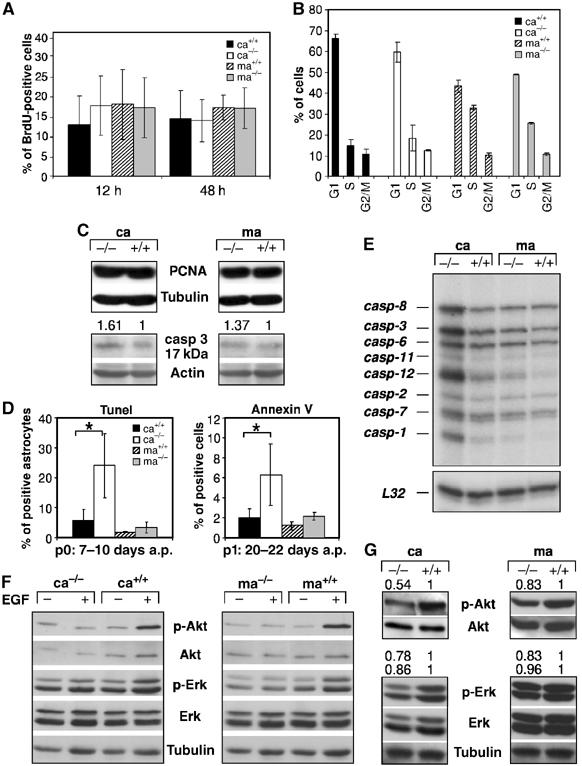
Increased apoptosis and reduced Akt activation in mutant cortical astrocytes. (A) Quantification of the number of BrdU-positive astrocytes (18 days a.p.) labeled for 12 and 24 h. Data represent the mean±s.e.m. of the number of positive cells counted in randomly chosen fields of three independent samples. (B) FACS analysis on primary astrocytes (18 days a.p.) pulsed for 12 h with BrdU showing the number of cells in the various stages of the cell cycle. Data represent the mean±s.e.m. of three independent samples. (C) Western blot analysis showing expression of the proliferating cell nuclear antigen (PCNA) and the active 17 kDa isoform of caspase-3 (casp-3). Actin served as loading control and the numbers indicate protein expression levels relative to controls normalized to actin. (D) Tunel and AnnexinV staining performed at the indicated time point after initial preparation (a.p.). For AnnexinV, data represent the mean±s.e.m. of the number of positive cells measured by FACS in three independent experiments. For Tunel staining, the data represent the percentages of Tunel and GFAP double-positive astrocytes after counting 700–1400 GFAP-positive astrocytes from randomly chosen fields of four independent experiments. (E) Expression of caspases in primary 80% confluent astrocytes of the indicated genotypes by RNase protection analysis. L32 mRNA served as loading control. (F) Akt and Erk 1/2 activation in cortical and midbrain astrocytes starved (−) for 18 h in medium containing 0.1% serum and stimulated with 20 ng/ml EGF (+) for 5 min. (G) Western blot analysis showing Akt and Erk 1/2 phosphorylation in 80% confluent astrocyte cultures of the indicated genotypes. Tubulin and total Akt and Erk served as loading controls. The numbers indicate protein expression levels relative to controls normalized to total Akt or Erk. Ca: cortical astrocytes; ma: midbrain astrocytes. *P<0.05.
Western blot analysis of downstream EGFR signaling components revealed that in wild-type cortical and midbrain astrocytes, EGF stimulation led to Erk 1/2 phosphorylation and to substantial activation of Akt, a kinase which positively regulates pathways leading to cell survival (Figure 2F). Erk 1/2 activation was only moderately reduced in exponentially growing mutant cortical and midbrain astrocytes (Figure 2G). In contrast, Akt phosphorylation was more severely impaired in EGFR−/− cortical astrocytes compared to mutant midbrain astrocytes (Figure 2G), showing that in cortical astrocytes efficient Akt activation depends on a functional EGFR signaling pathway. Therefore, reduced Akt activation together with upregulated caspase expression provides a molecular explanation for the increased apoptosis of EGFR-deficient cortical astrocytes.
Mutant cortical astrocytes do not support neuronal survival
It is well known that, unless grown in Neurobasal/B27 medium, neurons generally require co-culture with astrocytes to survive in culture (Banker, 1980). To test the possibility that reduced survival of cortical astrocytes also impairs their ability to keep neurons alive, we used an in vitro co-culture system, where astrocytes are plated on dishes and neurons are seeded on coverslips in serum-free medium with the addition of growth factors (Celis, 1998). Under these culture conditions, where physical contact between cells cannot be excluded, neurons can differentiate and survive due to the trophic support provided by astrocytes. When wild-type and EGFR mutant cortical neurons were co-cultured with wild-type cortical and midbrain astrocytes, neurons of both genotypes survived and extended long axons by 10 days in culture (Figure 3A, C, E and G). In contrast, most of the neurons, regardless of whether wild type or mutant had died in co-cultures with EGFR−/− cortical astrocytes (Figure 3B and D), whereas neurons co-cultured with EGFR−/− midbrain astrocytes survived perfectly well (Figure 3F and H). These results demonstrate that EGFR−/− cortical astrocytes are not able to support neuronal survival, whereas EGFR−/− midbrain astrocytes are fully capable to keep neurons alive.
Figure 3.
