

Abstract
We present a machine learning method (a hierarchical network of k-nearest neighbor classifiers) that uses an RNA sequence alignment in order to predict a consensus RNA secondary structure. The input to the network is the mutual information, the fraction of complementary nucleotides, and a novel consensus RNAfold secondary structure prediction of a pair of alignment columns and its nearest neighbors. Given this input, the network computes a prediction as to whether a particular pair of alignment columns corresponds to a base pair. By using a comprehensive test set of 49 RFAM alignments, the program KNetFold achieves an average Matthews correlation coefficient of 0.81. This is a significant improvement compared with the secondary structure prediction methods PFOLD and RNAalifold. By using the example of archaeal RNase P, we show that the program can also predict pseudoknot interactions.
Keywords: RNA, secondary structure, mutual information, machine learning, alignment
INTRODUCTION
Predicting the secondary structure of a set of RNA sequences remains a challenging task. The approaches used so far can be divided into three groups: thermodynamic approaches (they try to find the RNA secondary structure with the lowest or near lowest free energy according to an energy model), comparative approaches (sequence alignments are used to identify pairs of columns that exhibit compensatory base changes), and hybrid approaches that combine both thermodynamic and comparative information in order to compute a prediction.
Among the thermodynamic methods for RNA secondary structure prediction, dynamic programing is most often used. Early descriptions of dynamic programing algorithms for RNA secondary structure prediction can be found in (Nussinov et al. 1978; Waterman and Smith 1978; Nussinov and Jacobson 1980). A widely used program for secondary structure prediction is mfold (Zuker and Stiegler 1981; Jaeger et al. 1989; Zuker 1989, 2003; Walter et al. 1994; Mathews et al. 1999). It is based on a dynamic programming algorithm that uses energy parameters that take into account Watson-Crick and GU base pairs, various types of loops and terminal unpaired nucleotides and mismatches. One should also mention the program RNAstructure. Like mfold, it uses a dynamic programming algorithm with an energy model based on thermodynamic parameters (Mathews et al. 2004). We use for the work described in this paper the program RNAfold (Hofacker et al. 1994). This program uses energy rules described in (Mathews et al. 1999). It also uses an algorithm for computing the partition function resulting in probabilities assigned to base pairs (McCaskill 1990).
Dynamic programming algorithms have the advantage that they can find the global minimum free energy of an RNA sequence according to the energy model relatively fast. The disadvantages are that not all energy rules fit into the framework of dynamic programming, and pseudoknots are often not considered (although dynamic programming algorithms including pseudoknots have been described) (Rivas and Eddy 1999; Reeder and Giegerich 2004). In addition, kinetic properties of the RNA molecule are ignored (how easily is a state accessible from other states; is a certain state an energetic “trap” with a high energy barrier?). These limitations can, in principle, be overcome by choosing a global optimization scheme such as that used in genetic algorithms (Shapiro and Navetta 1994; Shapiro et al. 2001a,b).
Compensatory base changes in RNA are an important property of two columns of a sequence alignment that can be indicative of the two nucleotides forming a base pair. Different measures have been developed for finding pairs of columns with significant compensatory base changes. One important concept is the mutual information. It is a measure that indicates if two columns in an alignment are better described by a joint evolution model or by two independent evolution models. Several approaches have been described that deal with the problem of a small number of sequences, which leads to an overestimation of the mutual information due to sampling noise (Basharin 1959; Schneider et al. 1986). The concept of mutual information takes the alignment columns “as is” and does not take phylogenetic relationships between the sequences into account. This concept has been extended to explicitly use phylogenetic information (Muse 1995; Chen et al. 1999; Akmaev et al. 2000; Parsch et al. 2000).
Another method for RNA secondary structure prediction is the program PFOLD. It computes a consensus secondary structure given a sequence alignment. It uses an evolutionary model and a stochastic context-free grammar (SCFG) in order to compute a maximum likelihood secondary structure for the given alignment (Knudsen and Hein 1999, 2003).
Several approaches that combine thermodynamic and comparative information have been described: For example, the Bayesfold method (Knight et al. 2004) uses a Bayesian approach in order to compute base-pair probabilities given three sources of information (mutual information, fraction of complementary base pairs, and average RNAfold pairing probabilities). Juan and Wilson (1999) describe a method that scores a potential base pair by using a linear combination of terms originating from thermodynamic structure predictions (using RNAfold), a score for covariation in the alignment, and a correction term for loops of different lengths. Similarly, Hofacker et al. (2002) also use a linear combination of the average pairing energy associated with a base pair and a covariation score. This method is called RNAalifold and is available in the form of a Web server. Ruan et al. (2004a,b) present the ILM Web server in which the user can specify the relative weight of a thermodynamic and comparative score.
The prediction of paired residues using a machine learning approach is well established in the area of protein structure prediction. Fariselli et al. (2001), for example, use a neural network to combine input values of sequence similarity, correlated mutation, and predicted secondary structures in order to compute a contact prediction.
The task of the method presented in this article is to compute the RNA consensus secondary structure or, in other words, to predict if any two columns of an alignment correspond to a base pair. The basic approach is to combine different sources of information (mutual information, thermodynamic secondary structure prediction, fraction of complementary base pairs) in order to predict for each pair of columns whether they correspond to a base pair or not.
The new aspect of our method is that it also takes into account information from neighboring columns (Fig. 1 ▶). Furthermore, we present a novel method that involves nonlinear reweighting of superposed RNAfold probability matrices. Also, we use an innovative classifier system that turned out to be very robust: a hierarchical network of k-nearest neighbor classifiers (Fig. 2 ▶). We show that our classifier system turns out to be very successful in predicting RNA secondary structures and is able to predict pseudoknot interactions. The performance of our method is evaluated by using a comprehensive set of 49 RFAM alignments.
FIGURE 1.

Schematic plot of feature positions used as input for prediction with respect to base pairs i,j. Dark gray and black indicate positions used for mutual information and fraction of complementary pairs; light gray, feature positions used only for mutual information.
FIGURE 2.

Structure of network of k-nearest neighbor classifiers. The classifier network computes a prediction whether or not a given pair of columns of an alignment corresponds to a base pair of the consensus secondary structure. It needs a set of features derived from a sequence alignment and an RNAfold consensus probability matrix. A–I indicate classifiers of level 1. Each classifier uses three features derived from the alignment. J–M indicate classifiers of level two and three. Each classifier of that level has as input the output from three classifiers of the previous level. N indicates a final classifier that has as input (1) the output from the classifier of the previous level and (2) the RNAfold consensus probability value for the given pair of columns.
