

Abstract
The acute effects of smoking a low or high nicotine cigarette on hypothalamic-pituitary-adrenal (HPA) hormones, subjective responses and cardiovascular measures were studied in 20 healthy men who met DSM-IV criteria for nicotine dependence. Within 4 puffs (or 2 min) after cigarette smoking began, plasma nicotine levels and heart rate increased significantly (P<0.01), and peak ratings of “high” and “rush” on a Visual Analogue Scale (VAS) were reported. Reports of “high”, “rush” and “liking” and reduction of “craving” were significantly greater after smoking a high nicotine cigarette than a low nicotine cigarette (P<0.05). Peak plasma nicotine levels after high nicotine cigarette smoking (23.9 ± 2.6 ng/ml) were significantly greater than after low nicotine cigarette smoking (3.63 ± 0.59 ng/ml) (P<0.001). After smoking a low nicotine cigarette, ACTH, cortisol, DHEA and epinephrine did not change significantly from baseline. After high nicotine cigarette smoking began, plasma ACTH levels increased significantly above baseline within 12 min and reached peak levels of 21.88 ± 5.34 pmol/L within 20 min. ACTH increases were significantly correlated with increases in plasma nicotine (r=0.85; P<0.0001), DHEA (r=0.66; P=0.002), and epinephrine (r=0.86; P<0.0001). Cortisol and DHEA increased significantly within 20 min (P<0.05) and reached peak levels of 424 ± 48 nmol/L and 21.13 ± 2.55 ng/ml within 60 and 30 min, respectively. Thus cigarette smoking produced nicotine dose-related effects on HPA hormones, subjective and cardiovascular measures. These data suggest that activation of the HPA axis may contribute to the abuse-related effects of cigarette smoking.
Keywords: Cigarette smoking, nicotine, HPA axis, ACTH, cortisol, DHEA, epinephrine
Introduction
Cigarette smoking remains a leading cause of death and disease and is associated with over 440,000 premature deaths each year (CDC, 2000). Despite increasing social and legal pressures to restrict tobacco use, an estimated 46 million adults smoke cigarettes in the United States (CDC, 2004). Evidence that implicates cigarette smoking in respiratory and cardiovascular disease continues to accumulate (2004). The 2004 Surgeon General's Report on the health effects of smoking identifies additional cancers as well as several other diseases that are now thought to be causally related to cigarette smoking (2004). Although awareness of these health risks has reduced the overall prevalence of cigarette smoking, relapse rates during and after treatment are very high (Jaffe, 1990).
Cigarette smoking is generally acknowledged to be an addictive disorder characterized by tolerance and physical dependence (Jaffe, 1990; APA, 1994; Henningfield et al., 1995; Benowitz, 1996; CDC, 2001). Nicotine appears to be the primary addictive agent in cigarette smoking, and intravenous nicotine is self-administered by humans and experimental animals under a number of conditions (Goldberg et al., 1981; Spealman and Goldberg, 1982; Henningfield and Goldberg, 1983; Corrigall, 1999; Donny et al., 1999; Harvey et al., 2004). With nicotine, as with many abused drugs, speed of the injection is an important determinant of relative reinforcing efficacy (Balster and Schuster, 1973; Wakasa et al., 1995; Abreu et al., 2001). The reinforcing effects of nicotine are mediated, in part, by stimulating nicotinic acetylcholine receptors on mesolimbic dopamine neurons to release dopamine (Fu et al., 2000; Gerasimov et al., 2000; Watkins et al., 2000). Nicotine also stimulates rapid release of adrenocorticotropin hormone (ACTH) in rodents following i.p. or i.c.v. administration and microinjection into catecholaminergic regions, and these effects are blocked by the nicotine antagonist, mecamylamine (Sharp and Beyer, 1986; Matta et al., 1990, 1993b, 1998). Hypothalamic corticotropin releasing-hormone (CRH) cannot be measured in venous blood, but one index of CRH activation is the release of ACTH from the anterior pituitary, followed by an increase in cortisol in humans and corticosterone in rodents. There is also evidence from preclinical studies that nicotine activates CRH neurons in extra-hypothalamic regions that mediate responses to stress (Matta et al., 1993a, 1997).
Although the interactions between the hypothalamic-pituitary-adrenal (HPA) axis and the abuse-related effects of drugs are poorly understood, HPA axis activation is thought to contribute to drug abuse at several phases of the addictive process (Koob and Le Moal, 2001; Sinha, 2001; Marinelli and Piazza, 2002; Contoreggi et al., 2003; Heinrichs and Koob, 2004). For example, it has been suggested that high CRH levels may contribute to the anxiety and irritability often associated with nicotine withdrawal (Watkins et al., 2000). It is generally agreed that CRH stimulation of ACTH is one component of an integrated physiological response to “stress” (Nemeroff, 1996; Tsigos and Chrousos, 2002). Yet paradoxically, stimulation of ACTH is also a consistent effect of a highly reinforcing drug, cocaine. Increases in ACTH are temporally concordant with reports of positive subjective effects by cocaine abusers (Mendelson et al., 1992, 2002; Mello and Mendelson, 2002). Although these opposite subjective effects of HPA activation seem counter-intuitive, it has been suggested that control of a predictable stressor may diminish its aversive effects (Goeders, 2002a and b). Cocaine, skydiving, roller coaster riding and similar risk-taking behaviors may fall into this category (Mendelson et al., 2002). B. F. Skinner's concept of a reinforcer as anything that maintains behavior leading to its self-administration provides a useful semantic description of these behaviors.
Several lines of evidence suggest that stimulation of CRH, as indicated by increases in ACTH and cortisol, may contribute to the reinforcing effects of cocaine (Goeders, 1997, 2002a and b; Mello and Mendelson, 2002; Mendelson et al., 2002). For example, the adrenal hormone corticosterone is critically important for the acquisition and maintenance of cocaine self-administration in rats (Goeders and Guerin, 1996; Mantsch et al., 2000). Consistent with the hypothesis that ACTH and corticosterone may contribute to cocaine's reinforcing effects, administration of a CRH-1 antagonist decreased i.v. cocaine self-administration by rats, with minimal effects on food-maintained responding (Goeders and Guerin, 2000). CRH antagonists are also associated with alleviation of stress-induced anxiety (Habib et al., 2000; Ayala et al., 2004). Yet, in rhesus monkeys, with normal glucocorticoid levels, drug-induced increases in ACTH did not appear necessary to maintain cocaine self-administration (Broadbear et al., 1999a and b).
The relevance of these studies of cocaine to nicotine is suggested by the several similarities between the abuse-related and hormonal effects of these drugs. Both cocaine and nicotine produce rapid onset, positive subjective effects of relatively brief duration (Jones et al., 1999; Mendelson et al., 2003; Sobel et al., 2004). Both cocaine and nicotine activate the mesolimbic dopamine system and increase extracellular dopamine levels, although by different mechanisms (Kuhar et al., 1991; Watkins et al., 2000). In addition to similar subjective and cardiovascular effects, both i.v. cocaine and cigarette smoking produced rapid increases in luteinizing hormone (LH) that were significantly correlated with the rapid increases in plasma levels of cocaine and nicotine (Mendelson et al., 2003).
The present study was designed to explore the interactions between HPA axis hormones and the abuse-related effects of cigarette smoking. There has been relatively little attention to the temporal covariance between the endocrine, cardiovascular and subjective effects of cigarette smoking. Early clinical endocrine studies of the hormonal effects of cigarette smoking were limited by the lack of sensitive and precise radioimmunoassays for ACTH (Pomerleau et al., 1983). Moreover, samples for ACTH and/or cortisol analysis usually were collected at relatively infrequent intervals, or as single daily samples (Seyler et al., 1984, 1986; Baron et al., 1995; Pickworth and Fant, 1998; del Arbol et al., 2000), and plasma nicotine levels were seldom measured. Because ACTH is released from corticotropes in the anterior pituitary in rapid bursts of short duration (3-10 min) (Iranmanesh et al., 1990), frequent blood sample collection is important to accurately measure drug effects on hormone release patterns (Teoh et al., 1994; Sarnyai et al., 1995). Moreover, rapid sample collection is essential to assess the temporal relationship between anterior pituitary and adrenal hormones, cardiovascular measures and the subjective reports during cigarette smoking.
One goal of this study was to assess the temporal covariance between reports of subjective effects, changes in anterior pituitary and adrenal hormones, plasma nicotine levels and cardiovascular measures during as well as after cigarette smoking. A second goal was to determine if cigarette smoking under controlled conditions produces a rapid perturbation of the HPA axis in nicotine-dependent men, similar to the effects of nicotine in rodents (Sharp and Beyer, 1986; Matta et al., 1990, 1993b). A third goal was to determine if the subjective, hormonal and cardiovascular effects of cigarette smoking were nicotine dose-related. The effects of smoking a low and high nicotine cigarette were compared under identical experimental conditions, and all dependent measures were sampled at 2 min intervals during and immediately following cigarette smoking.
Materials and Methods
Subjects
Twenty healthy adult men were recruited through newspaper advertisements and provided written informed consent for pre-study screening procedures and for participation in this study. The study was approved by the Institutional Review Board of the McLean Hospital. All men fulfilled American Psychiatric Association Diagnostic and Statistical Manual (DSM-IV) criteria for current nicotine dependence (305.1). Volunteers with any lifetime DSM-IV Axis 1 disorder other than nicotine dependence were excluded. Men who were seeking treatment for nicotine dependence, or who were wearing a nicotine patch were also excluded. All men selected for this study were in good physical health and had normal medical and laboratory screening examinations. Subjects in the high and low nicotine groups did not differ significantly with respect to age, years of smoking, number of cigarettes smoked per day and body mass index (BMI) (Table 1). Ten men were randomly assigned to smoking a high nicotine cigarette and ten men were assigned to smoking a low nicotine cigarette. A between subjects design was used, because it was difficult to recruit nicotine-dependent men under 35 years of age. Moreover, an interval of at least 5 weeks between successive studies was required because of the volume of blood withdrawn for plasma nicotine and hormone analyses. It is usually difficult to maintain contact with subjects over this period.