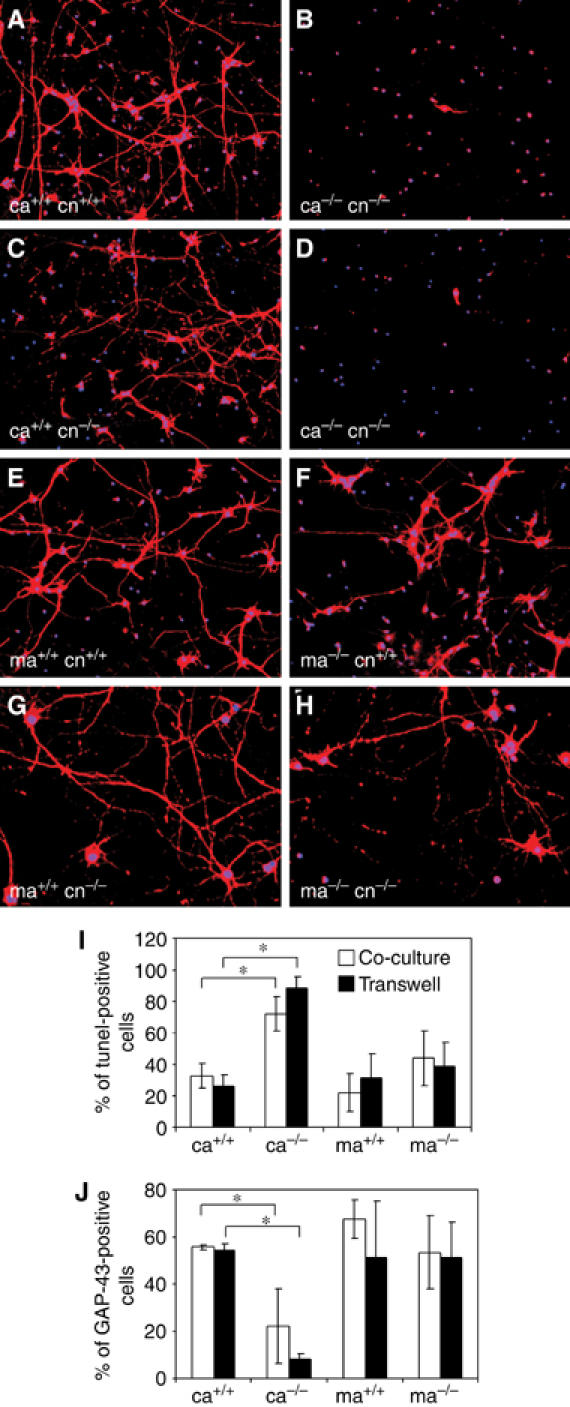
Neurons do not survive in co-culture with mutant cortical astrocytes. Cortical neurons (cn) from EGFR+/+ (A, B, E, F) and EGFR−/− (C, D, G, H) brains were co-cultured with wild-type (A, C) and EGFR−/− (B, D) cortical astrocytes (ca) as well as with wild-type (E, G) and mutant (F, H) midbrain astrocytes (ma). Immunofluorescence stainings were performed 10 days after co-culture using GAP-43 (red) as a neuronal marker and DAPI (blue) as a nuclear counterstain. (I, J) Neuronal survival measured by Tunel staining (I) and by the numbers of GAP-43-positive neurons (J) in co-cultures and transwell cultures 8 days after plating. In transwell cultures, astrocytes are plated on transwell dishes, which only allow diffusion of soluble factors. Data represent the mean±s.e.m. of the number of positive cells counted in randomly chosen fields of three independent experiments. Ca: cortical astrocytes; ma: midbrain astrocytes. *P<0.05.
A transwell culture system, which only allows the passage of secreted molecules, was used to test whether cell–cell contact between astrocytes and neurons is required for neuronal survival in co-culture. Transwell-cultured neurons survived and extended long axons in the presence of conditioned medium from cortical and midbrain EGFR+/+ as well as midbrain EGFR−/− astrocytes, suggesting that cell–cell contact was not necessary (Figure 3I and J). However, conditioned medium from EGFR−/− cortical astrocytes was unable to support neuronal survival. The number of Tunel-positive neurons was high with low numbers of surviving GAP-43-positive neurons (Figure 3I and J), suggesting that in these co-cultures the concentration of neurotrophic factors becomes limiting likely due to increased death of mutant cortical astrocytes. These results recapitulate the situation in vivo, where degeneration is only detected in the cortex but not in the midbrain of EGFR knockout mice, thereby strengthening the hypothesis that defects in cortical astrocytes are responsible for the neurodegeneration.
Increased apoptosis of EGFR−/− cortical astrocytes negatively affects neuronal survival
The reduced survival of EGFR−/− cortical astrocytes appears to compromise their ability to keep neurons alive in culture and therefore loss of neurons occurs as a consequence of astrocyte defects. This predicts that neuronal survival may be better at earlier co-culture time points when the number of EGFR−/− cortical astrocytes is higher. Therefore, the morphology and survival of cortical neurons with different astrocyte populations was assessed at various time points. On day 4 after co-culture, neurons cultured with wild-type and EGFR−/− cortical astrocytes appeared morphologically similar (Figure 4A and B) and the number of neurons extending axons was comparable in all co-cultures (Figure 4E). However, significant differences were detected between these cultures at day 8. While neurons cultured with wild-type cortical astrocytes extended many long axons, very few GAP-43-positive neurons were present in co-cultures with EGFR−/− cortical astrocytes (Figure 4C–E). Most surviving neurons had short axons and few cell–cell contacts (Figure 4D). Tunel staining revealed that on day 4 the number of apoptotic neurons was already increased in co-cultures with EGFR−/− cortical astrocytes (Figure 4F). This difference became more prominent by day 8 where more than 70% of neurons were apoptotic. These results indicate that mutant cortical astrocytes can initially trigger neuronal survival. However, at later time points, neuronal integrity is lost most likely due to increased death of EGFR−/− cortical astrocytes which cannot provide the necessary trophic support to prevent neuronal apoptosis.
Figure 4.
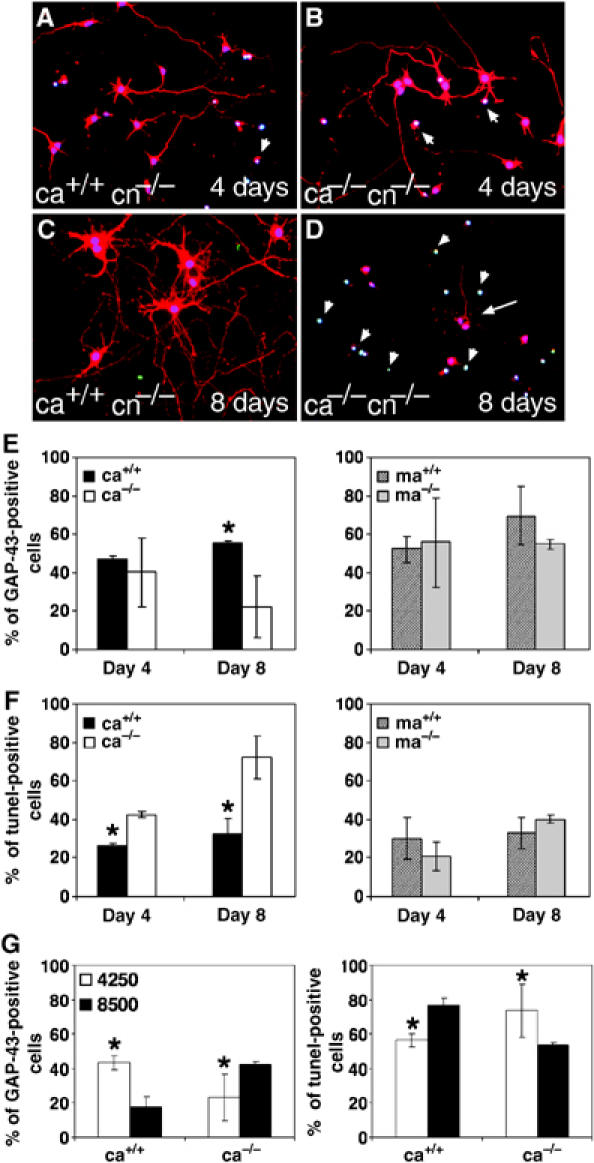
Neurons co-cultured with mutant cortical astrocytes undergo apoptosis. Immunofluorescence and Tunel staining of cortical neurons (cn) after 4 and 8 days of co-culture with wild-type (A, C) and EGFR−/− (B, D) cortical astrocytes. Stainings were performed using GAP-43 (red) as neuronal marker, DAPI (blue) as a nuclear counterstain and Tunel (green) for apoptotic neurons. Arrowheads point to apoptotic nuclei and arrow in (D) points to a surviving neuron. (E, F) Quantification of the number of GAP-43 (E)- and Tunel (F)-positive neurons present at various time points in co-cultures with cortical (ca) or midbrain (ma) astrocytes. (G) Percentage of GAP-43- and Tunel-positive neurons present in co-cultures with different concentrations of cortical astrocytes (4250 astrocytes/cm2=regular concentration). The data represent the mean±s.e.m. of the number of positive cells counted in randomly chosen fields of two independent experiments. Each experiment was performed in triplicates *P<0.05.