ALGORITHM
Mutual information with small sample correction
The mutual information of two alignment columns is a measure of correlated mutations. The information R of a set of characters (here a column in the alignment) is the decrease in uncertainty H after reading that set of characters. The mutual information ΔRij of two alignment columns i and j is the information from that column pair taken together minus the information of the alignment columns taken separately. The formula used is
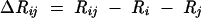 |
with
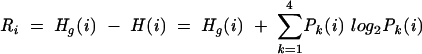 |
and
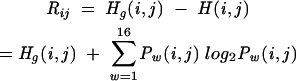 |
Pk(i) denotes the probability of finding a character of type k at position i; Pw(i,j) is the probability of finding one of the 16 possible character pairs w at positions i and j. The probabilities are approximated by frequencies (Pk(i): number of found characters of type k divided by the total number of non-gap characters in column i; Pw(i,j): number of two-character words found of type w divided by the total number of non-gap characters in columns i and j). Sequences that contain a gap character at either position i or j (or both) are ignored for the computation of Pw(i,j) as well as Pk(i) and Pk(j). The value Hg(i) represents the expected uncertainty at position i in the alignment. In the limit of an infinite number of sequences with the possible characters having the same probability of occurrence, Hg is equal to 2 bits for four characters (single column) and 4 bits for 16 characters (two combined columns).
For a small number of sequences, the expected uncertainty is smaller, because it is more likely that the characters in the alignment column are not evenly distributed. We correct for this “sampling noise” in a fashion similar to that described in (Schneider et al. 1986, Stephens and Schneider 1992). For more than a given number of sequences (50 sequences in the case of single columns, 500 sequences in the case of column pairs), we use the approximate correction term 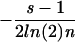 with s = 4 for a single column and s = 16 for two columns (the alphabet sizes are s = 4 because of the four-letter alphabet ACGU and s = 16 because of the 16 possible base pairs). The variable n in this case is the number of sequences that have a nongap character at position i (and j). For alignment columns with <50 nongap characters (500 character pairs in the case of the two-column uncertainty), we compute the expected uncertainty as follows: The algorithm iteratively generates random sequences and computes the corresponding uncertainty. This loop is terminated if the standard deviation of the resulting mean uncertainty is <0.01. This procedure approximates the expected uncertainty for a small number of sequences better than the approximate correction term and is computationally feasible even for a 16-letter alphabet. We find that for >70 sequences, the difference between the result of the random sequence method and the approximate correction term is <0.01 bits. The current implementation generates random sequences by using the same probability (one of four) for each nucleotide type. It might be interesting to extend this algorithm to consider the nucleotide frequencies that are observed in the alignment.
with s = 4 for a single column and s = 16 for two columns (the alphabet sizes are s = 4 because of the four-letter alphabet ACGU and s = 16 because of the 16 possible base pairs). The variable n in this case is the number of sequences that have a nongap character at position i (and j). For alignment columns with <50 nongap characters (500 character pairs in the case of the two-column uncertainty), we compute the expected uncertainty as follows: The algorithm iteratively generates random sequences and computes the corresponding uncertainty. This loop is terminated if the standard deviation of the resulting mean uncertainty is <0.01. This procedure approximates the expected uncertainty for a small number of sequences better than the approximate correction term and is computationally feasible even for a 16-letter alphabet. We find that for >70 sequences, the difference between the result of the random sequence method and the approximate correction term is <0.01 bits. The current implementation generates random sequences by using the same probability (one of four) for each nucleotide type. It might be interesting to extend this algorithm to consider the nucleotide frequencies that are observed in the alignment.
k-nearest neighbor algorithm
We use a machine learning algorithm to make a prediction whether any two nucleotides form a base pair or not. This can be viewed as a classification problem. The method used in our approach is a hierarchical network of k-nearest neighbor classifiers. The k-nearest neighbor algorithm is a well-known classification method. If one wants to classify (i.e., determine the “class” of) a query vector, one simply determines which class most of the “k” known feature vectors that are closest to the query vector belong to. Even more, the counts for the different classes obtained from the closest known vectors can serve as an approximation to the probability that the query vector belongs to a certain class. We use as a distance measure the Euclidean distance between two vectors.
Let there be, for example, two classes “A” and “B” and a value of k = 10. If there is a query vector q, one first determines the 10 vectors of the training data (a set of vectors for which one knows to which class they belong) that are closest to q. If seven of the closest vectors belong to class A, we assign an approximate probability of 70% to the chance that query vector q belongs to class A. One problem with this approach is that the k closest training vectors get an equal weight when the probability that the query vector belongs to a certain class is estimated. Intuitively, it makes sense to weight training vectors that are closer to the query vector higher than training vectors that are further away. This leads to the use of (empirical) distance-weighting functions that determine the weight of the individual “votes” of the k-nearest neighbors.
A k-nearest neighbor algorithm with a distanceweighting function is a very effective inductive inference method (Mitchell 1997). It can be used to classify feature vectors. As a distance weighting function, we use a Gaussian function with a standard deviation of 0.2. We set the parameter k equal to 15 for all k-nearest neighbor classifiers except for the classifier named “M” in Figure 2 ▶, which uses a value of k = 10. The distance weighting function makes the classifiers less sensitive to the chosen value of k. Also, we later replace for some classifiers the k-nearest neighbor value computation with a look-up table, which allows us to use k = 1 (see section Noise Reduction of Classifiers). We use the “voting-results” of the k-nearest neighboring training vectors with respect to a query vector as an estimate of the confidence of the classification.
The first level of classifiers forms a prediction by using a vector of features. The features are values derived from the alignment that describe a pair of alignment columns and its neighboring columns. The used features are as follows: (1) the mutual information of two alignment columns; (2) the fraction of nucleotides that form complementary base pairs (in this article, complementary base pairs are defined by Watson-Crick base pairs and also GU base pairs); and (3) a nonlinear consensus of the RNAfold prediction probability matrices (described in section Thermodynamic Consensus; also called NL-RNAfold in this article).