TABLE 1:
SUBJECT CHARACTERISTICS
| Age (Years) * | Years of Smoking * | Cigarettes Smoked/Day * | BMI (KG/M2) (x̄ ± S.E.) * | Baseline CO Levels * | Baseline Plasma Nicotine Levels * | |
|---|---|---|---|---|---|---|
| Demographic data, carbon monoxide levels and baseline plasma nicotine levels were analyzed using a one-factor ANOVA | ||||||
| High Nicotine Cigarette Group (N=10) | 25.6 ± 1.1 | 6.1 ± 0.7 | 16.5 ± 1.0 | 24.1 ± 0.6 | 3.4 ± 0.8 ppm | 1.61 ± 0.20 ng/ml |
| Low Nicotine Cigarette Group (N=10) | 25.2 ± 0.8 | 5.0 ± 0.3 | 15.0 ± 0.5 | 24.8 ± 0.4 | 3.7 ± 0.7 ppm | 1.20 ± 0.18 ng/ml |
| ANOVA Results | *P = 0.77 (df=1, F = 0.87) | *P = 0.12 (df=1, F = 2.73) | *P = 0.16 (df=1, F = 2.15) | *P = 0.34 (df=1, F = 0.96) | *P = 0.71 (df=1, F = 0.76) | *P = 0.15 (df=1, F = 2.1) |
The study procedures were explained during initial screening and again on the study day, and any questions or concerns were discussed. Subjects were admitted to the clinical research ward on the morning of the study day, and all studies were conducted at the same time of day. Subjects were asked to provide a urine specimen for drug testing and levels of breath carbon monoxide (CO) and breath alcohol were measured. Subjects were told they would be asked to smoke cigarettes with a low or high nicotine content under controlled conditions. After the study was completed, subjects remained on the clinical research ward for two or more hours. Lunch was provided and vital signs were measured at 30 min intervals. When subjects were medically stable and comfortable, they were discharged and transportation was provided. Subjects were paid for their participation in the study, consistent with NIH regulations.
Smoking Abstinence Requirements
Cigarette smokers were asked to abstain from cigarettes and caffeinated beverages after midnight on the night before the study. Carbon monoxide (CO) levels were measured with a Vitalograph Breath CO Monitor (Vitalograph, Inc., Lenexa, Kansas 66215) to assess compliance with smoking abstinence requirements. Cigarette smokers with a CO level above 10 ppm were not allowed to participate in the study and, as shown in Table 1, average CO levels were below 4 ppm. It is important for subject safety, as well as to avoid confounding of the dependent variables, to ensure that subjects have not used any drugs before cigarette smoking. On the morning of each study day, urines were collected and analyzed with a Triage® screen. The Triage® Panel for Drugs of Abuse (Biosite Diagnostics, San Diego, CA) is a rapid multiple immunoassay system for the qualitative detection of the major metabolites of these drugs of abuse in urine at the following cut-off concentrations as recommended by the Substance Abuse and Mental Health Services Administration: Phencyclidine 25 ng/mL, Benzodiazepines 300 ng/mL, Benzoylecgonine, a metabolite of Cocaine, 300 ng/mL, Amphetamines 1000 ng/mL, Tetrahydrocannabinol 50 ng/mL, Opiates 300 ng/mL and Barbiturates 300 ng/mL.
Nicotine Dose Selection
A commercially available, high nicotine cigarette and a low nicotine cigarette were studied. The high nicotine cigarette (Marlboro Red, Phillip Morris brand) contained 15.48 mg of nicotine and 16 mg of tar based on analysis by the Massachusetts Department of Public Health (MDPH, 1998). In our previous studies, smoking a Marlboro Red cigarette under the conditions described below produced peak plasma nicotine levels above 20 ng/ml (Mendelson et al., 2003). The low nicotine cigarettes contained 1.1 mg of nicotine and 2.8 mg of tar based on analyses provided by the manufacturer. We refer to this cigarette as low nicotine rather than as de-nicotinized, because most “de-nicotinized” cigarettes contain trace amounts of nicotine (< 0.06 – 0.16 mg) (Pickworth et al., 1999; Shahan et al., 1999; Robinson et al., 2000). Low nicotine cigarettes were acquired from Murty Pharmaceuticals Inc. (Lexington, KY 40509).
Nicotine Administration Procedures
These studies were carried out on a clinical research ward. Subjects were studied in a semi-supine position, and cigarettes were administered using a controlled smoking procedure designed to standardize puff volume and duration of inhalation (Griffiths et al., 1982). Every 30 sec, subjects were asked to take one puff from a cigarette, hold the smoke for 5 sec, and exhale at the end of 5 sec. Subjects took 24 puffs over a 12 min smoking period with an inter-puff interval of 25 sec. This is equivalent to approximately two cigarettes. A new lighted cigarette was presented after every four puffs so that changes in cigarette length would not influence puff duration (Nemeth-Coslett and Griffiths, 1984a and b).
Cardiovascular Measures and Safety Precautions
Heart rate, blood pressure and electrocardiograms (EKG) were continuously monitored with a Hewlett-Packard EKG monitor (Model 78 352A) for ten min prior to cigarette smoking and for two hours following cigarette administration. A physician certified in cardiopulmonary resuscitation was present during each study, and a cardiac defibrillator and appropriate emergency treatment medications were located in the study room.
Sample Collection Procedures
Baseline samples for analysis of ACTH, cortisol, DHEA, epinephrine and plasma nicotine levels were collected 10 min before the onset of cigarette smoking, then at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 25, 30, 40, 50, 60, 80, and 120 min following the onset of smoking a high or low nicotine cigarette. This rapid sampling frequency was based upon our previous observations that nicotine levels in plasma increase rapidly after smoking begins and reach peak levels within 14 min (Mendelson et al., 2003). The catheter site and collection tubes were covered during the smoking studies so that smoke in the air would not contaminate plasma samples. Blood samples for hormone analysis and for nicotine analysis were collected in vacutainer tubes without preservative. All samples were iced immediately, centrifuged and plasma or serum was removed and frozen at -70° C for nicotine and hormone analyses.
Subjective Effects Measures
Subjects rated their reactions to smoking on a Visual Analogue Scale (VAS) that ranged from zero to 100. Subjects were asked to rate how “high” they felt, if they felt a “rush,” how much they “liked” nicotine and how much they “craved” cigarettes. Subjects were also asked to rate feelings of “sick,” “jittery,” “bad feeling,” “dizzy” and “alert.” Subjects were asked to rate these subjective effects before smoking and every two minutes for the first 20 min after cigarette smoking began, then at 25, 30, 40, 50, 60, 70, 80 and 120 min following smoking onset.
Assay Procedures
Adrenocorticotropin Hormone (ACTH): Plasma ACTH concentrations were measured in duplicate using an immunoradiometric assay (IRMA) kit purchased form Nichols Institute (San Juan Capistrano, CA). The assay sensitivity was 0.37 pmoL/L and the intra- and interassay C.V.s were 3.9% and 5.6%, respectively.
Cortisol: Plasma cortisol was determined in duplicate by the GammaCoat RIA method, using kits purchased from DIASORIN Corporation (Stillwater, MN). The assay sensitivity was 5.5 nmol/L and the intra- and interassay C.V.s were 5.9% and 9.6%, respectively.
Dehydroepiandrosterone (DHEA): Plasma DHEA concentrations were determined in duplicate by Coated Tubes RIA method using kits purchased from Diagnostic Systems Laboratories, Inc. (Webster, TX). The assay sensitivity was 0.4 ng/ml and intra- and interassay C.V.s were 5.8% and 10.2%, respectively.
Epinephrine: Plasma epinephrine concentrations were measured in duplicate by a double antibody RIA method, on previously extracted and acylated samples, using kits purchased from American Laboratory Products Company (Windham, NH). The assay sensitivity was 7.6 pg/ml and the intra- and interassay C.V.s were 4.3% and 6.0%, respectively.
Plasma Nicotine Analysis: Plasma nicotine levels were measured in duplicate using a gas chromatography-mass spectrometry method described by Jacob and co-workers (Jacob et al., 2000). The nicotine assay sensitivity was 1.0 ng/ml and the intra- and interassay CV's were 4.7% and 11.5%, respectively. Nicotine assays were conducted in the laboratory of Dr. Peton Jacob III at the University of California at San Francisco.
Data Analysis
Comparisons between the subjective and physiological effects of high and low nicotine cigarettes were analyzed with ANOVA for repeated measures. Within each group, changes in each dependent variable from baseline were assessed with a one factor repeated measures ANOVA. If significant main effects were detected, one-way ANOVAS were performed to determine which points differed significantly from baseline. Plasma nicotine levels, hormone levels, subjective measures and cardiovascular parameters in the low and high nicotine groups were analyzed using a two factor (dose group x time) repeated measures ANOVA with time post-smoking as the repeated factor. If significant main effects were detected, one-way ANOVA's were performed to identify the time points that differed significantly between the two dose groups. The statistical significance of temporal covariance between ACTH, cortisol, DHEA and epinephrine and plasma nicotine levels were determined by Pearson Product Moment Correlation analyses. The statistical significance of the temporal covariance between subjective measures and heart rate were also evaluated with a Pearson Product Moment Correlation analyses.