The number of astrocytes present in the co-cultures is critical for the survival of neurons (Banker, 1980) and too high astrocyte concentrations can compromise neuronal survival because medium nutrients are used up much faster thereby becoming limiting for neurons. We next investigated whether increasing the number of astrocytes would improve neuronal survival in the presence of mutant cortical astrocytes. As expected, increasing from 4250 (regular) to 8500 astrocytes/cm2 (high) decreased the survival rate of neurons with wild-type astrocytes (Figure 4G). However, high concentrations of EGFR−/− cortical astrocytes significantly improved neuronal survival, and after 8 days the number of living neurons was comparable to co-cultures with wild-type astrocytes at regular astrocyte concentrations (Figure 4G). These results show that doubling the number of EGFR−/− cortical astrocytes in the co-cultures can improve their capacity to keep neurons alive in co-culture.
Molecular analysis of neuronal loss in co-cultures with EGFR−/− cortical astrocytes
We next investigated whether secreted factors become limiting due to increased death of EGFR−/− cortical astrocytes or due to inefficient production. Vascular endothelial growth factor (VEGF) can be controlled by EGFR signaling and was recently implicated in several neurodegenerative disorders (Storkebaum and Carmeliet, 2004). However, VEGF secretion was not impaired in EGFR−/− cortical astrocytes (Figure 5A), thereby excluding its involvement. Also IGF-1, a well-known neuronal survival factor (Leinninger and Feldman, 2005), was abundantly secreted by mutant cortical astrocytes (Figure 5B).
Figure 5.
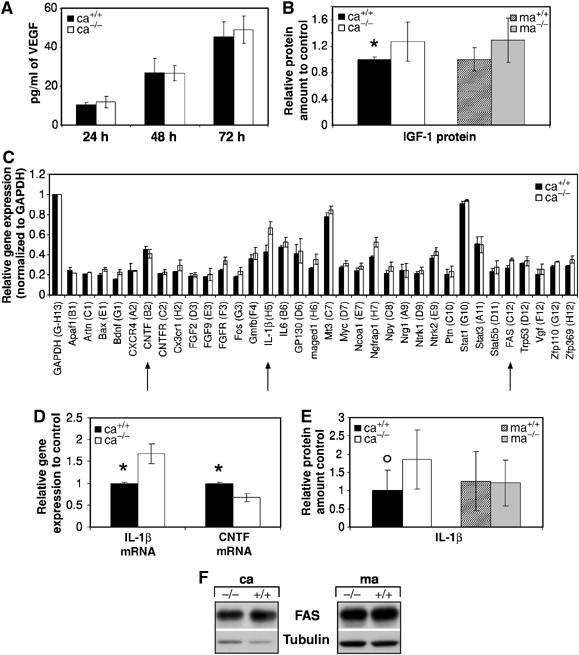
Mechanisms of neuronal loss in co-cultures with EGFR−/− cortical astrocytes. ELISA showing VEGF (A) and IGF-1 (B) protein levels in the supernatants of astrocyte cultures (18–20 days a.p.) of the indicated genotypes. The data represent the mean±s.e.m. of six independent samples. (C) Relative expression of each gene in wild-type and EGFR−/− cortical astrocytes after normalization to the house-keeping gene GAPDH. Normalization to Ppia gave similar results (data not shown). Only genes with signals above background are displayed. The data represent the mean±s.e.m. of the normalized values for each gene of two array experiments with two independent RNA batches. (D) Real-time quantitative RT–PCR analysis showing expression of CNTF and IL-1β. Similar results were obtained in a second independent RNA batch. Data represent the mean±s.e.m. of the relative expression levels to controls (ca+/+) after normalization with the house-keeping gene PBGD measured in two independent experiments. (E) IL-1β protein levels measured by ELISA in the supernatants of astrocyte cultures (18–20 days a.p.) of the indicated genotypes. The data represent the mean±s.e.m. of four independent samples. (F) Western blot analysis showing Fas expression in astrocytes of the indicated genotypes. Tubulin served as loading control. *P<0.05, °P=0.05.