There is a rationale for each of the different features: The mutual information is a measure of how mutations in the two columns are correlated. The fraction of complementary nucleotides is also an important feature, because one expects in a pair of alignment columns that correspond to a base pair, a bias toward complementary pairings. Figure 2 ▶ shows the architecture of the classifier network. The network considers in the first stage the mutual information and the fraction of complementary base pairs, while the results from RNAfold are only considered in the last stage.
The classifier system computes a prediction as to whether any two nucleotides i and j form a base pair (the indices denote the column numbers in the given alignment). It uses a 14-dimensional feature vector that describes the columns i and j and its four nearest neighboring diagonal and anti-diagonal columns. The mutual information and the fraction of complementary base pairs are given for the central element (columns i, j) and the four nearest anti-diagonal neighboring elements (columns i + 2, j − 2; i + 1, j − 1; i, j; i − 1, j + 1; i − 2, j + 2).
Only mutual information is stored in the feature vector for the four diagonal positions i + 2, j + 2; i + 1, j + 1; i − 1, j + 1; i − 2, j + 2. The rationale for also using diagonal elements is that it provides a possible “base line” in the sometimes quite noisy mutual information matrix. The idea is that the algorithm is specifically looking for anti-diagonal “stripes” corresponding to stems of the secondary structure. Providing the algorithm with diagonal elements gives it the chance to distinguish “amorphous” areas with high mutual information (maybe due to gaps and sampling noise) from patterns that resemble anti-diagonal lines. Using the diagonal elements leads to a total of 5 × 2 + 4 = 14 possible features. The positions that are chosen as features are shown in Figure 1 ▶. We only have a few hundred positive training feature vectors that correspond to a true base pair. Because of this small number of training cases, it is problematic to use a machine learning algorithm with a 14-dimensional feature vector (danger of overfitting and memorizing effects). Therefore we use the procedure described in the next paragraph to reduce the number of dimensions of the feature vectors used for training.
The 14 features of the original feature vectors yield 14 × 13 × 12/3! = 364 possible triplets of features. We further require that every feature triplet contains either the mutual information or the fraction of complementary nucleotides associated with the columns i,j. Lastly, no two chosen classifiers are allowed to have two features in common. Given these constraints, we identify nine classifiers with the help of the AdaBoost algorithm. In short, the AdaBoost algorithm is an iterative method to find an optimal set of “weak learners” from a set of classifiers. The algorithm chooses in the first iteration the classifier with the highest prediction accuracy (smallest number of misclassified training vectors). The training vectors that were misclassified with the chosen classifier are given a higher weight for the subsequent iteration. For the next iteration, the classifier with the next highest accuracy is chosen; again the accuracy value is biased to give the training vectors that were misclassified in the previous iterations a higher weight. For a more detailed description of the algorithm, see Freund and Schapire (1996). We obtain in this way in the first stage nine k-nearest neighbor classifiers. Each of these initial classifiers generates a prediction (in the form of an estimated probability) as to whether or not an observed feature triplet corresponds to a true base pair.
The predictions of the nine classifiers of the initial stage are pooled in a hierarchy of levels of classifiers. Each level consists of n/3 classifiers, n being the number of classifiers from the previous level. Each classifier uses as input the output of three randomly chosen classifiers from the previous level. Each level consists of three times less classifiers than the previous level, until at the third level there is only one classifier left that computes the final prediction not using thermodynamics. The RNAfold consensus value together with that resulting value forms the input of the last k-nearest neighbor classifier. In this way the system avoids ever encountering a feature vector that has more than three dimensions. A schematic view of this network of classifiers can be seen in Figure 2 ▶.
Many schemes for a fast implementation of the k-nearest neighbor algorithm have been developed. They concentrate on devising a data structure for the training data that allows one to find the k-nearest neighbors faster than a linear “brute-force” search. We use for the k-nearest neighbor search the ANN library using the k-d tree algorithm (Arya and Mount 1993; http://www.cs.umd.edu/~mount/ANN/).
Thermodynamic consensus
Our system also uses what we call an RNAfold consensus probability matrix: It is a matrix generated from superposed aligned probability matrices generated by RNAfold. We use the program RNAfold from the Vienna RNA package, version 1.5 (Hofacker et al. 1994). The consensus matrix is computed as follows: Our system runs RNAfold for each sequence of the alignment provided by the user. RNAfold computes a base-pair probability matrix for each sequence by using a partition function (McCaskill 1990). The resulting probability matrices are aligned according to the sequence alignment, averaged, and stored in a result matrix. Each element of this result matrix is weighted with a factor equal to a logistic function applied to the fraction f of sequences that have a nonzero prediction probability at that position:
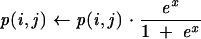 |
with
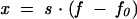 |
The center point f0 of the logistic function is set to 2/3, corresponding to 66.7% of the sequences having a nonzero prediction probability at that point. The scaling factor s is set to 0.1. This consensus score turned out to be very effective for computing the secondary structure. While the logistic function does not have a sharp cutoff, it down-regulates potential base pairs that appear in <66.7% of the sequences. We call this method “NL-RNAfold” as opposed to “L-RNAfold,” which corresponds to a simple average of the aligned probability matrices. The information from the RNAfold consensus probability matrix is used only at the last stage of the KNetFold prediction cascade (see Figs. 2 ▶, 5 ▶). Results of using the nonlinear as well as the linear consensus matrix as a predictor can be seen in Table 1 ▶ (labeled NL-RNAfold and L-RNAfold).
FIGURE 5.

U2 spliceosomal RNA: example of processing matrices of features to a final secondary structure prediction. The matrices shown correspond to possible interactions of the positions of the first sequence in the RFAM seed alignment for RF00004. The 5′ end of the alignment corresponds to the lower left corner of the shown matrices. The matrices are as follows: Mutual Information, mutual information between two alignment columns; Complementary, fraction of Watson-Crick and GU base pairs; RNAfold, RNAfold consensus probability matrix; Intermediate, output matrix of classifier network not using RNAfold; Prediction, final prediction produced by the classifier network; and Reference, reference structure provided by RFAM.
TABLE 1.