Estimates of time to peak plasma concentrations (Tmax), and half-life (t1/2) of plasma nicotine and hormone data were calculated with GraphPad Prism software (version 4, San Diego, CA) by fitting the equation for one phase exponential decay to the data. These pharmacokinetic parameters were analyzed with an ANOVA to determine whether there were any significant differences between plasma nicotine and HPA hormones after smoking a high nicotine cigarette. Low nicotine cigarette smoking produced minimal changes in HPA axis hormones, so pharmacokinetic analyses were not performed.
Results
Plasma Nicotine Levels (Figure 1, Table 1)
Figure 1.
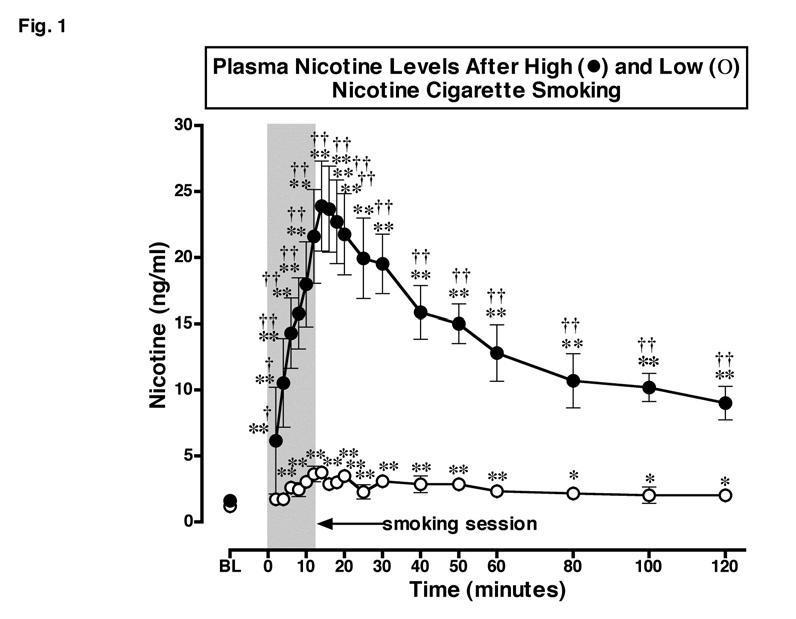
: Plasma Nicotine Levels After Smoking a Low or High Nicotine Cigarette. Plasma nicotine levels after smoking a high nicotine cigarette (filled circles) and a low nicotine cigarette (open circles) are shown on the left ordinates. Time (min) is shown on the abscissae. Points above BL were collected 10 min before cigarette smoking began at time 0. The 12 min cigarette smoking period is indicated by a grey rectangle. Each data point is the average (± S.E.M.) of 10 subjects. Statistical analyses indicated significant changes in plasma nicotine levels from baseline in both low [df=18, F=6.96, P=.03)], and high [df=18, F=21.8, P<.0001] nicotine cigarette groups. Significant changes from the pre-smoking baseline are indicated by asterisks (* = P < 0.05; ** = P < 0.01). Daggers indicate points at which plasma nicotine levels after smoking a low nicotine cigarette differed from plasma nicotine levels after smoking a high nicotine cigarette († = P < 0.05; †† = P <0.01) [df=1, F= 23.9, P=.0004].
There were no significant differences between groups in baseline nicotine levels or baseline CO levels (Table 1). Plasma nicotine levels increased significantly within 2 min, or 4 puffs on a high nicotine cigarette, and remained significantly above baseline throughout the 120 min sampling period (P = 0.009-0.0001). Peak plasma nicotine levels of 23.90 ± 2.68 ng/ml were detected within 14 min, then plasma nicotine levels gradually decreased to 9.01 ± 1.25 ng/ml at the end of the sampling period. Plasma nicotine levels also increased significantly above baseline levels after smoking a low nicotine cigarette (P = 0.03-0.0003). Peak plasma nicotine levels of 3.63 ± 0.59 ng/ml were detected within 12 min after low nicotine cigarette smoking began. At the end of the sampling period plasma nicotine levels averaged 2.02 ± 0.31 ng/ml. Plasma nicotine levels were significantly higher after high nicotine cigarette smoking than after low nicotine cigarette smoking throughout the 120 min sampling period (P = 0.05-0.0001) (Figure 1).
Pharmacokinetic analysis of plasma nicotine levels confirmed that the Cmax was significantly higher after smoking a high nicotine cigarette than after smoking a low nicotine cigarette (P < 0.0001). However, there were no significant differences in the time to reach maximum plasma levels of nicotine (Tmax), or in the nicotine half-life (T1/2) after low and high nicotine cigarette smoking. The Tmax was 15.2 ± 1.5 min and 14.6 ± 1.2 min after smoking low and high nicotine cigarettes, respectively. The T1/2 was 49.8 ± 13.3 and 62.9 ± 11.5 min after smoking low and high nicotine cigarettes, respectively.
Hypothalamic-Pituitary-Adrenal Hormones after Low and High Nicotine Cigarette Smoking (Figure 2, Table 2)
Figure 2.

: ACTH, Cortisol, DHEA and Epinephrine Levels after Smoking a Low or High Nicotine Cigarette. Hormone levels after smoking a high nicotine cigarette (filled circles) and a low nicotine cigarette (open circles) are shown on the left ordinates. Time (min) is shown on the abscissae. Points above BL were collected 10 min before cigarette smoking began at time 0. The 12-min cigarette-smoking period is indicated by a grey rectangle. Each data point is the average (± S.E.M.) of 10 subjects. Statistical analyses indicated significant changes from baseline in ACTH levels [df=18, F= 8.8, P=.005], epinephrine levels [df=18, F=3.9, P=.05], DHEA levels [df=18, F=8.7, P=.0006], and cortisol levels [df=18, F=9.0, P=.002] after high dose nicotine. ACTH (pmol/L) and epinephrine (pg/ml) are shown in the left column; cortisol (nmol/L) and DHEA (ng/ml) are shown in the right column. Asterisks indicate points that were significantly different from baseline (* = P< 0.05; ** = P< 0.01). Daggers indicate points at which hormone levels were significantly different after high nicotine cigarette smoking than after low nicotine cigarette smoking († = P< 0.05; †† = P< 0.01); ACTH [df=1, F=7.0, P=.016], Epinephrine [df=1, F=5.8, P=.026], DHEA [df=1, F=4.8, P=.048], and cortisol [df=1, F=5.5, P=.03].
TABLE 2:
TIME COURSE OF NICOTINE, ACTH, EPINEPHRINE, CORTISOL AND DHEA AFTER HIGH NICOTINE CIGARETTE SMOKING
| Tmax | T1/2 | |
|---|---|---|
| Significant differences from plasma nicotine are indicated by astrisks | ||
| Nicotine (ng/ml) | 14.6 ± 1.2 min | 62.9 ± 11.5 min |
| ACTH (pmol/L | 20.9 ± 1.2 min*** | 15.8 ± 1.6 min** |
| Epinephrine (pg/ml) | 22.3 ± 6.9 min | 19.1 ± 8.1 min** |
| Cortisol (nmol/L) | 47.0 ± 4.2 min*** | 29.7 ± 10.5 min |
| DHEA (ng/ml) | 37.0 ± 3.9 min*** | 24.0 ± 9.3 min* |
p = 0.01
p = 0.001
p <0.001
Baseline levels of ACTH, cortisol, epinephrine and DHEA did not differ significantly before low and high nicotine cigarette smoking. However, high nicotine cigarette smoking produced rapid and significant increases in all four measures. Low nicotine cigarette smoking did not alter ACTH or epinephrine levels, but both cortisol and DHEA levels decreased after smoking. The temporal profiles of these changes are described separately for each measure.
Adrenocorticotropin Hormone (ACTH) Levels After Low and High Nicotine Cigarette Smoking (Figure 2): Before smoking began, average baseline ACTH levels were equivalent in the low and high nicotine cigarette groups. After low nicotine cigarette smoking, ACTH levels did not change significantly from baseline, and were significantly lower than after high nicotine cigarette smoking between 14 min and 60 min (P= .029-.002). After smoking a high nicotine cigarette, ACTH levels increased significantly above baseline within 12 min and peak ACTH levels of 21.88 ± 5.34 pmol/L were measured at 20 min when plasma nicotine levels averaged 21.76 ± 2.10 ng/ml. The increase in ACTH was significantly correlated with the increase in plasma nicotine levels shown earlier in Figure 1 (r = 0.85; P < 0.0001). ACTH remained significantly higher than baseline for 60 min (P = 0.048-0.008). At 80 min, ACTH levels were equivalent in the low and high nicotine groups.
Cortisol Levels After Low and High Nicotine Cigarette Smoking (Figure 2): Baseline cortisol levels were equivalent before low and high nicotine cigarette smoking. Cortisol levels decreased significantly from baseline at 80, 100, and 120 min after smoking a low nicotine cigarette (P = 0.03-0.02), and were significantly lower than after high nicotine cigarette smoking between 25 and 120 min (P = 0.016-0.0003). Cortisol increased significantly within 18 min and remained significantly above baseline for 80 min (P = 0.05-0.006). Cortisol reached peak levels of 424 ± 48 nmol/L within 60 min, i.e., 40 min after peak ACTH levels were detected.