Gene microarrays (GEArray) containing 96 neurotrophins and receptors were employed to analyze differential gene expression between wild-type and EGFR−/−cortical astrocytes (Figure 5C, Supplementary Figure 1). A factor slightly reduced in EGFR−/− cortical astrocytes was ciliary neurotrophic factor (CNTF), a neuroprotective factor produced by glial cells (Figure 5C) (Murphy et al, 1997). This result was independently validated by qRT–PCR (Figure 5D). However, addition of this factor to co-cultures with mutant cortical astrocytes did not improve neuronal survival (Table I). Moreover, addition of other factors such as EGF, NGF, bFGF, IL-6, T3 or a mixture thereof to co-cultures was not able to restore the ability of EGFR−/− cortical astrocytes to keep neurons alive (Table I). The expression of the proinflammatory cytokine IL-1β was significantly upregulated in EGFR−/− cortical astrocytes by GEArray and qRT–PCR analysis (Figure 5C and D). A slight increase of IL-1β could be detected in the supernatants of EGFR−/− cortical astrocytes (Figure 5E), raising the possibility that increased production of this cytokine might have neurotoxic effects. However, addition of IL-1β to co-cultures with wild-type cortical astrocytes did not affect neuronal survival (Table I). Moreover, addition of conditioned medium from EGFR−/− cortical astrocytes to neurons co-cultured with wild-type cortical and midbrain astrocytes also did not influence neuronal survival excluding that toxic factors are present in EGFR−/− cortical supernatants (data not shown). Several other genes such as nerve growth factor receptor associated protein 1 (Ngfrap1) and FAS, both of which are involved in the induction of apoptosis, were upregulated in EGFR−/− cortical astrocytes (Figure 5C); however, FAS protein levels were comparable to controls (Figure 5G). These results show that the expression of many neurotrophins and their receptors is not altered in EGFR−/− cortical astrocytes, but that the expression of several proapoptotic genes is upregulated.
Table 1.
Role of trophic factors in co-cultures with cortical astrocytes
| Genotype |
Growth factors | Neuronal survival | |
|---|---|---|---|
| Neurons | Astrocytes | ||
| cn−/− | ca+/+ | — | +++ |
| cn−/− | ca−/− | — | — |
| cn−/− | ca−/− | EGF | — |
| cn−/− | ca−/− | bFGF | — |
| cn−/− | ca−/− | IL-6 | — |
| cn−/− | ca−/− | T3 | — |
| cn−/− | ca−/− | NGF | — |
| cn−/− | ca−/− | CNTF | — |
| cn−/− | ca−/− | MIX | — |
| cn−/− | ca+/+ | — | +++ |
| cn−/− | ca+/+ | IL-1β | +++ |
| cn−/− | ca−/− | IL-1β | — |
| Mutant cortical neurons (cn−/−) were co-cultured with wild-type (ca+/+) or EGFR−/− (ca−/−) cortical astrocytes. Upper panel: The indicated growth factors were added alone or in combination (MIX). Co-cultures with wild-type astrocytes served as controls. Lower panel: Neurons co-cultured with wild-type cortical astrocytes in the absence or presence of IL-1β (10 U/ml). Co-cultures with mutant cortical astrocytes in the presence of IL-1β were used as controls. Neuronal survival was scored by counting the number of neurons present after 12 days. —, neuronal survival <25%; +, neuronal survival 25–50%; ++, neuronal survival 50–75%; +++, neuronal survival >75%. | |||
Ras expression in postmitotic neurons rescues cortical degeneration in EGFR−/− mice
We next investigated whether providing a strong neuroprotective signal in neurons could rescue the deficiency of EGFR−/− cortical astrocytes. It was shown that transgenic mice expressing an activated form of Ras (RasV12) in postmitotic neurons (Syn-Ras) can induce such neuroprotective mechanisms (Heumann et al, 2000). Therefore, these mice were crossed into an EGFR−/− background. Like EGFR−/− mice, EGFR−/− mice carrying the Syn-Ras transgene did not survive beyond P20, suggesting that the lethality observed in EGFR−/− mice is not caused by the brain degeneration. Interestingly, brains from EGFR−/− Syn-Ras mice appeared normal at P16, whereas at this time in EGFR−/− mice parts of the cortex had already degenerated (Figure 6A–C). Histological analyses of EGFR−/− Syn-Ras brains revealed neuronal hypertrophy and expansion of the cortical layers as previously described for Syn-Ras transgenic mice (Figure 6E and F) (Heumann et al, 2000). However, no signs of neuronal degeneration were noticeable, whereas the cortex of EGFR−/− brains had already degenerated (Figure 6D and E). In addition, no apoptotic neurons could be detected in the cortex by Tunel staining, whereas many apoptotic cells were present in EGFR−/− cortex (Figure 6G–I). These results demonstrate that neuroprotective processes induced by increased Ras signaling are sufficient to prevent cortical degeneration.
Figure 6.
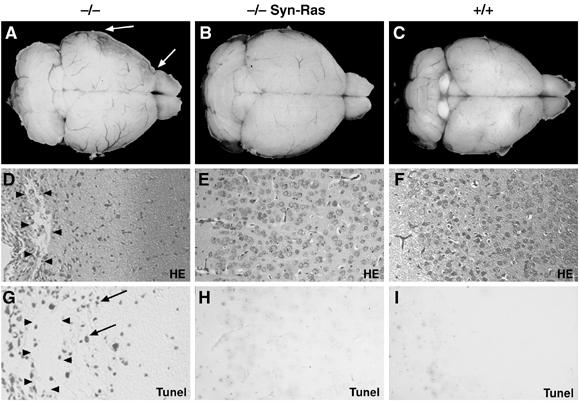
Expression of RasV12 in neurons rescues the brain degeneration in EGFR−/− mice. Dorsal view of the whole brain of (A) EGFR−/− mice with cortical degeneration (arrows), (B) EGFR−/− mice expressing the RasV12 transgene in postmitotic neurons (−/− Syn-Ras) and (C) control mice isolated at postnatal day 16. (D–F) Histological sections showing extensive degeneration evidenced by large cysts (arrowheads) in EGFR−/− cortex (D), and normal architecture in EGFR−/− Syn-Ras (E) and wild-type littermate control cortex (F). (G–I) Tunel staining showing apoptotic neurons in EGFR−/− cortex (arrows) (G) but not in EGFR−/− Syn-Ras (H) and control cortex (I). Magnification: D–I, × 20.
To exclude that the neurodegeneration in EGFR−/− Syn-Ras mice is rescued due to ectopic expression of RasV12 in astrocytes, Ras activity and the expansion of astrocytes was measured. No increase in Ras-GTP could be detected in cortical astrocytes from Syn-Ras transgenic mice (Figure 7A) and in culture a similar expansion defect as EGFR−/− cortical astrocytes was observed (Figure 7B), suggesting that RasV12 is not expressed in astrocytes.
Figure 7.