Table showing results for a set of 49 RFAM seed alignments
| 49 Alignments with 20–40 sequences | 49 Alignments with five sequences | |||||
| Matthews | Selectivity (%) | Sensitivity (%) | Matthews | Selectivity (%) | Sensitivity (%) | |
| KnetFold | 0.811 ± 0.032 | 82.15 ± 3.33 | 81.07 ± 3.18 | 0.742 ± 0.042 | 76.22 ± 4.21 | 73.08 ± 4.14 |
| NL-RNAfold | 0.726 ± 0.035 | 69.80 ± 3.82 | 77.41 ± 3.15 | 0.742 ± 0.042 | 76.22 ± 4.21 | 73.08 ± 4.14 |
| PFOLD | 0.705 ± 0.041 | 82.34 ± 4.02 | 62.65 ± 4.37 | 0.670 ± 0.045 | 78.08 ± 4.69 | 59.36 ± 4.53 |
| RNAalifold | 0.578 ± 0.051 | 67.51 ± 5.73 | 50.87 ± 4.77 | 0.660 ± 0.051 | 72.63 ± 5.33 | 61.34 ± 5.03 |
| L-RNAfold | 0.641 ± 0.045 | 63.35 ± 4.52 | 66.22 ± 4.37 | 0.591 ± 0.045 | 59.53 ± 4.61 | 60.10 ± 4.39 |
| Intermediate | 0.234 ± 0.053 | 30.40 ± 6.58 | 19.29 ± 4.72 | 0.009 ± 0.010 | 2.04 ± 2.04 | 0.49 ± 0.49 |
The results show the average Matthews correlation coefficient computed with the script provided as part of BRaliBase I (Gardner and Giegerich 2004). The row values are as follows: KNetFold, method presented in this article; NL-RNAfold, result from superposed RNAfold probability matrices using a nonlinear weighting scheme (see Algorithm section); PFOLD, prediction according to PFOLD program; RNAalifold, results from the RNAalifold server; L-RNAfold, results using average RNAfold probability matrices; and Intermediate, intermediate prediction results not using thermodynamic information (corresponds to output of classifier M in Fig. 2 ▶ and plot “Intermediate” in Fig. 5 ▶).
Noise reduction of classifiers
We found that it is important to reduce the noise of the individual classifiers by using a “monotonization” procedure: All classifiers of level two or higher should react to an increase of one of its input values (an element of the input vector that is a prediction from a classifier from a previous level) with an increased or constant output value. By using a set of query vectors representing a cube [0,1] × [0,1] × [0,1] with step size of 0.025, we queried the response of three classifiers from level two (labeled “J”, “K” and “L” in Fig. 2 ▶) and one classifier from level 4 (labeled “N” in Fig. 2 ▶). We found that the responses of the individual classifiers are often quite noisy, because an increase of an element of the input vector could sometimes result in a decrease in the output value of the classifier. We reduced that problem by applying a monotonization procedure. The output of the query data was subject to an iterative procedure that makes the response of the classifier more monotonic. This procedure is best explained for a one-dimensional function of integer values. For a nonmonotonic function with one variable, one can obtain two different monotonic increasing functions: One function can be obtained by replacing the values of f(n) for which f(n) < f(n − 1) with its “left” neighbor f(n − 1); the other function can be obtained by replacing the values of f(n) for which f(n) > f(n + 1) with its “right” neighbor f(n + 1). Taking an average of those two functions results in a function with reduced nonmonotonic behavior. This concept can readily be extended to higher dimensions. The resulting “smoothed” classifier responses are used in lookup tables for the classifiers. The lookup tables replace the initial “raw” k-nearest neighbor training data. We found that these smoothed classifiers result in a higher prediction accuracy.
Treatment of gaps in the alignment
The basic idea of the classifier system is to detect anti-diagonal lines in different square matrices. The anti-diagonal patterns potentially correspond to stems in the consensus secondary structure. If an alignment contains a lot of gaps, the anti-diagonal lines can become discontinuous, making it harder for the classifier to detect them. We deal with this problem in the following way: The training is performed on a set of “collapsed” alignments (by collapsing we mean removing all alignment columns that correspond to a gap with respect to a certain sequence; see section Test and Training Data). Testing of the method is performed on alignments that are not collapsed. However, to reduce the effective number of gaps in an alignment, the following procedure is performed: A set of 10 sequences is randomly chosen from the sequences of the original alignment. The alignment is collapsed with respect to each of the chosen sequences (all columns are removed that correspond to a gap in the chosen sequence). The KNetFold method is applied to each of the 10 different collapsed alignments individually. A prediction of a collapsed alignment (a square matrix with scores) is expanded to the original alignment size by introducing rows and columns with zeros at the positions that correspond to a gap in the chosen sequence. The 10 resulting prediction matrices are in a last step averaged to form the final prediction matrix.
Applied filters
Several filters are applied to the resulting output of the classifier system:
The growth-filter is applied to the output of the classifier called “M” in Figure 2 ▶. If the output of that classifier is for a position i,j > 0.2 and a neighboring position (either position [i + 1,j − 1] or [i − 1,j + 1]) with a smaller classifier score has a fraction of complementary base pairs >0.5, then the prediction matrix element of that neighboring position is set equal to the matrix element of the position i,j. This filter is iteratively applied until no changes in the prediction matrix can bemade. This filter is designed to handle cases of long stems with very few positions with compensatory base changes.
A “winner-takes-all” filter allows one nucleotide to base pair only with one other nucleotide. If a nucleotide is predicted to be base-pairingwithmore than one nucleotide, only the highest scoring interaction of that nucleotide is kept in the final prediction. This filter can be deactivated, in case one is interested in finding alternative conformations.
Optionally, predicted stems can be required to have at least a minimum length of two base pairs. The default is a minimum stem length of only one base pair.
As a final step, a cutoff is applied such that all elements of the prediction matrix that are higher than the cutoff are considered a predicted base pair. We determined cutoffs for the methods KNetFold, NL-RNAfold, and L-RNAfold that correspond to a maximum average Matthews correlation coefficient (AMCC) of the training set.
Test and training data
We use the “full” alignments and consensus secondary structures provided by RFAM, version 6.1 (Griffiths-Jones et al. 2003, 2005), for training the system. The training set consists of the 43 alignments shown in Table 2 ▶ (for RF00002, the RFAM seed alignment instead of the full alignment is used because the full alignment consists of >60000 sequences). These are alignments with −30 sequences, with the length of the first sequence being <500 nucleotides. If the alignment consists of >40 sequences, only 40 representative sequences of each alignment are used. The representative sequences are constructed by iteratively removing the sequences with the highest sequence similarity with respect to another sequence of the alignment until only 40 sequences are left. The alignments of the training set are “collapsed”; the columns that correspond to a gap in the first sequence of the alignment are removed. The idea of preferring collapsed alignments with fewer gaps is that the objective is to train a classifier to recognize small continuous anti-diagonal “stripes” in square matrices. Using alignments with a lot of gaps can lead to discontinuous anti-diagonal lines, which in turn could worsen the prediction accuracy of the classifier. The predictions for the test set (described below) are made for alignments that were not collapsed. However, as described in the section Treatment of Alignment Gaps, the prediction algorithm uses collapsed alignments as an intermediate step to deal with the problem of “gappy” alignments.