Dehydroepiandrosterone (DHEA) Levels After Low and High Nicotine Cigarette Smoking (Figure 2): Baseline DHEA levels were equivalent before low and high nicotine cigarette smoking. After low nicotine cigarette smoking, DHEA levels decreased significantly from baseline within 20 min and remained below baseline throughout the study (P = 0.043-0.007). DHEA levels were significantly lower after low nicotine cigarette smoking than after high nicotine cigarette smoking. Significant differences in DHEA levels between the low and high nicotine cigarette groups were detected at 20 min and persisted throughout the sampling period (P = 0.03-0.0003). After high nicotine cigarette smoking, DHEA increased significantly above baseline within 20 min, and peak levels of 21.13 ± 2.55 ng/ml were detected at 30 min. DHEA remained significantly above baseline until 60 min after high nicotine cigarette smoking (P = 0.044-0.005). Increases in DHEA and cortisol were significantly correlated (r = 0.92; P < 0.0001). DHEA was also significantly correlated with plasma nicotine levels (r= 0.46; P = 0.049).
Epinephrine Levels After Low and High Nicotine Cigarette Smoking (Figure 2): Baseline epinephrine levels were equivalent before low and high nicotine cigarette smoking. Epinephrine levels did not change significantly from baseline after low nicotine cigarette smoking, and were significantly lower than after high nicotine cigarette smoking between 8 min and 25 min after smoking began (P = 0.047-0.008). At the end of the sampling period, epinephrine averaged 48.6 ± 8.4 pg/ml and 43.0 ± 13.1 pg/ml after high and low nicotine smoking, respectively. Epinephrine began to increase immediately during high nicotine cigarette smoking and levels were significantly above baseline within 14 min (P = 0.043). Peak epinephrine levels of 115.2 ± 37.0 pg/ml were detected at 18 min after smoking began (P = 0.048) and epinephrine remained significantly above baseline for 8 min, i.e., until 20 min after smoking began. The increases in epinephrine were significantly correlated with the increases in ACTH (r = 0.86; P < 0.0001). Epinephrine was also significantly correlated with plasma nicotine levels (r = 0.83; P < 0.0001) after high nicotine cigarette smoking.
Comparison of the Time Course of Plasma Nicotine, ACTH, Epinephrine and DHEA (Table 2): Pharmacokinetic analyses indicated that after smoking a high nicotine cigarette, nicotine reached maximum plasma levels (Tmax) significantly faster than ACTH, cortisol and DHEA (P <0.0001). The half-life (T1/2) of plasma nicotine was significantly longer than the half-life of ACTH, epinephrine or DHEA (P <0.01-0.001). Taken together with Figures 1 and 2, these data suggest that the rapid increase in plasma nicotine levels stimulates ACTH and epinephrine release. Cortisol and DHEA began to increase during the ascending limb of the increase in ACTH and reached peak levels within 16 to 26 min after peak levels of ACTH were detected.
Cardiovascular Effects of Low and High Nicotine Cigarette Smoking (Figure 3)
Figure 3.

: Cardiovascular Measures after Smoking a Low or High Nicotine Cigarette. Heart rate (bpm), systolic and diastolic blood pressure (mm/Hg) after smoking a high nicotine cigarette (filled circles) and a low nicotine cigarette (open circles) are shown on the left ordinates in the first, second and third rows, respectively. Time (min) is shown on the abscissae. Points above BL were collected 10 min before smoking began at time 0. The 12-min cigarette-smoking period is indicated by a grey rectangle. Each data point is the average (± S.E.M.) of 10 subjects. Statistical analyses indicated significant changes from baseline in heart rate [df=18, F= 18, P<.0001], systolic blood pressure [df=11, F=6.1, P=.001], and diastolic blood pressure [df=11, F=7.0, P=.0006] after high nicotine cigarettes. Asterisks indicate points that were significantly different from baseline (* = P< 0.05; ** = P< 0.01). Daggers indicate points that were significantly different after high nicotine cigarette smoking than after low nicotine cigarette smoking († = P< 0.05; †† = P< 0.01); Heart Rate [df=1, F=6.3, P=.021], Systolic Blood Pressure [df=1, F=5.9, P=.026], and Diastolic Pressure [df=1, F=5.8, P=.03].
Heart Rate: There were no significant differences in heart rate between groups before smoking began. Heart rate increased significantly within 2 min after cigarette smoking began in both nicotine dose groups (P <0.0001-0.0005). However, heart rate was significantly higher after the high nicotine cigarette smoking than after the low nicotine cigarette smoking between 2 min and 16 min (P = 0.048-0.001). After high nicotine cigarette smoking, peak heart rates averaging 92.4 ± 2.3 bpm were detected within 10 min (P <0.0001), and heart rate remained significantly above baseline for 30 min (P = 0.032-0.0001). After low nicotine cigarette smoking, peak heart rates of 80.2 ± 2.6 bpm were detected within 12 min (P <0.002). Heart rate remained significantly above baseline levels for 20 min (P = 0.037-0.0003).
Systolic Blood Pressure: Baseline systolic blood pressure did not differ before low and high nicotine cigarette smoking. Systolic blood pressure increased significantly from baseline at 5 and 10 min after smoking a high nicotine cigarette (P = 0.002-0.0005), but there were no significant changes after smoking a low nicotine cigarette. Systolic blood pressure was significantly higher after smoking a high than a low nicotine cigarette at 5 and 10 min after smoking began (P = 0.03-0.009).
Diastolic Blood Pressure: Baseline diastolic blood pressure did not differ before low and high nicotine cigarette smoking. Diastolic blood pressure increased significantly from baseline at 5 and 10 min after high nicotine cigarette smoking began (P = 0.001-0.0005) and at 10 min after low nicotine cigarette smoking began (P=0.003), but there were no significant differences in diastolic blood pressure between the two groups.
Subjective Effects of Low and High Nicotine Cigarette Smoking (Figure 4).
VAS ratings on four subjective effects measures during and after smoking a low or a high nicotine cigarette are shown in Figure 4. Baseline levels of these subjective measures did not differ significantly between the high and low nicotine cigarette groups. When high nicotine cigarette smoking began, VAS ratings of “high,” “rush” and “nicotine liking” increased significantly within the first 2 min or four puffs, and reached peak levels within 4 to 6 min. These VAS ratings were significantly inter-correlated during the first 20 min after smoking began (r=0.96-0.98; P<0.0001). Increases in ratings of “high,” “rush” and “nicotine liking” were also significantly correlated with increases in heart rate (r = 0.86-0.88; P <0.0001). After low nicotine cigarette smoking, the VAS ratings of “high,” “rush” and “nicotine liking” also increased significantly (P = 0.002-0.026), but these increases were significantly less than after high nicotine cigarette smoking (P <0.05-0.001).
Figure 4.

: Reports of Subjective Effects After Smoking a Low or High Nicotine Cigarette. Subjective ratings on a Visual Analogue Scale (VAS) (0-100) are shown on the left ordinates and time (min) is shown on the abscissae. Points above BL were collected 10 min before smoking began at time 0. Each data point is the average (± S.E.M.) of 10 subjects. The 12 min cigarette smoking period is indicated by a grey rectangle. Asterisks indicate points that were significantly different from baseline (* = P< 0.05; ** = P< 0.01). Statistical analyses indicated significant changes from baseline in reports of “high” after high nicotine cigarettes [df=18, F=16.9, P<.0001] and low nicotine cigarettes [df=18, F=5.2, P=.01; reports of “liking” after high nicotine cigarettes [df=18, F=14.1, P<.0001] and low nicotine cigarettes [df=18, F=3.9, P=.05]; reports of “rush” after high nicotine cigarettes [df=18, F=13.2, P<.0001] and low nicotine cigarettes [df=18, F=5.1, P=.009; and reports of “craving“ after high nicotine cigarettes [df=18, F=8.1, P=.0007] and after low nicotine cigarettes [df=18, F=5.6, P=.007]. Daggers indicate points that were significantly different after high nicotine cigarette smoking than after low nicotine cigarette smoking († = P< 0.05; †† = P<0.01) “high “ [df=1, F=4.5 P=.049], “like”[df=1, F=6.2, P=.023], “rush” [df=1, F=6.3, P=.02], and “craving” [df=1, F=5.6, P=.04].
In contrast, VAS ratings of “craving” for nicotine were high in both groups before cigarette smoking began, and decreased significantly during smoking. High nicotine cigarette smoking decreased “craving” scores significantly more than low nicotine cigarette smoking between 16 and 50 min after smoking began (P = 0.05-0.032). “Craving” ratings decreased to a nadir of 2.0 ± 1.3 mm within 18 min after high nicotine cigarette smoking began, and remained significantly below baseline for 100 min (P <0.03-0.0003). “Craving” ratings decreased to a nadir of 20.0 ± 8.6 mm within 16 min after low nicotine cigarette smoking began and remained significantly below baseline for 30 min (P <0.02-0.003). There were no significant changes from baseline in VAS ratings of “sick,” “jittery,” “bad feeling” and “alert,” in either the high and low nicotine cigarette groups (data not shown). The high nicotine cigarette group reported significantly higher ratings of dizziness than the low nicotine cigarette group during the first 8 min after smoking began (P <0.01) (data not shown).
Discussion
We believe that this is the first study to use rapid sampling procedures to examine the covariance between endocrine, cardiovascular and subjective measures both during and after cigarette smoking. Cigarette smoking produced nicotine dose-dependent increases in anterior pituitary and adrenal hormones as well as cardiovascular and subjective responses in nicotine dependent men. The low nicotine cigarette did not increase pituitary or adrenal hormones, and increases in heart rate and VAS ratings of positive subjective effects were significantly lower than after smoking a high nicotine cigarette. Although smoking behavior, defined in terms of duration of cigarette exposure and number and duration of puffs, was equivalent for high and low nicotine cigarettes, nicotine plasma levels were significantly higher after smoking high nicotine cigarettes. Thus, it is likely that the significant differences in hormonal and subjective effects were attributable to the relative dose of nicotine. The remainder of this discussion examines the temporal interactions between cigarette smoking and changes in HPA axis hormones, VAS ratings of subjective effects and cardiovascular measures. Some possible implications of these findings for the neurobiology of nicotine's abuse-related effects are considered.