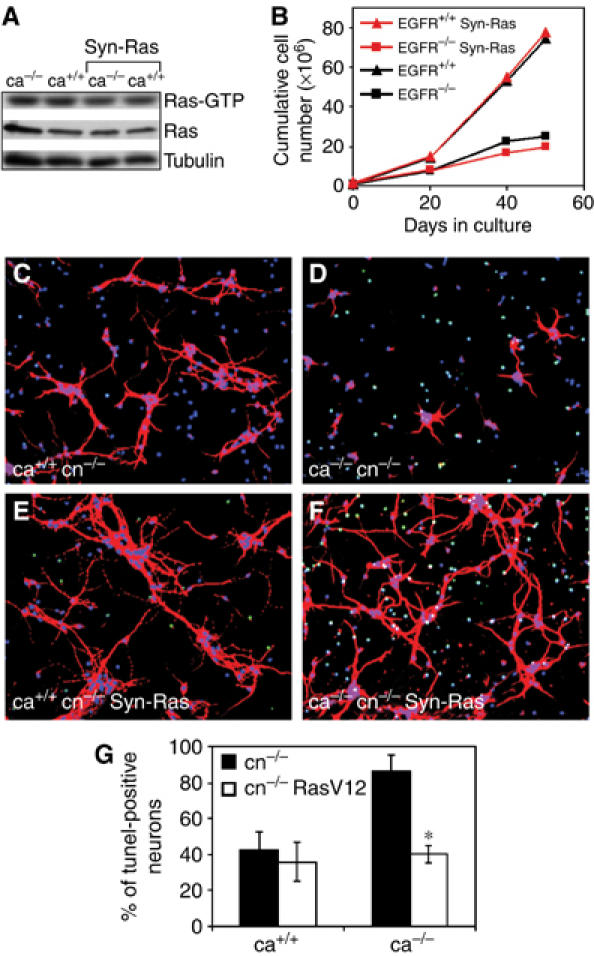
EGFR−/− Syn-Ras transgenic neurons survive in co-cultures with EGFR−/− astrocytes. (A) Western blot analysis showing levels of Ras-GTP in cultured astrocytes of the indicated genotypes. (B) Cumulative cell number of primary astrocytes demonstrating that EGFR−/− Syn-Ras transgenic astrocytes display a growth defect comparable to EGFR−/− astrocytes. (C–F) Cortical EGFR−/− (C, D) and EGFR−/− Syn-Ras (E, F) neurons (cn) co-cultured with wild-type (C, E) and EGFR−/− (D, F) cortical astrocytes (ca). Immunofluorescence stainings were performed 12 days after co-culture using GAP-43 (red) as neuronal marker, DAPI (blue) as a nuclear counterstain and Tunel (green) for apoptotic neurons. (G) The percentage of Tunel-positive neurons present in the indicated co-cultures at day 12 is shown. Data represent the mean±s.e.m. of the number of apoptotic cells counted in randomly chosen fields of six independent samples. *P<0.05.
To address whether RasV12 expression in neurons is sufficient to prevent their degeneration in vitro, co-cultures between EGFR−/− Syn-Ras cortical neurons and mutant or wild-type cortical astrocytes were established. EGFR−/− Syn-Ras neurons appeared morphologically similar to EGFR−/− neurons co-cultured with wild-type cortical astrocytes (Figure 7C and E). Interestingly, RasV12-expressing neurons appeared also perfectly healthy and survived when co-cultured with mutant cortical astrocytes, whereas most wild-type neurons had died (Figure 7D and F). Tunel staining revealed that the number of apoptotic EGFR−/− Syn-Ras neurons co-cultured with EGFR−/− astrocytes was low and comparable to neurons co-cultured with wild-type astrocytes (Figure 7G). These results show that elevated Ras activity in postmitotic neurons can prevent neuronal degeneration both in vivo and in vitro.
Discussion
In this study, we identified two functionally different astrocyte populations which show different dependence on EGFR signaling. In the absence of EGFR, midbrain astrocytes are unaffected, whereas EGFR−/− cortical astrocytes display increased apoptosis mediated by an Akt- and caspase-dependent mechanism. As a consequence, EGFR-deficient cortical astrocytes cannot support neuronal survival in co-culture experiments. In contrast, midbrain EGFR mutant astrocytes can fully support neuronal survival. This recapitulates the situation in EGFR−/− mice, where neurodegeneration occurs only in the cortex and olfactory bulbs sparing other parts of the brain (Sibilia et al, 1998). Expression of several neurotrophins and their receptors is not altered in EGFR−/− cortical astrocytes, suggesting that neuronal loss occurs as a consequence of increased astrocyte apoptosis rather than impaired secretion of specific factors.
Interestingly, neuron-specific expression of an activated form of Ras can rescue the neurodegenerative phenotype in vivo. This rescue is not mediated by astrocytes, since RasV12 is not expressed in these cells. In addition, neurons from Syn-Ras transgenic mice survive well in co-culture with EGFR−/− cortical astrocytes. Therefore, the Syn-Ras transgene may be compensating for the lack of EGFR signaling in neurons, the lack of trophic support from cortical astrocytes or both. However, our results show that EGFR is dispensable in neurons. EGFR−/− cortical neurons survive very well when co-cultured with wild-type astrocytes or when cultured in Neurobasal/B27 that promotes the survival of neurons in the absence of astrocytes (Brewer et al, 1993). Moreover, deletion of the EGFR in neurons does not lead to neuronal death in vivo (BW, manuscript in preparation). These results suggest that EGFR expression in neurons is not essential for their growth and survival.
Therefore, we propose that expression of RasV12 in postmitotic neurons rather than compensating for EGFR deficiency activates either proneurotrophic or antiapoptotic pathways, which in turn prevent neurons from undergoing apoptosis. Ras is a downstream mediator of neurotrophic signaling (Borasio et al, 1993) and elevated levels of neuropeptides have been observed in mice carrying the Syn-Ras transgene (Heumann et al, 2000). Moreover, constitutive activation of Ras can render neurons independent of growth factor treatment (Vogel et al, 1995) and neurons from Syn-Ras transgenic mice do not undergo apoptosis after injury (Heumann et al, 2000).
We found that neurons irrespective of whether they are wild type or mutant cannot survive, when co-cultured with EGFR−/− cortical astrocytes. However, neurons can survive very well if cultured with EGFR mutant astrocytes from the midbrain, suggesting that astrocytes from different brain regions display different requirements for EGFR signaling. Cortical and midbrain astrocytes from wild-type brains expressed similar amounts of EGFR and there was also no difference in EGFR ligand expression regardless of their genotype, demonstrating that EGFR ligands are still expressed in the absence of functional EGFR. EGFR seems to be essential for the survival of cortical astrocytes, yet is not necessary for the endurance of midbrain astrocytes.
