

Abstract
Pyrrolo-C (PC), or 3-[β-D-2-ribofuranosyl]-6-methylpyrrolo[2,3-d]pyrimidin-2(3H)-one, is a fluorescent analog of the nucleoside cytidine that retains its Watson-Crick base-pairing capacity with G. Due to its red-shifted absorbance, it can be selectively excited in the presence of natural nucleosides, making it a potential site-specific probe for RNA structure and dynamics. Similar to 2-aminopurine nucleoside, which base-pairs with uridine (or thymidine), PC’s fluorescence becomes reversibly quenched upon base-pairing, most likely due to stacking interactions with neighboring bases. To test its utility as an RNA probe, we examined PC’s fluorescent properties over a wide range of ionic strengths, pH, organic cosolvents, and temperatures. Incorporation of PC into a single-stranded RNA results in an ~60% reduction of fluorescence intensity, while duplex formation reduces the fluorescence by ~75% relative to the free ribonucleoside. We find that the fluorescence intensity of PC is only moderately affected by ionic strength, pH, and temperature, while it is slightly enhanced by organic cosolvents, making it a versatile probe for a broad range of buffer conditions. We demonstrate two applications for PC: fluorescent measurements of the kinetics of formation and dissociation of an RNA/DNA complex, and fluorescent monitoring of the thermal denaturation of the central segment of an RNA duplex. Taken together, our data showcase the potential of pyrrolo-C as an effective fluorescent probe to study RNA structure, dynamics, and function, complementary to the popular 2-aminopurine ribonucleoside.
Keywords: fluorescence anisotropy, fluorescence quenching, melting transition, pyrrolo-cytosine ribonucleoside, RNA folding kinetics
INTRODUCTION
Fluorescence spectroscopy is a powerful tool for probing the structure and conformational dynamics of biopolymers, as well as their interactions, under a wide range of solution conditions in vitro and in vivo. For example, fluorescence spectroscopy has successfully been utilized to directly observe and quantify conformational change and catalytic function of nucleic acids in solution (Millar 1996; Walter and Burke 2000; Klostermeier and Millar 2001; Walter et al. 2001, 2002; Ha 2004; Zhuang 2005). Unlike proteins, which in many cases carry intrinsic fluorophores such as tryptophan, nucleic acids must be extrinsically labeled, typically with fluorescent organic dyes, due to the low fluorescence quantum yield of their natural components. Even though a plethora of such dyes has been developed (see, e.g., http://probes.com/), a risk is that they sterically disrupt nucleic acid structure and possibly function. One solution to this problem is the use of fluorescent base analogs as alternative, less invasive probes of nucleic acid structure, dynamics, and function.
Fluorescent base analogs are valuable in monitoring nucleic acid structural dynamics because they behave similarly to the natural nucleobases in their interactions with other biomolecules (Millar 1996; Jameson and Eccleston 1997; Rist and Marino 2002). They typically do not disrupt duplex formation, making them useful probes of basepairing and base-stacking with neighboring natural bases. Fluorescent base analogs generally have large Stokes shifts and their fluorescence is sensitive to their environment (Jean and Hall 2001), making them good probes of local conformational changes that occur within nucleic acids (Millar 1996; Jameson and Eccleston 1997; Rist and Marino 2002; Walter et al. 2002; Berry et al. 2004). A number of fluorescent base analogs have been incorporated into nucleoside phosphoramidites and thus synthetic nucleic acids. One of the most popular fluorescent nucleobases is 2-aminopurine, an analog of the purines adenine and guanine (Ward et al. 1969; Doudna et al. 1990; Fujimoto et al. 1996; Millar 1996; Rist and Marino 2002). It specifically Watson-Crick base-pairs with thymine or uracil and has proven its utility as a probe to monitor local conformational changes in nucleic acid structure, for example, in catalytic RNA and upon formation of nucleic acid–protein complexes (Millar 1996; O’Neill and Barton 2002; Walter et al. 2002; Patel and Bandwar 2003; Roy 2003; Bradrick and Marino 2004; Clerte and Hall 2004). Several fluorescent pyrimidine analogs also have been developed, although they have not yet reached a similar level of popularity (Inoue et al. 1985; Wu et al. 1990; Godde et al. 1998; Wilhelmsson et al. 2001).
A recent addition to the arsenal of fluorescent pyrimidine analogs available for probing nucleic acids is 3-[β-D-2-ribofuranosyl]-6-methylpyrrolo[2,3-d]pyrimidin-2(3H)- one, in short pyrrolo-C (PC) (Berry et al. 2004; Fig. 1A ▶). Its 2′-deoxy ribose variant has been successfully used, for example, in the characterization of the transcription bubble in elongation complexes of T7 RNA polymerase (Liu and Martin 2001, 2002), in a biosensor for metal ions (Chen and He 2004), and to characterize local structure in a double-stranded DNA (Johnson et al. 2005) or DNA/RNA hybrid (Dash et al. 2004). Its unperturbed Watson- Crick face allows PC to base-pair specifically with G, similar to an unmodified cytidine (Fig. 1A ▶). PC is of relatively small size, which is unlikely to perturb the structure of an A-type RNA helix. In addition, it promises to have potent applications in RNA structural analysis as it shares many of the same fluorescence characteristics that make 2-aminopurine ribonucleoside a versatile probe (Berry et al. 2004). PC is highly fluorescent, with excitation and emission maxima of 350 and 460 nm, respectively, relatively far from those of RNA and protein, making it a suitable probe for both RNAs and RNA–protein complexes. Furthermore, the fluorescence of PC is significantly quenched in duplex DNA and RNA, most likely through base-stacking interactions with surrounding nucleotides, rendering it sensitive to its local environment (Liu and Martin 2001, 2002; Berry et al. 2004; Chen and He 2004; Dash et al. 2004).
FIGURE 1.