Nicotine Plasma Levels after Smoking High and Low Nicotine Cigarettes
Nicotine plasma levels increased significantly above baseline within 2 min (4 puffs) and reached peak levels within 12 to 14 min after cigarette smoking began. This time course was similar to that reported after 10 min of smoking at 1 puff per min (Tmax = 11.9 ± 1.9 min) (Gourlay and Benowitz, 1997). Nicotine plasma levels and pharmacokinetic profiles in the present study also were similar to those previously reported under similar smoking conditions (Mendelson et al., 2003). Although the low nicotine cigarette produced measurable levels of nicotine (less than 4 ng/ml), these levels were significantly lower than peak plasma levels produced by high nicotine cigarettes (over 20 ng/ml). Moreover, the small but significant increase in plasma nicotine occurred later (after 6 min or 12 puffs) after smoking a low than a high nicotine cigarette and remained at relatively stable, low levels for 120 min. In a study designed to determine the threshold dose at which subjects could discriminate nicotine nasal spray from placebo, nicotine-dependent smokers were able to distinguish a dose of 3 μg/kg from placebo (Perkins et al., 2001). This dose of nicotine nasal spray produced nicotine plasma levels of 2.6 ng/ml, or slightly lower than the low nicotine cigarette in the present study. Despite differences in routes of nicotine administration (nasal spray and smoking), these data converge to suggest that very low doses of nicotine are discriminable.
Subjective Responses to Smoking High and Low Nicotine Cigarettes
High, Rush and Liking: Significant increases in ratings of positive subjective effects occurred within 4 puffs over 2 min after smoking both a low and a high nicotine cigarette. However, VAS ratings were significantly higher during high than low nicotine cigarette smoking, and this suggests that subjects could detect the relative nicotine levels within 4 puffs. These data are consistent with the failure of low yield cigarettes to reduce smoking behavior (Benowitz, 2001). It is well established that smokers adjust their smoking topography and smoke more cigarettes to try to compensate for a low nicotine yield (Benowitz, 2001).
VAS ratings were highest during the initial ascending limb of the plasma nicotine curve, then gradually diminished during the smoking period as plasma nicotine approached peak levels. In contrast, after smoking a low nicotine cigarette, positive VAS ratings remained relatively stable across the 12 min smoking period. Interestingly, the time course of increases in VAS ratings of positive subjective effects was similar during smoking a high nicotine cigarette and after intravenous nicotine administration. When intravenous nicotine was infused over 10 sec., peak VAS ratings of High, Rush and Liking (Jones et al., 1999) and Strength of Drug Effect (Soria et al., 1996) occurred within two min. In the present study, VAS ratings of High, Rush and Liking began to decrease within 4 min after high nicotine cigarette smoking began. The relatively rapid decrease in reports of positive subjective effects during smoking occurred while plasma nicotine levels were increasing. A similar rapid decrease in positive subjective reports was also observed after intravenous nicotine administration, although plasma nicotine levels were not measured (Soria et al., 1996; Jones et al., 1999). It is unlikely that the decrease in positive VAS ratings reflected an increase in the aversive properties of smoking, because ratings of Sick, Jittery and Bad Feeling did not change significantly from baseline.
One limitation of the present study is that nicotine and hormone levels were measured in venous rather than arterial blood. Arterial nicotine levels are 6 to 10 fold higher than venous nicotine levels (Benowitz, 1996). It is estimated that nicotine from cigarette smoke reaches peak levels in the arterial circulation within 20 sec (Rose et al., 1999). Although significant increases in subjective ratings and plasma nicotine were detected within 4 puffs in the present study, arterial nicotine, CRH and ACTH levels may have increased significantly after a single puff.
Craving: Ratings of cigarette craving decreased rapidly during and after smoking in both the low and high nicotine cigarette groups. Smoking a high nicotine cigarette reduced ratings of craving significantly more than smoking a low nicotine cigarette, and craving scores remained significantly below baseline for much longer (100 min vs 30 min). Decreases in cigarette craving often are reported after smoking low nicotine cigarettes that contain <0.06 to 0.1 mg of nicotine (Gross et al., 1997; Pickworth et al., 1999; Robinson et al., 2000; Rose et al., 2000). The salient effects of low nicotine cigarettes on reports of craving, liking, and reduction of acute withdrawal symptoms are usually interpreted as evidence that complex sensory cues are important in maintaining cigarette smoking behavior (Butschky et al., 1995; Gross et al., 1997; Pickworth et al., 1999; Shahan et al., 1999; Rose et al., 2000). In the present study, the greatest reductions in ratings of craving occurred within the first 20 min after smoking, then craving ratings began to increase. The extent to which increases in craving may prompt the next smoking episode is poorly understood. However, during several weeks residence on a clinical research ward, men usually initiated smoking within 20 to 30 min after the last cigarette (Mello et al., 1985; Mutschler et al., 2002).
Cigarette Smoking, Heart Rate and the HPA Axis
Heart Rate: The rate and magnitude of heart rate increases after cigarette smoking were similar to those reported after intravenous nicotine administration (Soria et al., 1996; Jones et al., 1999). After high nicotine cigarette smoking, the ascending limb of the plasma nicotine curve was highly correlated with significant increases in ACTH, epinephrine and heart rate. However, significant increases in heart rate occurred within 2 min after smoking began, whereas significant increases in epinephrine were detected 12 min later. These data are consistent with previous reports, and are usually interpreted to suggest that the initial effects of nicotine on heart rate are mediated by release of norepinephrine in the cardiovascular system, rather than release of epinephrine from the adrenal medulla (Gourlay and Benowitz, 1997).
ACTH and Epinephrine: Increases in ACTH after cigarette smoking have been reported under a number of conditions where samples for analysis were collected once a day or at relatively infrequent intervals, often without concurrent plasma nicotine measurement (Seyler et al., 1984, 1986; Baron et al., 1995; Pickworth and Fant, 1998; Coiro and Vescovi, 1999; del Arbol et al., 2000). The present study confirms the generality of earlier findings and examines ACTH and epinephrine levels at 2 min intervals during as well as after smoking. ACTH and epinephrine began to increase within 6 min after high nicotine cigarette smoking began and reached peak levels within 4 to 6 min after peak nicotine plasma levels were detected. This time course was similar to our previous report of the temporal covariance between ACTH, epinephrine and plasma cocaine levels, after i.v. cocaine administration in men (Mendelson et al., 2002). In that study, peak levels of ACTH, epinephrine, euphoria and plasma cocaine were measured within 8 to 10 min after i.v. cocaine injection. We interpreted those data to suggest that the HPA hormone increases may be significant concomitants of the abuse-related effects of cocaine in humans (Mendelson et al., 2002). As described earlier, corticotropin-releasing-hormone (CRH) from the hypothalamus stimulates ACTH release from the anterior pituitary. Antagonism of CRH release by a CRH-1 antagonist selectively reduced i.v. cocaine self-administration by rats (Goeders and Guerin, 2000). The possible influence of HPA axis activation, as indicated by ACTH and cortisol increases, on the abuse-related effects of cocaine has been discussed in several reviews (Goeders, 1997, 2002a and b; Mello and Mendelson, 2002). The similar patterns of HPA axis activation and subjective responses after i.v. cocaine and smoked nicotine are provocative.
Cortisol and DHEA: It is also interesting to consider the possible contribution of cortisol and DHEA to the abuse-related effects of cigarette smoking. Cortisol began to increase significantly when ACTH levels were highest, and reached peak levels between 30 and 60 min after smoking when ACTH levels were decreasing. This time course is consistent with established feedback relationships between ACTH and cortisol (Yen et al., 1999). The increases in cortisol and DHEA were significantly correlated, and each had a similar half-life after high nicotine cigarette smoking. These data are consistent with previous reports that smokers have higher basal cortisol and DHEA levels than non-smokers (Pomerleau et al., 1992; Field et al., 1994; del Arbol et al., 2000; al'Absi et al., 2003). Cigarette smoking usually induces an increase in plasma cortisol levels (Cryer et al., 1976; Winternitz and Quillen, 1977; Spohr et al., 1979; Wilkins et al., 1982; Seyler et al., 1984; Gossain et al., 1986; see for review Pickworth and Fant, 1998), and cortisol may remain elevated as a consequence of repeated cigarette smoking throughout the day. It has also been suggested that nicotine may inhibit the diurnal decrease in plasma cortisol levels (Pomerleau et al., 1992). The contribution of high cortisol levels to the maintenance of smoking behavior is unclear, but decreases in cortisol on the first day of smoking abstinence were associated with a higher rate of relapse during the first week (al'Absi et al., 2004). The importance of cortisol for the reinforcing effects of cocaine has been clearly demonstrated in rodents (Goeders, 2002a).