Apoptosis of mutant cortical astrocytes in culture was observed starting 5–7 days after initial plating which corresponds to the time after birth when degeneration is detected in the cortex of mutant mice. Since reduced numbers of astrocytes have been found in EGFR−/− cortex (Sibilia et al, 1998), these observations suggest that astrocyte apoptosis might be the initial trigger for neuronal death in vivo. Costainings with GFAP and Tunel on cortical sections isolated between postnatal days 1 and 8 (P1–P8) identified apoptotic cells around P4 in the cortex of EGFR−/− brains that dramatically increased in numbers by P8 (Supplementary Figure 2). Owing to the low expression of GFAP in vivo and the close interaction between astrocytes and neurons, the identity of the apoptotic cells could not be determined.
In the absence of EGFR, increased levels of apoptosis were observed exclusively in cortical astrocytes and Akt activation was impaired. It is therefore likely that cortical astrocytes are more dependent on EGFR signaling and Akt activation for their survival, whereas survival of midbrain astrocytes can be mediated by other signaling pathways. We also observed increased expression of caspases in cortical astrocytes lacking EGFR. Caspases involved in the initiation of apoptosis such as caspase-8 as well as those responsible for the execution of apoptosis such as caspase-3, -6 and -7 were upregulated. Moreover, increased levels of the cleaved active form of caspase-3 were also detected in cortical astrocytes, thereby demonstrating that the apoptotic machinery is fully activated in these cells. Induction of apoptosis and activation of caspase cascades has also been reported for colon cancer cells treated with anti-EGFR monoclonal antibodies, an effect that could be reverted by pre-exposure of cells with caspase inhibitors (Liu et al, 2000). It has been proposed that activated Akt might be responsible for inhibiting the activity of caspase-3 and -9 thereby preventing apoptosis (Grant et al, 2002). Since in cortical astrocytes lacking EGFR, Akt activation is reduced, it is likely that the inhibitory activity on caspases is relieved thereby inducing the apoptotic program.
Astrocytes secrete factors not only involved in neuronal survival but also in neuronal development and synaptogenesis (van den Pol and Spencer, 2000; Song et al, 2002; Slezak and Pfrieger, 2003). Our results show that, except for CNTF, the expression of several genes encoding neurotrophic factors and their receptors was not altered in mutant cortical astrocytes. However, addition of CNTF alone or in combination with other neurotrophic factors to co-cultures with mutant cortical astrocytes did not improve neuronal survival, suggesting that lack of one specific factor is not responsible for neuronal death. The proinflammatory cytokine IL-1β, one of the factors produced within the first hours after brain injury and proposed to induce neuronal death (Allan and Rothwell, 2001), was significantly overexpressed in EGFR−/− cortical astrocytes, implying that it might be involved in promoting neuronal death. However, addition of IL-1β to co-cultures with wild-type astrocytes did not affect neuronal survival suggesting that IL-1β alone is not sufficient to induce neuronal death. We speculate that increased apoptosis of mutant cortical astrocytes leads to reduced concentrations of several secreted neurotrophic factors below the levels required for the survival of neurons. The observation that neurons in the initial phases of co-culture survive in the presence of mutant cortical astrocytes suggests that secreted factors can initially trigger neuronal survival but that at later stages these factors become limiting. In fact, increasing the number of mutant cortical astrocytes in co-cultures improves neuronal survival. These results demonstrate that an overall decline in factors is likely responsible for neuronal death.
Our results demonstrate that functionally different subtypes of astrocytes exist in distinct regions of the mouse brain, which can be genetically distinguished by their dependence on EGFR signaling. These differences in astrocyte composition and function provide a mechanism for the region-specific neurodegeneration in EGFR−/− mice and may explain the predominant fronto-temporal localization of neurodegenerative diseases in humans. Identifying ways of how to promote astrocyte survival will be a challenging task for the future to design new therapies aimed at preventing and curing neurodegenerative disorders.
Materials and methods
Mice
All mice employed in this study were maintained in a mixed C57Bl/6 × MF1 background, where EGFR−/− offspring can survive up to postnatal day 20. EGFR+/− were intercrossed to obtain homozygous mutants (EGFR−/−). EGFR+/− mice were crossed to EGFR+/− mice carrying the Synapsin-RasV12 (Syn-Ras) transgene to obtain EGFR−/− Syn-Ras offspring. EGFR−/− offspring with or without the Syn-Ras transgene were identified by their open eyes and their genotype was confirmed by PCR analysis (Sibilia and Wagner, 1995).
Astrocyte cultures
Cortical astrocytes were isolated from the hemispheres of newborn brains. Midbrain astrocytes were isolated from the brain part that was left after removing the cortices, olfactory bulbs, hippocampi, striatum, cerebellum and hindbrain. Astrocytes were prepared as previously described (Sibilia et al, 1998) and seeded at 20 000 cells/cm2 in high glucose DMEM (Invitrogen) supplemented with 10% Horse Serum (Invitrogen), 1 mM penicillin/streptomycin (Invitrogen) and 2 mM glutamine (Invitrogen). Cultures were split at ratios from 1:2 to 1:4 when confluent. Astrocytes at days 18–20 after initial preparation (a.p.) were pulsed with 10 μM Bromdeoxyuridine (Sigma) for 12 or 24 h, fixed in 70% EtOH and processed further for immunofluorescence staining or FACS analysis using anti-BrdU FITC-conjugated antibody (Becton Dickinson) and propidium iodide. For AnnexinV staining, astrocytes were trypsinized and labeled with FITC-conjugated AnnexinV antibody (BD Pharmingen) according to the manufacturer's protocol. The amount of dead cells was determined by propidium iodide (PI) staining. The fraction of cells positive for AnnexinV and negative for PI, which represent cells in early apoptosis, was determined by FACS analysis.
Neuronal cultures
Neurons were prepared from E16.5 embryos (day of plug=E0.5) as previously described (Celis, 1998). After dissection, cortices were digested in Trypsin-EDTA (Invitrogen) containing 7 mM Hepes (pH 7.3) and 1 mM penicillin/streptomycin for 15 min and washed three times with Hepes-buffered HBSS, followed by titration with a fire-polished Pasteur pipette. Cells were counted using Trypan-Blue (Sigma) exclusion and 1 × 105 cells were plated in a 3 cm dish containing a poly-L-Lysine-coated (0.5 mg/ml; Sigma) coverslip (Marienfeld No.1, 20 × 20) in MEM-HS (10% Horse Serum (Invitrogen), 1 × MEM (Invitrogen), 2 mM glutamine (Invitrogen), 0.6% glucose (Sigma), 0.22% NaHCO3 (Sigma), 10 × MEM nonessential amino acids (Invitrogen), 5 × MEM essential amino acids (Invitrogen)). On the next day, neurons were transferred to astrocyte dishes to set up the co-cultures.