TABLE 2.
The 43 RFAM entries used for training
| RF00002 | RF00003 | RF00004 | RF00008 | RF00011 | RF00012 | RF00015 | RF00017 | RF00019 | RF00020 |
| RF00026 | RF00031 | RF00032 | RF00037 | RF00045 | RF00048 | RF00050 | RF00061 | RF00094 | RF00100 |
| RF00102 | RF00106 | RF00162 | RF00163 | RF00164 | RF00167 | RF00168 | RF00169 | RF00171 | RF00175 |
| RF00176 | RF00181 | RF00198 | RF00199 | RF00209 | RF00210 | RF00214 | RF00215 | RF00216 | RF00233 |
| RF00250 | RF00260 | RF00374 |
The set consists of alignments with at least 30 sequences with a length not >500 nucleotides.
The training is performed as follows: For each alignment of the training set, feature vectors are computed for all possible pairs of alignment columns. The feature vectors of all alignments form the initial set of training vectors. This set of training vectors is the training data for the classifiers of the first level. However, each classifier uses only a three-dimensional projection of the training data and uses a clustering algorithm such that no >40 feature vectors are <0.0001 to a representative vector of that cluster. This prevents a large number of virtually identical vectors from dominating the k-nearest-neighbor training vector set. Each vector of the training data set is used as a sample input vector for the network in which the first level of classifiers have been defined as described above. The results of the first level classifiers are then used as training vectors for the classifiers of the second level and so forth.
For evaluating the performance of our method, we generate 10 different subsets of the training data set and produce 10 different sets of training vectors. For each alignment to be predicted, a training vector set that does not contain information about the current target alignment is chosen. In this way we avoid memorizing effects, because testing and training sets are not overlapping.
The test set consists of the 49 RFAM alignments shown in Table 3 ▶. The alignments are “seed” alignments taken from RFAM, version 7.0. Because the PFOLD Web server currently has a limit that allows not >40 sequences to be submitted, we used only 40 representative sequences of each alignment in order to fairly compare our system with PFOLD. The representative sequences are constructed as described above. If an alignment consists of <40 sequences, no sequences are removed. The 49 RFAM entries correspond to alignments consisting of at least 20 sequences and a file size of <10 kilobytes. The file size restriction is a requirement of the RNAalifold server and is applied after an optional “thinning” of the alignment to 40 representative sequences. The lengths of the alignments of the test set range between 26 and 242 nucleotides.
TABLE 3.
The 49 RFAM entries used for testing
| RF00001 | RF00005 | RF00008 | RF00015 | RF00031 | RF00032 | RF00037 | RF00041 | RF00045 | RF00048 |
| RF00049 | RF00062 | RF00066 | RF00095 | RF00097 | RF00098 | RF00102 | RF00105 | RF00163 | RF00164 |
| RF00167 | RF00169 | RF00175 | RF00181 | RF00198 | RF00199 | RF00214 | RF00215 | RF00233 | RF00238 |
| RF00250 | RF00260 | RF00309 | RF00374 | RF00375 | RF00376 | RF00386 | RF00389 | RF00436 | RF00451 |
| RF00465 | RF00467 | RF00468 | RF00469 | RF00480 | RF00481 | RF00485 | RF00497 | RF00506 |
The entries represent alignments with at least 20 sequences and a file size of <10 kilobytes when using not >40 representative sequences.
RESULTS
Computing cost
The majority of the computing time is spent applying the network of classifiers to each possible pair of columns of the alignment. The classifier system itself does not change, so we expect a quadratic increase of compute time with respect to alignment length. We found for the set of 49 alignments an average computing time of 92 sec per run (using an Intel Xeon 3GHz Linux PC; not counting the run-time of RNA-fold). This corresponds to an average time span of 920 sec if one uses 10 iterations over different collapsed alignments (see section Treatment of Alignment Gaps).
By fitting a quadratic function to the obtained run-times (as a function of the length of the alignment), we obtain an approximate formula for the run-time t measured in seconds: t = 0.0046 × length2. This corresponds to a run-time of 46 sec for an alignment with a length of 100 nucleotides and a run-time of 1150 sec for an alignment with a length of 500 nucleotides. These time spans have to be multiplied by the number of collapsed alignments that are used as an intermediate step (this number is set to 10 for the results presented in this article).
Prediction accuracy
When comparing the predicted base pairs to a reference secondary structure (in our case the consensus secondary structure provided by RFAM), one can compute the number of true-positive, true-negative, false-positive, and false-negative base-pair predictions (tp, tn, fp, fn, respectively). We use as accuracy measures the Matthews correlation coefficient (MCC), as well as the selectivity and the sensitivity according to Gardner and Giegerich (2004). The definitions for the sensitivity X and the selectivity Y are
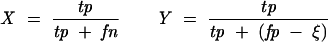 |
The selectivity Y is also often called specificity (Baldi et al. 2000), but we follow in this article the nomenclature according to Gardner and Giegerich (2004). The difference compared with the standard definition of the selectivity is the term ζ. This term represents base pairs that are not present in the reference structure but are nonetheless considered compatible. Compatible base pairs are defined as predicted base pairs that are not true positives but are also not inconsistent (with one or both of the bases being part of another base pair in the reference structure) or contradicting (non-nested with respect to the reference structure). The number of compatible base pairs ζ reduces the effective number of false positives and makes the selectivity measure more “forgiving.” The definition of the MCC is modified in a similar fashion (the standard definition of the MCC does not contain the term ζ):
 |
The MCC can be approximated by the geometric mean of sensitivity and selectivity for the purpose of RNA secondary structure comparison (Gorodkin et al. 2001). To compute these values, we use the perl script “compare_ ct.pm” made available by Gardner and Giegerich (2004) on the BRaliBase I homepage. We multiply the obtained values for selectivity and sensitivity by a factor of 100 to obtain percentage values.