DHEA is an adrenal androgen precursor of testosterone (Yen et al., 1999). DHEA formulations are sold over the counter as dietary supplements, and DHEA is believed to improve a sense of wellbeing and sexuality in the elderly, and in persons with adrenal insufficiency. Although the evidence is conflicting (see for review, Spark, 2002), two placebo-controlled clinical trials support the notion that DHEA treatment improves mood and alleviates depression (Morales et al., 1994; Schmidt et al., 2005). In a 6 month, placebo-controlled, cross-over trial, DHEA administration at physiological levels (50 mg, P.O.) consistently increased energy and improved mood and feelings of wellbeing in both men and women (Morales et al., 1994). In a 6 week, placebo-controlled cross-over trial in men and women with major or minor depression, DHEA administration (90 mg/day for 3 weeks; 450 mg/day for 3 weeks) significantly improved mood and depression ratings in comparison to pre-treatment baseline and placebo conditions (Schmidt et al., 2005). In the present study, peak levels of DHEA after smoking a high nicotine cigarette (21.13 ± 2.55 ng/ml or 73.32 ± 8.84 nmol/L or 2113.0 ng/dl) were over four times higher than average DHEA levels achieved after 3 months of DHEA replacement in men (14.72 ± 1.4 nmol/L) (Morales et al., 1994), and over twice as high as average DHEA levels after 6 weeks of high dose DHEA treatment (1047.2 ± 709.1 ng/dl) (Schmidt et al., 2005). Taken together, these data suggest that increases in DHEA levels may contribute to the mood elevating effects reported after cigarette smoking.
The effects of cigarette smoking and intravenous cocaine on DHEA were also similar. The significant increases in DHEA and cortisol after smoking a high nicotine cigarette observed in the present study were similar in time course and magnitude to the increases in DHEA and cortisol measured after i.v. cocaine in men (Mendelson et al., 2002). It is interesting to note the temporal covariance between changes in DHEA and cortisol levels and craving for nicotine. Nicotine craving increased sharply between 60 and 80 min after cigarette smoking began, when both cortisol and DHEA were decreasing. Although relatively little is known about the interactions between DHEA and cocaine or nicotine, DHEA administration produced a dose-dependent enhancement of cocaine conditioned place preference in mice (Romieu et al., 2003).
Cigarette Smoking and the HPA Axis: These data reported in the present study are consistent with the interpretation that the hormonal milieu may modulate the abuse-related effects of cigarette smoking. The rapid increases in positive subjective ratings, and the rapid release of HPA axis hormones were nicotine dose-related. The temporal covariance between plasma nicotine levels and HPA axis activation parallels patterns reported after i.v. cocaine administration under similar experimental conditions (Mendelson et al., 2002). HPA axis activation is also a response to “stress” variously defined, and there has been considerable interest in the relationship between “stress” and vulnerability to drug addiction (Koob and Le Moal, 2001; Sinha, 2001; Grammatopoulos and Chrousos, 2002; Marinelli and Piazza, 2002; Heinrichs and Koob, 2004), as well as other psychiatric disorders (see for review Tsigos and Chrousos, 1994, 2002). HPA axis hormones, as well as gonadal steroid hormones, appear to influence some behavioral responses to cocaine (Goeders, 1997; Goeders, 2002a and b; Mello and Mendelson, 2002). Studies of the generality of these findings to women at the follicular and luteal phases of the menstrual cycle are in progress (Mendelson et al., 2004). Although much remains to be learned about the interactions between cigarette smoking and the HPA axis, medications that mimic or attenuate the hormonal effects of cigarette smoking may be useful for treatment of this addictive disorder.
Acknowledgments
This research was supported in part by grants R01-DA15067, P01-DA14528, K05-DA00064, K05-DA00101 and T32-DA07252. We are grateful to Murty Pharmaceuticals Inc., Lexington, KY 40509, for generously supplying the de-nicotinized cigarettes used in this study. We thank Alicja Skupny and Howard Gelles for excellent technical assistance in conducting the hormone analyses. We also thank Dr. Peton Jacob III, University of California at San Francisco for conducting the nicotine assays. Preliminary data were presented to the American College of Neuropsychopharmacology in 2003.
References
- U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion. Office on Smoking and Health; Atlanta, GA: 2004. U.S. Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General; p. 941. [Google Scholar]
- Abreu ME, Bigelow GE, Fleisher L. Effect of intravenous injection speed on responses to cocaine and hydromorphone in humans. Psychopharmacology. 2001;154:76–84. doi: 10.1007/s002130000624. [DOI] [PubMed] [Google Scholar]
- al'Absi M, Hatsukami DK, Davis GL, Wittmers LE. Prospective examination of effects of smoking abstinence on cortisol and withdrawal symptoms as predictors of early smoking relapse. Drug Alc. Depend. 2004;73:267–278. doi: 10.1016/j.drugalcdep.2003.10.014. [DOI] [PubMed] [Google Scholar]
- al'Absi M, Wittmers LE, Erickson J, Hatsukami DK, Crouse B. Attenuated adrenocortical and blood pressure responses to psychological stress in ad libitum and abstinent smokers. Pharmacol. Biochem. Behav. 2003;74:401–410. doi: 10.1016/s0091-3057(02)01011-0. [DOI] [PubMed] [Google Scholar]
- APA . Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition American Psychiatric Association; Washington, D.C.: 1994. p. 886. [Google Scholar]
- Ayala AR, Pushkas J, Higley JD, Ronsaville D, Gold PW, Chrousos GP, et al. Behavioral, adrenal, and sympathetic responses to long-term administration of an oral corticotropin-releasing hormone receptor antagonist in a primate stress paradigm. J. Clin. Endocrinol. Metab. 2004;89:5729–5737. doi: 10.1210/jc.2003-032170. [DOI] [PubMed] [Google Scholar]
- Balster RL, Schuster CR. Fixed-interval schedule of cocaine reinforcement: Effect of dose and infusion duration. J. Exper. Anal. Behav. 1973;20:119–129. doi: 10.1901/jeab.1973.20-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron JA, Comi RJ, Cryns V, Brinck-Johnsen T, Mercer NG. The effect of cigarette smoking on adrenal cortical hormones. J. Pharmacol. Exp. Ther. 1995;272:151–155. [PubMed] [Google Scholar]
- Benowitz NL. Pharmacology of nicotine: Addiction and therapeutics. Annu. Rev. Pharmcol. Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- Benowitz NL.Compensatory smoking of low-yield cigarettes Risks associated with smoking cigarettes with low machine-measured yields of tar and nicotine. Smoking and tobacco control monograph No. 13 200139–63.U.S. Department of Health and Human Services, National Insitutes of Health, National Cancer Institute, NIH Pub. No. 02-5074; Bethesda, MD: October 2001 [Google Scholar]
- Broadbear JH, Winger G, Cicero TJ, Woods JH. Effects of self-administered cocaine on plasma adrenocorticotropic hormone and cortisol in male rhesus monkeys. J. Pharmacol. Exp. Ther. 1999a;289:1641–1647. [PubMed] [Google Scholar]
- Broadbear JH, Winger G, Woods JH. Cocaine-reinforced responding in rhesus monkeys: Pharmacological attenuation of the hypothalamic-pituitary-adrenal axis response. J. Pharmacol. Exp. Ther. 1999b;290:1347–1355. [PubMed] [Google Scholar]
- Butschky MF, Bailey D, Henningfield JE, Pickworth WB. Smoking without nicotine delivery decreaes withdrawal in 12-hour abstinent smokers. Pharmacol. Biochem. Behav. 1995;50:91–96. doi: 10.1016/0091-3057(94)00269-o. [DOI] [PubMed] [Google Scholar]
- CDC Declines in lung cancer rates - California, 1988-1997. Morbidity and Mortality Weekly Report. 2000;49:1066–1069. [PubMed] [Google Scholar]
- CDC . National Center for Chronic Disease Prevention and Prevention and Health Promotion. Office on smoking and Health; Washington, D.C.: 2001. Women and Smoking: A report of the Surgeon General; p. 675. [Google Scholar]
- CDC Adult cigarette smoking in the United States: Current estimates. National Center for Chronic Disease Prevention and Health Promotion. Tobacco Information and Prevention Source (TIPS) 2004;15:20–58. [Google Scholar]
- Coiro V, Vescovi PP. Effect of cigarette smoking on ACTH/cortisol secretion in alcoholics after short- and medium-term abstinence. Alcohol Clin. Exp. Res. 1999;23:1515–1518. [PubMed] [Google Scholar]
- Contoreggi C, Herning RI, Na P, Gold PW, Chrousos G, Negro PJ, et al. Stress hormone responses to corticotropin-releasing hormone in substance abusers with severe comorbid psychiatric disease. Biol Psychiatry. 2003;54:873–878. doi: 10.1016/s0006-3223(03)00167-7. [DOI] [PubMed] [Google Scholar]