Co-cultures between neurons and astrocytes
Confluent astrocytes (12–16 days after initial preparation) were trypsinized and replated at 30 000 cells/3 cm dish (equivalent to 4250 cells/cm2) in high glucose DMEM (Invitrogen) supplemented with 10% Horse Serum (Invitrogen), 1 mM penicillin/streptomycin (Invitrogen) and 2 mM glutamine (Invitrogen). After 1–2 days, the medium was changed to N2-medium (1 × MEM (Invitrogen), 1 × pyruvate (Sigma), 2 mM glutamine (Invitrogen), 0.6% glucose (Sigma), 0.22% NaHCO3 (Sigma), 1 × N2 supplement (Invitrogen), 0.1% ovalbumin (Sigma)). After 20 h, coverslips containing neurons were added to 3 cm dishes containing astrocytes. Every 5–7 days, half of the medium of the co-cultures was replaced with fresh N2 medium.
ELISA
Subconfluent astrocytes at 16–22 days a.p. were cultured in N2-MEM for 4 days. Supernatants were collected every 24 h and ELISA kits (R&D Systems, Quantikine) were employed according to the manufacturer's recommendations to determine levels of secreted VEGF (MMV00), IGF-1 (MG100), IL-1β (MLB00B) and TNFα (MTA00).
Immunofluorescence and Tunel staining
For immunofluorescence staining, neurons or astrocytes were cultured on coverslips, fixed in 4% paraformaldehyde and blocked with 0.2% bovine serum albumin (BSA; Sigma) in PBS. If required, cells were permeabilized with 1% sodium citrate and 0.1% Triton-X 100. Antibodies were diluted in 0.2% BSA/PBS: GFAP (1:100, DAKO), GAP-43 (1:200, Sigma), Nestin (1:4, Developmental Studies Hybridoma Bank). Secondary antibodies: Alexa goat anti-mouse 488, Alexa goat anti-rabbit 488, Alexa goat anti-mouse 594, Alexa goat anti-rabbit 594, all from Molecular Probes and diluted 1:800. For detecting BrdU, cells were fixed with 70% ethanol, dried, treated with 1 M NaOH, and incubated with anti-BrdU antibody (Becton Dickinson) for 30 min and washed three times with PBS. Coverslips were mounted using the Vectashield mounting medium containing DAPI (H-1200). Tunel staining was performed on neurons or astrocytes using the In Situ Cell Death Detection Kit II, FITC (Boehringer Mannheim) according to the manufacturer's recommendation. Immunofluorescence staining with anti-GFAP or anti-GAP-43 antibodies was performed after the Tunel reaction.
Histology
Mice were killed and brains were fixed in 4% paraformaldehyde and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin (Sigma) according to standard procedures. GFAP immunoflurescence and Tunel staining were performed as previously described (Sibilia et al, 1998) using the In Situ Cell Death Detection Kit II, AP or FITC (Boehringer Mannheim).
Western blot analysis
Cells were lysed, cleared by centrifugation and processed for Western blotting as previously described (Sibilia et al, 2000). The following primary antibodies were employed at the recommended dilutions: phosphotyrosine clone 4G10 (Upstate Biotechnology), EGFR (UBI), phospho-Akt (Cell Signaling), Akt (Santa Cruz), actin (Sigma), tubulin (Sigma), Erk1/2 (Santa Cruz), phospho p44/42 MAPK (New England Biolabs), PCNA (Santa Cruz), caspase-3 (Santa Cruz), Fas (Santa Cruz). HRP-coupled secondary antibodies were obtained from DAKO. Signals were visualized using ECL detection kit (Amersham).
RBD pull-down assay
Cells were lysed in lysis buffer (Sibilia et al, 2000) containing 25 μg GST-Raf1-(RBD) and 100 μM GDP. Lysates were centrifuged at 4°C for 15 min at 14 000 r.p.m. and the supernatants transferred to tubes containing GSH-sepharose and rotated at 4°C for 30 min. After centrifugation, the pellets were washed 2 × with lysis buffer and then processed for Western blot analysis.
RNase protection assay (RPA)
Total RNA was isolated from 80% confluent astrocyte cultures with Trizol™ reagent (Invitrogen) and 8 μg of RNA were used for each RPA reaction. RPA was performed using the RiboQuant multiprobe RNase protection assay systems mApo-1 and mJun/Fos (PharMingen) according to the manufacturer's recommendations.
Real-time and RT–PCR analysis
cDNA was obtained from total RNA by reverse transcription with SUPERSCRIPT™ First-Strand Synthesis System (Invitrogen). RT–PCR conditions and primers for HB-EGF, TGFα and Tubulin were previously described (Sibilia et al, 2000). Quantitative real-time RT–PCR was performed and analyzed with Light Cycler (Roche) as previously described (Sibilia et al, 2000) by employing the porphobilinogen deaminase (PBGD) gene as an internal loading control. Primers: EGF1, TCTGCAGATGCTCAGAAGGC; EGF2, GTCATCATCTACGACCGGACA; CNTF253f, GCAATCACCTCTGACCCTTC; CNTF481r, ACGGTAAGCCTGGAGGTTCT; IL-1β582f, CAAAATACCTGTGGCCTTGG; IL-1β782r, TACCAGTTGGGGAACTCTGC; PBGD199f, TCCCTGAAGGATGTGCCTAC; PBGD337r, GCACTTTTCTCTGGCAAGGT.
Microarray experiments and analysis
GEArray gene expression array systems (MM-018, SuperArray, Beteshda, MD) consisting of spotted cDNA fragments encoding 96 mouse neurotrophin and receptor genes as well as control sequences (PUC18, glyceraldehydes 3 phosphate dehydrogenase (GAPDH), peptidylpropyl isomerase A (Ppia) and β-actin) were employed to compare gene expression between wild-type and EGFR−/− cortical astrocytes. Total RNA was isolated with the Trizol™ method (Invitrogen) and further processed for microarray hybridization according to the manufacturer's instructions. The arrays were visualized by autoradiography and the hybridization signals were scanned by ImageJ (NCBI) to obtain digital numbers for the density. The normalized value for each gene was calculated by dividing the value of each gene to the average value of the house-keeping genes GAPDH or Ppia.