We applied our prediction method to all 49 alignments of our data set (see section Test and Training Data). In Table 1 ▶ we show the results for alignments consisting of 20– 40 sequences and for alignments consisting of five sequences each. We also plot in Figure 3 ▶ the prediction accuracy for all 49 test cases. Each curve is sorted such that the “best” values are plotted leftmost. Shown are graphs of the results of different prediction schemes: “A,” our full network of k-nearest neighbor classifiers (“KNet-Fold”); “B,” the nonlinear RNAfold consensus (“NL-RNAfold”); “C,” PFOLD; “D,” RNAalifold; and “E,” the intermediate results corresponding to the output of classifier “M” in Figure 2 ▶ and the “Intermediate” matrix in Figure 5 ▶. One can see that KNetFold has for this data set the highest prediction accuracy.
FIGURE 3.

Prediction accuracy for the test set of 49 RFAM alignments. A indicates method presented in this article (KNetFold); B, nonlinear RNAfold consensus probability matrix (NL-RNAfold); C, PFOLD Web server; D, RNAalifold; E, intermediate result (corresponds to output of classifier M in Figure 2 ▶ and “Intermediate” in Table 1 ▶). The data shown correspond to the results of RFAM alignments in the test set. For each method, the highest prediction accuracies are plotted leftmost. If the original RFAM alignment contained >40 sequences, a “thinned” alignment consisting of 40 representative sequences was used instead.
The average Matthews correlation coefficient (AMCC) using the set of 49 alignments (with 20–40 sequences each) is, with our system, 0.811 ± 0.032 (cf. Table 1 ▶, left side). This canbe comparedwith the value of 0.726 ± 0.035 for the nonlinear RNAfold consensus (called here NL-RNAfold). A pair-wise t-test of the MCCs obtained with KNetFold and with NL-RNAfold results in a single-sided P-value of 1.1 × 10−6. In other words, the improvement of the KNetFold results compared with the NL-RNAfold results is statistically significant. The result of NL-RNAfold is significantly higher compared with the result of the average RNAfold probability matrix method (called here L-RNAfold), with 0.641 ± 0.045. The PFOLD results for these alignments correspond to an AMCC of 0.705 ± 0.041. RNAalifold yields an AMCC of 0.578 ± 0.051.
We also compute results for the same set of 49 RFAM entries with only five representative sequences per alignment (Table 1 ▶, right side). By using this second set of alignments with a smaller number of sequences, we wanted to see how the prediction accuracies of the various methods are affected by the number of sequences in the alignment. Not surprisingly, the prediction accuracies are generally lower compared with the results for the alignments that contain 20–40 sequences. In the case of KNetFold, the AMCC is 0.742 ± 0.042, that is a difference of 0.069 compared with the results obtained when using the set of larger alignments. This result is identical to the results obtained from NL-RNAfold. The method NL-RNAfold is remarkably resilient to a decrease in the number of sequences in the alignment (there is a difference of −0.016 AMCC between the two sets of alignments). The program RNAalifold exhibits an interesting behavior: Its results are more accurate for the alignment set consisting of only five sequences (average Matthews coefficient of 0.660 ± 0.051) compared with the alignment set consisting of 20–40 sequences (average Matthews coefficient of 0.578 ± 0.051).
Furthermore, we investigated how the homology level of alignments affects the prediction accuracy. We divided the set of 49 alignments into a high-homology set (>80% average pairwise sequence similarity) and a low-homology set (<80% average pairwise sequence similarity). The resulting AMCC is 0.793 ± 0.049 for the high-homology set and 0.825 ± 0.043 for the low-homology set.
How does the prediction accuracy depend on the length of the alignment? We plotted the Matthews coefficients obtained from the alignments of the test set as a function of the alignment length (data not shown). Visual inspection of the plot revealed no obvious dependency of the prediction accuracy as a function of alignment length. Therefore, it appears that the accuracy is more dependent on the set of sequences being studied than their length. However, we did note that all “perfect” predictions (MCC of 1.0) correspond to alignments with a length of <120 nucleotides.
Another point that deserves attention is the quality of the used “standard of truth” of the consensus secondary structures provided by RFAM. An RFAM entry can have either the status “Predicted” or “Published.” An RFAM consensus secondary structure with the Published status has been reported in a publication. A published structure might be based on experimental results, but this is not a requirement. On average, one expects RFAM entries with the status Published to be more “trustworthy” than are entries with the status Predicted. Our test set of 49 alignments consists of 34 entries with the status Published and 15 entries with the status Predicted. We computed the average prediction accuracy for both subsets independently. We obtain an AMCC of 0.834 ± 0.033 for RFAM entries with the status Published and of 0.759 ± 0.073 for RFAM entries with the status Predicted. This difference in prediction accuracy might be due to inaccuracies in the predicted consensus structures provided by RFAM or due to the fact the RFAM entries that do not correspond to published secondary structures correspond to “harder” cases.
Example: Secondary structure prediction of archaeal RNase P
As an example of a secondary structure prediction, we present the results for archaeal RNase P. We use the “seed” alignment according to RFAM entry RF00373 (Brown 1999). A secondary structure prediction for RNase P (projected onto the sequence of Methanobacterium thermoautotrophicum ΔH; accession code GenBank AF295979) (Smith et al. 1997) is shown in Figure 4 ▶. The helices are labeled according to previous publications (Haas et al. 1994; Harris et al. 2001). The results are in good agreement with the structure published in Harris et al. (2001), which we use here as a reference structure. Compared with that published structure, most predicted helices are identical. Two pseudoknot interactions (P4 and P6) are predicted, in agreement with the reference structure. The helix P17 (G222:A234, length 3) is not predicted by our program. In addition to the reference structure, our program predicts four single base-pair interactions that are shown in Figure 4 ▶ as solid lines.
FIGURE 4.

RNA secondary structure prediction for archaeal RNase P (sequence of Methanobacterium thermoautotrophicum ΔH, GenBank: AF295979). The labeling of the helices is according to references Haas et al. (1994) and Harris et al. (2001). Most helices are in agreement with the structure published in Harris et al. (2001). Two pseudoknot interactions are predicted. Part of the picture was generated with the help of the program STRUCTURELAB (Shapiro and Kasprzak 1996).