- Corrigall WA. Nicotine self-administration in animals as a dependence model. Nicotine Tob. Res. 1999;1:11–20. doi: 10.1080/14622299050011121. [DOI] [PubMed] [Google Scholar]
- Cryer PE, Haymond MW, Santiago JV, Shah SD. Norepinephrine and epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. N. Engl. J. Med. 1976;295:573–577. doi: 10.1056/NEJM197609092951101. [DOI] [PubMed] [Google Scholar]
- del Arbol JL, Munoz JR, Ojeda L, Cascales AL, Irles JR, Mianda MT, et al. Plasma concentrations of beta-endophin in smokers who consume different numbers of cigarettes per day. Pharm. Biochem. Behav. 2000;67:25–28. doi: 10.1016/s0091-3057(00)00291-4. [DOI] [PubMed] [Google Scholar]
- Donny EC, Caggiula AR, Mielke MM, Booth S, Gharib MA, Hoffman A, et al. Nicotine self-administration in rats on a progressive ratio schedule of reinforcement. Psychopharmacology (Berl.) 1999;147:135–142. doi: 10.1007/s002130051153. [DOI] [PubMed] [Google Scholar]
- Field AE, Colditz GA, Willett WC, Longcope C, McKinlay JB. The relation of smoking, age, relative weight, and dietary intake to serum adrenal steroids, sex hormones, and sex hormone-binding globulin in middle-aged men. J. Clin. Endocrinol. Metab. 1994;79:1310–1316. doi: 10.1210/jcem.79.5.7962322. [DOI] [PubMed] [Google Scholar]
- Fu Y, Matta SG, Gao W, Brower VG, Sharp BM. Systemic nicotine stimulates dopamine release in nucleus accumbens: Re-evaluation of the role of N-methyl-D-aspartate receptors in the ventral tegmental area. J. Pharmacol. Exp. Ther. 2000;294:458–465. [PubMed] [Google Scholar]
- Gerasimov MR, Franceschi M, Volkow ND, Rice O, Schiffer WK, Dewey SL. Synergistic interactions between nicotine and cocaine or methylphenidate depend on the dose of dopamine transporter inhibitor. Synapse. 2000;38:432–437. doi: 10.1002/1098-2396(20001215)38:4<432::AID-SYN8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Goeders NE. A neuroendocrine role in cocaine reinforcement. Psychoneuroendocrinology. 1997;22:237–259. doi: 10.1016/s0306-4530(97)00027-9. [DOI] [PubMed] [Google Scholar]
- Goeders NE. The HPA axis and cocaine reinforcement. Psychoneuroendocrinology. 2002a;27:13–33. doi: 10.1016/s0306-4530(01)00034-8. [DOI] [PubMed] [Google Scholar]
- Goeders NE. Stress and cocaine addiction. J. Pharmacol. Exp. Ther. 2002b;301:785–789. doi: 10.1124/jpet.301.3.785. [DOI] [PubMed] [Google Scholar]
- Goeders NE, Guerin GF. Role for corticosterone in intravenous cocaine self-administration in rats. Neuroendocrinology. 1996;64:337–348. doi: 10.1159/000127137. [DOI] [PubMed] [Google Scholar]
- Goeders NE, Guerin GF. Effects of the CRH receptor antagonist CP-154,526 on intravenous cocaine self-administration in rats. Neuropsychopharmacology. 2000;23:577–586. doi: 10.1016/S0893-133X(00)00148-2. [DOI] [PubMed] [Google Scholar]
- Goldberg SR, Spealman RD, Goldberg DM. Persistent behavior at high rates maintained by intravenous self-administration of nicotine. Scinece. 1981;214:573–575. doi: 10.1126/science.7291998. [DOI] [PubMed] [Google Scholar]
- Gossain VV, Sherma NK, Srivastava L, Michelakis AM, Rovner DR. Hormonal effects of smoking--II: Effects on plasma cortisol, growth hormone, and prolactin. Am. J. Med. Sci. 1986;291:325–327. doi: 10.1097/00000441-198605000-00007. [DOI] [PubMed] [Google Scholar]
- Gourlay SG, Benowitz NL. Arteriovenous differences in plasma concentration of nicotine and catecholamines and related cardiovascular effects after smoking, nicotine nasal spray, and intravenous nicotine. Clin. Pharmacol. Ther. 1997;62:453–463. doi: 10.1016/S0009-9236(97)90124-7. [DOI] [PubMed] [Google Scholar]
- Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol. Metab. 2002;13:436–444. doi: 10.1016/s1043-2760(02)00670-7. [DOI] [PubMed] [Google Scholar]
- Griffiths RR, Henningfield JE, Bigelow GE. Human cigarette smoking: Manipulation of number of puffs per bout, interbout interval and nicotine dose. J. Pharmacol. Exp. Ther. 1982;220:256–265. [PubMed] [Google Scholar]
- Gross J, Lee J, Stitzer ML. Nicotine-containing versus de-nicotinized cigarettes: Effects on craving and withdrawal. Pharmacol. Biochem. Behav. 1997;57:159–165. doi: 10.1016/s0091-3057(96)00309-7. [DOI] [PubMed] [Google Scholar]
- Habib KE, Weld KP, Rice KC, Pushkos J, Champoux M, Listwak S, et al. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc. Natl. Acad. Sci. 2000;97:6079–6084. doi: 10.1073/pnas.97.11.6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey DM, Yasar S, Heishman SJ, Panlilio LV, Henningfield JE, Goldberg SR. Nicotine serves as an effective reinforcer of intravenous drug-taking behavior in human cigarette smokers. Psychopharmacology (Berl) 2004;175:134–142. doi: 10.1007/s00213-004-1818-6. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311:427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Goldberg SR. Control of behavior by intravenous nicotine injections in human subjects. Pharmacol Biochem Behav. 1983;19:1021–1026. doi: 10.1016/0091-3057(83)90409-4. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Schuh LM, Jarvik ME. Pathophysiology of Tobacco Dependence. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology. The Fourth Generation of Progress. Raven Press; New York: 1995. pp. 1715–1729. [Google Scholar]
- Iranmanesh A, Lizarralde G, Short E, Veldhuis JD. Intensive venous sampling paradigms disclose high frequency adrenocorticotropin release episodes in normal men. J. Clin. Endocrinol. Metab. 1990;71:1276–1283. doi: 10.1210/jcem-71-5-1276. [DOI] [PubMed] [Google Scholar]
- Jacob P, Wu S, Yu L, Benowitz NL. Simultaneous determination of mecamylamine, nicotine, and cotinine in plasma by gas chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2000;23:653–661. doi: 10.1016/s0731-7085(00)00343-5. [DOI] [PubMed] [Google Scholar]
- Jaffe JH. Tobacco smoking and nicotine dependence. In: Wonnacott S, Russell MAH, Stolerman IP, editors. Nicotine Psychopharmacology Molecular, Cellular, and Behavioural Aspects. Oxford University Press; New York: 1990. pp. 1–37. [Google Scholar]
- Jones HE, Garrett BE, Griffiths RR. Subjective and physiological effects of intravenous nicotine and cocaine in cigarette smoking cocaine abusers. J. Pharmacol. Exp. Ther. 1999;288:188–197. [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Ritz MC, Boja JW. The dopamine hypothesis of the reinforcing properties of cocaine. Trends. Nerosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- Mantsch JR, Schlussman SD, Ho A, Kreek MJ. Effects of cocaine self-administraiton on plasma corticosterone and prolactin in rats. J. Pharmacol. Exp. Ther. 2000;294:239–247. [PubMed] [Google Scholar]
- Marinelli M, Piazza PV. Interaction between glucocorticoid hormones, stress and psychostimulant drugs. Eur J Neurosci. 2002;16:387–394. doi: 10.1046/j.1460-9568.2002.02089.x. [DOI] [PubMed] [Google Scholar]
- Matta SG, Foster CA, Sharp BM. Nicotine stimulates the expression of cFos protein in the parvocellular paraventricular nucleus and brainstem catecholaminergic regions. Endocrinology. 1993a;132:2149–2156. doi: 10.1210/endo.132.5.8386611. [DOI] [PubMed] [Google Scholar]
- Matta SG, Foster CA, Sharp BM. Selective administration of nicotine into catecholaminergic regions of rat brainstem stimulates adrencorticotropin secretion. Endocrinology. 1993b;133:2935–2942. doi: 10.1210/endo.133.6.8243321. [DOI] [PubMed] [Google Scholar]
- Matta SG, McAllen KM, Sharp BM. Role of the fourth cerebroventricle in mediating rat plasma ACTH responses to intravenous nicotine. J. Pharmacol. Exp. Ther. 1990;252:623–630. [PubMed] [Google Scholar]
- Matta SG, Valentine JD, Sharp BM. Nicotinic activation of CRH neurons in extrahypothalamic regions of the rat brain. Endocrine. 1997;7:245–253. doi: 10.1007/BF02778147. [DOI] [PubMed] [Google Scholar]
- Matta SG, Yitong F, Valentine JD, Sharp BM. Response of the hypothalamo-pituitary-adrenal axis to nicotine. Psychoneuroendocrinology. 1998;23:103–113. doi: 10.1016/s0306-4530(97)00079-6. [DOI] [PubMed] [Google Scholar]
- MDPH Massachusetts Department of Public Health 1997 Cigarette Nicotine Disclosure Report as required by Massachusetts General Laws Chapter 307B 1998. CMR 660,000, January 16, 1998
- Mello NK, Lukas SE, Mendelson JH. Buprenorphine effects on cigarette smoking. Psychopharmacology. 1985;86:417–424. doi: 10.1007/BF00427902. [DOI] [PubMed] [Google Scholar]
- Mello NK, Mendelson JH. Cocaine, hormones and behavior: Clinical and preclinical studies. In: Pfaff DW, Arnold AP, Etgen AM, Fahrbach SE, Rubin RT, editors. Hormones, Brain and Behavior. Academic Press; New York: 2002. pp. 665–745. [Google Scholar]