Statistical analysis
Each experiment was performed in duplicates or triplicates and results were confirmed in at least three independent experiments. The statistical significance of the data was determined by applying the two-tailed Student's t-test. The difference was considered statistically significant at P⩽0.05.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We are grateful to M Hammer for maintaining our mouse colonies, to Gertraud Steniczka for help with genotyping. We thank Drs Denise Barlow, Jürgen Knoblich and Frank Heppner for critical reading of the manuscript. BW was recipient of a DOC-Fellowship of the Austrian Academy of Sciences. MS thanks Boehringer Ingelheim International for supporting this project and acknowledges funding by the K-plus program Biomolecular Therapeutics (BMT), the Austrian National Bank ÖNB-10556, the Austrian Science Fund FWF-P18421 and EC program QLG1-CT-2001-00869. The IMP (EFW) is supported by Boehringer Ingelheim and the Austrian Industrial Research Promotion Fund (FFF).
References
- Allan SM, Rothwell NJ (2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2: 734–744 [DOI] [PubMed] [Google Scholar]
- Banker GA (1980) Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 209: 809–810 [DOI] [PubMed] [Google Scholar]
- Borasio GD, Markus A, Wittinghofer A, Barde YA, Heumann R (1993) Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic neurons. J Cell Biol 121: 665–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567–576 [DOI] [PubMed] [Google Scholar]
- Burrows RC, Lillien L, Levitt P (2000) Mechanisms of progenitor maturation are conserved in the striatum and cortex. Dev Neurosci 22: 7–15 [DOI] [PubMed] [Google Scholar]
- Burrows RC, Wancio D, Levitt P, Lilien L (1997) Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 19: 251–267 [DOI] [PubMed] [Google Scholar]
- Caric D, Raphael H, Viti J, Feathers A, Wancio D, Lillien L (2001) EGFRs mediate chemotactic migration in the developing telencephalon. Development 128: 4203–4216 [DOI] [PubMed] [Google Scholar]
- Celis JE (1998) Cell Biology: A Laboratory Handbook. San Diego: Academic Press [Google Scholar]
- Craig CG, Tropepe V, Morshead CM, Reynolds BA, Weiss S, van der Kooy D (1996) In vivo growth factor expansion of endogenous subependymal neural precursor cell populations in the adult mouse brain. J Neurosci 16: 2649–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone SA, Negro A, Trumpp A, Giovannini M, Lee KF (2003) Colonic epithelial expression of ErbB2 is required for postnatal maintenance of the enteric nervous system. Neuron 37: 29–40 [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003) AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868 [DOI] [PubMed] [Google Scholar]
- Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, Lu LH, Moore MW (1997) ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2- and heregulin-deficient mice. Development 124: 4999–5011 [DOI] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G (1995) Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor (see comments). Nature 378: 390–394 [DOI] [PubMed] [Google Scholar]
- Grant S, Qiao L, Dent P (2002) Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci 7: d376–d389 [DOI] [PubMed] [Google Scholar]
- Heumann R, Goemans C, Bartsch D, Lingenhohl K, Waldmeier PC, Hengerer B, Allegrini PR, Schellander K, Wagner EF, Arendt T, Kamdem RH, Obst-Pernberg K, Narz F, Wahle P, Berns H (2000) Transgenic activation of Ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J Cell Biol 151: 1537–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z (1998) Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J Neurosci Res 53: 697–717 [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C (1995) Requirement for neuregulin receptor erbB2 in neural and cardiac development (see comments). Nature 378: 394–398 [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Feldman EL (2005) Insulin-like growth factors in the treatment of neurological disease. Endocr Dev 9: 135–159 [DOI] [PubMed] [Google Scholar]
- Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF (2000) Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA 97: 1299–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Fang M, Schmidt M, Lu Y, Mendelsohn J, Fan Z (2000) Induction of apoptosis and activation of the caspase cascade by anti-EGF receptor monoclonal antibodies in DiFi human colon cancer cells do not involve the c-jun N-terminal kinase activity. Br J Cancer 82: 1991–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C (1995) Multiple essential functions of neuregulin in development (see comments). Nature 378: 386–390 [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R (1995) Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376: 337–341 [DOI] [PubMed] [Google Scholar]
- Murphy M, Dutton R, Koblar S, Cheema S, Bartlett P (1997) Cytokines which signal through the LIF receptor and their actions in the nervous system. Prog Neurobiol 52: 355–378 [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19: 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C (1997) Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389: 725–730 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110: 669–672 [DOI] [PubMed] [Google Scholar]
- Sibilia M, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, Schlessinger J, Wagner EF (2000) The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell 102: 211–220 [DOI] [PubMed] [Google Scholar]
- Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF (1998) A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J 17: 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibilia M, Wagner B, Hoebertz A, Elliott C, Marino S, Jochum W, Wagner EF (2003) Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 130: 4515–4525 [DOI] [PubMed] [Google Scholar]
- Sibilia M, Wagner EF (1995) Strain-dependent epithelial defects in mice lacking the EGF receptor (published erratum appears in Science 1995 Aug 18; 269 (5226): 909). Science 269: 234–238 [DOI] [PubMed] [Google Scholar]
- Slezak M, Pfrieger FW (2003) New roles for astrocytes: regulation of CNS synaptogenesis. Trends Neurosci 26: 531–535 [DOI] [PubMed] [Google Scholar]
- Song H, Stevens CF, Gage FH (2002) Astroglia induce neurogenesis from adult neural stem cells. Nature 417: 39–44 [DOI] [PubMed] [Google Scholar]
- Storkebaum E, Carmeliet P (2004) VEGF: a critical player in neurodegeneration. J Clin Invest 113: 14–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Goderie SK, Temple S (2005) Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron 45: 873–886 [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, Barnard JA, Yuspa SH, Coffey RJ, Magnuson T (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269: 230–234 [DOI] [PubMed] [Google Scholar]
- Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D (1999) Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 208: 166–188 [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Spencer DD (2000) Differential neurite growth on astrocyte substrates: interspecies facilitation in green fluorescent protein-transfected rat and human neurons. Neuroscience 95: 603–616 [DOI] [PubMed] [Google Scholar]
- Viti J, Feathers A, Phillips J, Lillien L (2003) Epidermal growth factor receptors control competence to interpret leukemia inhibitory factor as an astrocyte inducer in developing cortex. J Neurosci 23: 3385–3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF (1995) Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell 82: 733–742 [DOI] [PubMed] [Google Scholar]
- Wang K, Yamamoto H, Chin JR, Werb Z, Vu TH (2004) Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J Biol Chem 279: 53848–53856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D (1996) Is there a neural stem cell in the mammalian forebrain? Trends Neurosci 19: 387–393 [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2