DISCUSSION
It is instructive to visualize the flow of information starting from the input features to the final prediction. This is shown in Figure 5 ▶ for the example of U2 spliceosomal RNA (RFAM entry RF00004). The data correspond to a single run (no averaging over different collapsed alignments) using the RF00004 seed alignment that was collapsed with respect to its first sequence. One can see that even though the matrices corresponding to the mutual information and the fraction of complementary nucleotides (diagrams called in Fig. 5 ▶ “Mutual Information” and “Complementary”) are very “noisy,” the classifier system generates a reasonably cleaned up contact matrix (diagram called “Intermediate”) even without using the thermodynamic prediction generated by RNAfold.
It is clear that comparative information can improve the prediction accuracy of an energy-based RNA prediction algorithm. This is reflected in the results shown in Figure 3 ▶ (cf. curves A, B, and E) and in Table 1 ▶ (cf. rows “KNet-Fold,” “NL-RNAfold,” and “Intermediate”). Even though the prediction using only the mutual information and fraction of complementary base pairs does poorly compared with the complete KNetFold method, it increases the prediction accuracy when used in combination with the NL-RNAfold consensus.
The results in Table 1 ▶ also show that the part of the classifier system responsible for detecting compensatory information basically “does not work” if only five sequences are present in the alignment (see Table 1 ▶, row “Intermediate”). We found for this case, that the intermediate classifier (corresponding to the output of classifier “M” in Fig. 2 ▶ and the plot “Intermediate” in Fig. 5 ▶) does not detect any correct base pairs for all alignments of the test set (Matthews coefficient of 0.0) with the exception of tRNA (RFAM entry RF00005), which has a Matthews coefficient of 0.485. The overall KNetFold method can still be useful with a small number of sequences, but its accuracy is in this case similar to the accuracy of the non-linear superposition of RNAfold probability matrices.
For quite a few cases, the nonthermodynamic component of the system adds little new information. This corresponds to the part of curve E in Figure 3 ▶ with height zero. In these cases, no clear compensatory base changes could be identified. The causes might be that (1) the regions in question are so highly conserved that no mutual information can be detected, even though it corresponds to a complementary pair of nucleotides. (2) The two alignment columns in question correspond to a conserved G (or U) in one column and alternating C and U (or A and G) in the other column. This leads to a mutual information of zero (or close to zero), even though there is a bias toward complementary base pairs. For this case it might also be interesting to explore other compensatory scoring functions, such as the covariance score presented in Hofacker et al. (2002). (3) The sequences are too dissimilar so that no good sequence alignment can be obtained. (4) Not enough sequences are known, so that the mutual information data is very noisy due to sampling errors. (5) The missing comparative information might also be an inherent property of that biological system: It could mean that certain stems are really present in some strains and not in others. (6) For noncanonical base pairs, our distinction between complementary base pairs (AU, GC, and GU) and noncomplementary base pairs is arguably somewhat artificial. A probabilistic approach might improve the situation.
We could not identify a strong trend of the prediction accuracy as a function of sequence homology. We believe that this might be due to two counteracting effects: A high level of sequence homology might lead on average to higher-quality alignments. On the other hand, if the average pairwise sequence similarity is too high, it presumably leads to a lower amount of compensatory base changes. The question of prediction accuracy as a function of the homology level of the alignment warrants further investigation, however, because the extreme cases of alignments with close to perfect conservation, on one hand, and “spurious” alignments, on the other hand, are not part of our test set.
Another observation is that the nonlinear superposition of RNAfold probability matrices (“NL-RNAfold”) yields surprisingly good results. It is a relatively simple procedure to reweight the elements of the average RNAfold probability matrix with a logistic function, but it results in a significantly higher AMCC (cf. rows for L-RNAfold and NL-RNAfold in Table 1 ▶).
In summary, our system combines thermodynamic information and compensatory information to compute a secondary structure prediction (represented in Table 1 ▶ and Fig. 3 ▶ by methods “NL-RNAfold,” “Intermediate,” and “KNetFold,” respectively). The average prediction accuracy of the thermodynamic component (“NL-RNAfold”) of the system is much higher compared with the compensatory component. By using a network of k-nearest neighbor classifiers, we were nonetheless able to obtain a RNA secondary structure predictor that performs better than each of its components. The method has been evaluated with a set of 49 RFAM alignments and has been compared with the program PFOLD and RNAalifold. To the best of our knowledge, this represents currently the largest test data set used to compare RNA secondary structure prediction methods.
Conclusion
Different concepts of comparative sequence analysis (mutual information, fraction of complementary base pairs, and conserved thermodynamically predicted contacts) have been combined in one classifier system. We show that for a test set of 49 RFAM alignments, the resulting prediction accuracy is higher compared with the programs PFOLD, RNAalifold, and an RNAfold consensus. We also show that the method is able to predict pseudoknot interactions. We believe that state-of-the-art classifiers that combine thermodynamic and comparative information represent a powerful methodology that will form the basis of many future RNA secondary structure prediction approaches.
Acknowledgments
We wish to thank the Advanced Biomedical Computing Center (ABCC) at the NCI for their computing support. Also, we would like to thank Tom Schneider for fruitful discussions, Matthias Heiler for suggesting the AdaBoost algorithm, and Nayan Randad for verifying data in this manuscript. This work has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. NO1-CO-12400. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2164906.
REFERENCES
- Akmaev, V.R., Kelley, S.T., and Stormo, G.D. 2000. Phylogeneticically enhanced statistical tools for RNA structure prediction. Bioinformatics 16: 501–512. [DOI] [PubMed] [Google Scholar]
- Arya, S. and Mount, D.M. 1993. Algorithms for fast vector quantization. Proceedings of DCC ’93: Data compression conference (eds. J.A. Storer, and M. Cohn), pp. 381–390. IEEE Press, Snowbird, UT.
- Baldi, P., Brunak, S., Chauvin, Y., Anderson, C., and Nielsen, H. 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 16: 412–424. [DOI] [PubMed] [Google Scholar]
- Basharin, G.P. 1959. On a statistical estimate for the entropy of a sequence of independent random variables. Theory Probability Appl. 4: 333–336. [Google Scholar]
- Brown, J.W. 1999. The Ribonuclease P Database. Nucleic Acids Res. 27: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Carlini, D., Baines, J., Parsch, J., Braverman, J., Tanda, S., and Stephan, W. 1999. RNA secondary structure and compensatory evolution. Genes Genet. Syst. 74: 271–286. [DOI] [PubMed] [Google Scholar]
- Fariselli, P., Olmea, O., Valencia, A., and Casadio, R. 2001. Prediction of contact maps with neural networks and correlated mutations. Protein Eng. 14: 835–843. [DOI] [PubMed] [Google Scholar]
- Freund, Y. and Schapire, R.E. 1996. Experiments with a new boosting algorithm. Proceedings of the Thirteenth International Conference on Machine Learning, pp. 148–156. Morgan Kaufmann, San Francisco.