- Mendelson JH, Mello NK, Sholar MB, Siegel AJ, Mutschler N, Halpern J. Temporal concordance of cocaine effects on mood states and neuroendocrine hormones. Psychoneuroendocrinology. 2002;27:71–82. doi: 10.1016/s0306-4530(01)00036-1. [DOI] [PubMed] [Google Scholar]
- Mendelson JH, Sholar MB, Goletiani N,V, Siegel AJ, Mello NK. Menstrual cycle phase influences the effects of cigarette smoking on mood states and the HPA axis. Neuropsychopharmacology; ACNP 43rd Annual Meeting; San Juan, PR. December 2004.2004. p. S138. [Google Scholar]
- Mendelson JH, Sholar MB, Mutschler NH, Jaszyna-Gasior M, Goletiani N,V, Siegel AJ, et al. Effects of intravenous cocaine and cigarette smoking on luteinizing hormone, testosterone and prolactin in men. J. Pharmacol. Exp. Ther. 2003;307:339–348. doi: 10.1124/jpet.103.052928. [DOI] [PubMed] [Google Scholar]
- Mendelson JH, Teoh SK, Mello NK, Ellingboe J, Rhoades E. Acute effects of cocaine on plasma adrenocorticotropic hormones, luteinizing hormone and prolactin levels in cocaine-dependent men. J. Pharmacol. Exp. Ther. 1992;263:505–509. [PubMed] [Google Scholar]
- Morales AJ, Nolan JJ, Nelson JC, Yen SSC. Effects of replacement dose of dehydroepiandrosterone in men and women of advancing age. J Clin Endo Metab. 1994;78:1360–1367. doi: 10.1210/jcem.78.6.7515387. [DOI] [PubMed] [Google Scholar]
- Mutschler NH, Stephen BJ, Teoh SK, Mendelson JH, Mello NK. An inpatient study of the effects of buprenorphine on cigarette smoking in men concurrently dependent on cocaine and opioids. Nicotine and Tobacco Research. 2002;4:223–228. doi: 10.1080/14622200210124012. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: New findings and new directions. Med. Psychiatry. 1996;1:336–342. [PubMed] [Google Scholar]
- Nemeth-Coslett R, Griffiths RR. Determinants of puff duration in cigarette smokers: I. Pharmacol. Biochem. Behav. 1984a;20:965–971. doi: 10.1016/0091-3057(84)90024-8. [DOI] [PubMed] [Google Scholar]
- Nemeth-Coslett R, Griffiths RR. Determinants of puff duration in cigarette smokers: II. Pharmacol. Biochem. Behav. 1984b;21:903–912. doi: 10.1016/s0091-3057(84)80072-6. [DOI] [PubMed] [Google Scholar]
- Perkins KA, Fonte C, Sanders M, Meeker J, Wilson A. Threshold doses for nicotine discrimination in smokers and non-smokers. Psychopharmacology. 2001;155:163–170. doi: 10.1007/s002130000660. [DOI] [PubMed] [Google Scholar]
- Pickworth WB, Fant RV. Endocrine effects of nicotine administration, tobacco and other drug withdrawal in humans. Psychoneuroendocrinology. 1998;23:131–141. doi: 10.1016/s0306-4530(97)00075-9. [DOI] [PubMed] [Google Scholar]
- Pickworth WB, Fant RV, Nelson RA, Rohrer MS, Henningfield JE. Pharmacodynamic effects of new de-nicotinized cigarettes. Nicotine Tob. Res. 1999;1:357–364. doi: 10.1080/14622299050011491. [DOI] [PubMed] [Google Scholar]
- Pomerleau OF, Fertig JB, Seyler LE, Jaffe J. Neuroendocrine reactivity to nicotine in smokers. Psychopharmacology. 1983;81:61–67. doi: 10.1007/BF00439275. [DOI] [PubMed] [Google Scholar]
- Pomerleau OF, Flessland KA, Pomerleau CS, Hariharan M. Controlled dosing of nicotine via an intranasal nicotine aerosol delivery device (INADD) Psychopharmacology. 1992;108:519–526. doi: 10.1007/BF02247431. [DOI] [PubMed] [Google Scholar]
- Robinson ML, Houtsmuller EJ, Moolchan ET, Pickworth WB. Placebo cigarettes in smoking research. Exp. Clin. Psychopharmacol. 2000;8:326–332. doi: 10.1037//1064-1297.8.3.326. [DOI] [PubMed] [Google Scholar]
- Romieu P, Martin-Fardon R, Bowen WD, Maurice T. s 1Receptor-related neuroactive steroids modulate cocaine-induced reward. J. Neurosci. 2003;23:3572–3576. doi: 10.1523/JNEUROSCI.23-09-03572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Coleman RE. Arterial nicotine kinetics during cigarette smoking and intravenous nicotine administration: implications for addiction. Drug Alc. Depend. 1999;56:99–107. doi: 10.1016/s0376-8716(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Rose JE, Behm FM, Westman EC, Johnson M. Dissociating nicotine and nonnicotine components of cigarette smoking. Pharmacol. Biochem. Behav. 2000;67:71–81. doi: 10.1016/s0091-3057(00)00301-4. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, Mello NK, Mendelson JH, Nguyen P, Erös-Sarnyai M. Effects of cocaine and corticotropin-releasing factor (CRF) on pulsatile ACTH and cortisol release in ovariectomized rhesus monkeys. J. Clin. Endocrinol. Metab. 1995;80:2745–2751. doi: 10.1210/jcem.80.9.7673418. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Daly RC, Bloch M, Smith MJ, Danaceau MA, St. Clair LS, et al. Dehydroepiandrosterone monotherapy iin midlife-onset major and minor depression. Arch. Gen. Psychiatry. 2005;62:154–162. doi: 10.1001/archpsyc.62.2.154. [DOI] [PubMed] [Google Scholar]
- Seyler LE, Jr., Fertig J, Pomerleau O, Hunt D, Parker K. The effects of smoking on ACTH and cortisol secretion. Life Sci. 1984;34:57–65. doi: 10.1016/0024-3205(84)90330-8. [DOI] [PubMed] [Google Scholar]
- Seyler LE, Jr., Pomerleau OF, Fertig JB, Hunt D, Parker K. Pituitary hormone response to cigarette smoking. Pharmacol. Biochem. Behav. 1986;24:159–162. doi: 10.1016/0091-3057(86)90062-6. [DOI] [PubMed] [Google Scholar]
- Shahan TA, Bickel WK, Madden GJ, Badger GJ. Comparing the reinforcing efficacy of nicotine containing and de-nicotinized cigarettes: A behavioral economic analysis. Psychopharmacology. 1999;147:210–216. doi: 10.1007/s002130051162. [DOI] [PubMed] [Google Scholar]
- Sharp BM, Beyer HS. Rapid desensitization of the acute stimulatory effects of nicotine on rat plasma adrenocorticotropin and prolactin. J. Pharmacol. Exp. Ther. 1986;238:486–491. [PubMed] [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sobel B-FX, Sigmon SC, Griffiths RR. Transdermal nicotine maintenance attenuates the subjective and reinforcing effects of intravenous nicotine, but not cocaine or caffeine, in cigarette-smoking stimulant abusers. Neuropsychopharmacology. 2004;29:991–1003. doi: 10.1038/sj.npp.1300415. [DOI] [PubMed] [Google Scholar]
- Soria R, Stapleton JM, Gilson SF, Sampson-Cone A, Henningfield JE, London ED. Subjective and cardiovascular effects of intravenous nicotine in smokers and non-smokers. Psychopharmacology (Berl) 1996;128:221–226. doi: 10.1007/s002130050129. [DOI] [PubMed] [Google Scholar]
- Spark RF. Dehydroepiandrosterone: a springboard hormone for female sexuality. Fertility and Sterility. 2002;77:S19–S25. doi: 10.1016/s0015-0282(02)02987-4. [DOI] [PubMed] [Google Scholar]
- Spealman RD, Goldberg SR. Maintenance of schedule-controlled behavior by intravenous injections of nicotine in squirrel monkeys. J. Pharmacol. Exp. Ther. 1982;223:402–408. [PubMed] [Google Scholar]
- Spohr U, Hofmann K, Steck W, Harenberg J, Walter E, Hengen N, et al. Evaluation of smoking-induced effects on sympathetic, hemodynamic and metabolic variables with respect to plasma nicotine and COHb levels. Atherosclerosis. 1979;33:271–283. doi: 10.1016/0021-9150(79)90179-5. [DOI] [PubMed] [Google Scholar]
- Teoh SK, Sarnyai Z, Mendelson JH, Mello NK, Springer SA, Sholar JW, et al. Cocaine effects on pulsatile secretion of ACTH in men. J. Pharmacol. Exp. Ther. 1994;270:1134–1138. [PubMed] [Google Scholar]
- Tsigos C, Chrousos G. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002;53:865–871. doi: 10.1016/s0022-3999(02)00429-4. [DOI] [PubMed] [Google Scholar]
- Tsigos C, Chrousos GP. Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulationin psychiatric and autoimmune disorders. Endocrinol. Metab. Clinics of North America. 1994;23:451–466. [PubMed] [Google Scholar]
- Wakasa Y, Takada K, Yanagita T. Reinforcing effect as a function of infusion speed in intravenous self-administration of nicotine in rhesus monkeys. Jpn. J. Psychopharmacol. 1995;15:53–59. [PubMed] [Google Scholar]
- Watkins SS, Koob GF, Markou A. Neural mechanisms underlying nicotine addiction: acute positive reinforcement and withdrawal. Nicotine Tob. Res. 2000;2:19–37. doi: 10.1080/14622200050011277. [DOI] [PubMed] [Google Scholar]
- Wilkins JN, Carlson HE, Van Vunakis H, Hill MA, Gritz E, Jarvik ME. Nicotine from cigarette smoking increaes circulating levels of cortisol, growth hormone, and prolactin in male chronic smokers. Psychopharmacology (Berl) 1982;78:305–308. doi: 10.1007/BF00433730. [DOI] [PubMed] [Google Scholar]
- Winternitz WW, Quillen D. Acute hormonal response to cigarette smoking. J. Clin. Pharmacol. 1977;17:389–397. doi: 10.1002/j.1552-4604.1977.tb04621.x. [DOI] [PubMed] [Google Scholar]
- Yen SSC, Jaffe RB, Barbieri RL. Reproductive Endocrinology: Physiology, Pathophysiology and Clinical Management. 4th edition W. B. Saunders; Philadelphia: 1999. p. 839. [Google Scholar]