- Gardner, P.P. and Giegerich, R. 2004. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics 5: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorodkin, J., Stricklin, S.L., and Stormo, G.D. 2001. Discovering common stem-loop motifs in unaligned RNA sequences. Nucleic Acids Res. 29: 2135–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A., and Eddy, S.R. 2003. Rfam: An RNA family database. Nucleic Acids Res. 31: 439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy S.R., and Bateman A. 2005. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33: D121–D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, E.S., Brown, J.W., Pitulle, C., and Pace, N.R. 1994. Further perspective on the catalytic core and secondary structure of ribonuclease P RNA. Proc. Natl. Acad. Sci. 91: 2527–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, J.K., Haas, E.S., Williams, D., Frank, D.N., and Brown, J.W. 2001. New insight into RNase P RNA structure from comparative analysis of the archaeal RNA. RNA 7: 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker, I.L., Fontana, W., Stadler, P.F., Bonhoeffer, S., Tacker, M., and Schuster, P. 1994. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 125: 167–188. [Google Scholar]
- Hofacker, I.L., Fekete, M., and Stadler, P.F. 2002. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 319: 1059– 1066. [DOI] [PubMed] [Google Scholar]
- Jaeger, J.A., Turner, D.H., and Zuker, M. 1989. Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. 86: 7706– 7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan, V. and Wilson, C. 1999. RNA secondary structure prediction based on free energy and phylogenetic analysis. J. Mol. Biol. 289: 935–947. [DOI] [PubMed] [Google Scholar]
- Knight, R., Birmingham, A., and Yarus, M. 2004. BayesFold: Rational two degree folds that combine thermodynamic, covariation, and chemical data for aligned RNA sequences. RNA 10: 1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen, B. and Hein J.J. 1999. Using stochastic context free grammars and molecular evolution to predict RNA secondary structure. Bioinformatics 15: 446–454. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 31: 3423– 3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., and Turner, D.H. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288: 911–940. [DOI] [PubMed] [Google Scholar]
- Mathews, D.H., Disney, M.D., Childs, J.L., Schroeder, S.J., Zuker, M., and Turner, D.H. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. 101: 7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaskill, J.S. 1990. The equilibrium partition function and base pair binding probabilities for RNA secondary structures. Biopolymers 29: 1105–1119. [DOI] [PubMed] [Google Scholar]
- Mitchell, T. 1997. Machine learning. WCB/McGraw-Hill.
- Muse, S. 1995. Evolutionary analysis of DNA sequences subject to constraints on secondary structure. Genetics 139: 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov, R. and Jacobson, A.B. 1980. Fast algorithm for predicting the secondary structure of single stranded RNA. Proc. Natl. Acad. Sci. 77: 6309–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov, R., Pieczenik, G., Griggs, J.R., and Kleitman, D.J. 1978. Algorithm for loop matchings. SIAM J. Appl. Math. 35: 68–82. [Google Scholar]
- Parsch, J., Braverman, J., and Stephan, W. 2000. Comparative sequence analysis and patterns of covariation in RNA secondary structures. Genetics 154: 909–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder, J. and Giegerich, R. 2004. Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics 5: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas, E. and Eddy, S.R. 1999. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 285: 2053–2068. [DOI] [PubMed] [Google Scholar]
- Ruan, J., Stormo, G.D., and Zhang, W. 2004a. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 20: 58–66. [DOI] [PubMed] [Google Scholar]
- ———. 2004b. ILM: A web server for predicting RNA secondary structures with pseudoknots. Nucleic Acids Res. 32 (Web server issue): W146–W149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T., Stormo, G., Gold, L., and Ehrenfeuch, A. 1986. The information content of binding sites on nucleotide sequences. J. Mol. Biol. 188: 415–431. [DOI] [PubMed] [Google Scholar]
- Shapiro, B.A. and Kasprzak, W. 1996. STRUCTURELAB: A heterogeneous bioinformatics system for RNA structure analysis. J. Mol. Graphics 14: 194–205. [DOI] [PubMed] [Google Scholar]
- Shapiro, B.A., Bengali, D., Kasprzak, W., and Wu, J.C. 2001a. RNA folding pathway functional intermediates: Their prediction and analysis. J. Mol. Biol. 7: 27–44. [DOI] [PubMed] [Google Scholar]
- Shapiro, B.A., Wu, J.C., Bengali, D., and Potts, M.J. 2001b. The massively parallel genetic algorithm for RNA folding: MIMD implementation and population variation. Bioinformatics 17: 137–148. [DOI] [PubMed] [Google Scholar]
- Smith, D.R., Doucette-Stamm, L.A., Deloughery, C., Lee, H., Dubois, J., Aldredge, T., Bashirzadeh, R., Blakely, D., Cook, R., Gilbert, K., et al. 1997. Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: Functional analysis and comparative genomics. J. Bacteriol. 179: 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, R.M. and Schneider, T.D. 1992. Features of spliceosome evolution and function inferred from an analysis of the information at human splice sites. J. Mol. Biol. 228: 1124–1136. [DOI] [PubMed] [Google Scholar]
- Walter, A., Turner. D., Kim, J., Lyttle, M., Müller, P., Mathews, D., and Zuker M. 1994. Coaxial stacking of helices enhances binding of oligoribonucleotides. P. Natl. Acad. Sci. 91: 9218–9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman, M.S. and Smith, T.M. 1978. RNA secondary structure: A complete mathematical analysis. Math. Biosci. 42: 257–266. [Google Scholar]
- Zuker, M. 1989. On finding all suboptimal foldings of an RNA molecule. Science. 244: 48–52. [DOI] [PubMed] [Google Scholar]
- ———. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. and Stiegler, P. 1981. Optimal computer folding of large RNA sequences using thermodynamic and auxiliary information. Nucleic Acids Res. 9: 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]