Structure of pyrrolo-C (PC) ribonucleoside and incorporation into an RNA. (A) Chemical structure of PC ribonucleoside. (B) RNA oligonuclotides used in this study. The position of PC is highlighted in gray.
To explore and evaluate its suitability as fluorescent probe of RNA structure and dynamics, we now have incorporated PC into the middle of a single-stranded RNA (ssRNA) (Fig. 1B ▶). To study how the fluorescence of PC is affected by duplex formation, we used a complementary strand, termed “cssRNA,” to form the corresponding double- stranded RNA complex (dsRNA) (Fig. 1B ▶). We find that the steady-state fluorescence of PC in the single and double strands decreases by ~60% and ~75%, respectively, relative to the free ribonucleoside. Fluorescence of PC is only moderately affected by ionic strength and pH, while it is slightly enhanced by organic co-solvents, making it a probe suitable for a range of buffer conditions. Exploiting these observations, we demonstrate two applications for PC: (1) We fluorescently measure the kinetics of formation and dissociation of an RNA/DNA hybrid, and (2) we fluorescently monitor the thermal denaturation of the central segment of an RNA duplex and find good agreement with the major, but not a minor lower-temperature, melting transition observed by UV absorbance. Taken together, our data highlight the potential of PC as a fluorescent probe to study RNA structure, dynamics, and function, and suggest it as a suitable experimental counterpart to 2-aminopurine ribonucleoside.
RESULTS AND DISCUSSION
Fluorescence of PC decreases upon RNA duplex formation
Figure 1 ▶ gives the structure and hydrogen-bonding pattern of PC, along with the PC-containing dsRNA used in this study. PC in strand ssRNA is base-paired with guanosine (G) in the fully complementary strand cssRNA. Previous investigations of DNA and DNA/RNA hybrid duplexes by fluorescence spectroscopy suggest that PC undergoes normal Watson-Crick pairing with G and is well-stacked within the DNA helix in a manner similar to cytosine (Liu and Martin 2001, 2002; Berry et al. 2004; Chen and He 2004; Dash et al. 2004). To determine the fluorescence characteristics of PC in RNA, we obtained excitation and emission spectra of the ssRNA and its double-stranded form (dsRNA) (Fig. 1 ▶) under our standard buffer conditions (chosen to be close-to-physiologic: 50 mM Tris, 25 mM MES-NaOH, 25 mM acetic acid–NaOH at pH 7.5, 100 mM NaCl) at 25°C. We compared these spectra with those of free PC, obtained by complete degradation of ssRNA with 0.5 U of S1 nuclease over 1 h (Fig. 2 ▶). Relative to this free PC control, the fluorescence decreases by ~60% in the intact ssRNA and by ~75% in the RNA duplex. In addition, the PC excitation and emission maxima are slightly blue-shifted from 353 and 448 nm, respectively, in the free PC control to 352 and 447 nm, respectively, in ssRNA, and to 351 and 443 nm, respectively, in dsRNA (all values obtained by Gaussian fits to the spectra) (Fig. 2 ▶). Such a hypsochromic effect is expected for π→π* transitions in the increasingly hydrophobic environment provided by single- and double-stranded base-stacking (Lakowicz 1999). Similar results were obtained when comparing ssRNA and dsRNA in standard buffer with the directly available free PC as reference; in ssRNA the fluorescence intensity drops to 48% ± 2%, and drops in dsRNA to 31% ± 4% relative to free PC (Table 1 ▶).
FIGURE 2.

Pyrrolo-C fluorescence studies using 1 μM ssRNA and 1 μM dsRNA in standard buffer (50 mM Tris, 25 mM MES-NaOH, 25 mM acetic acid–NaOH at pH 7.5, 100 mM NaCl) at 25°C. Steady-state fluorescence emission (A) and excitation spectra (B) of ssRNA (solid line), dsRNA (dash-dotted line), and ssRNA after complete degradation with 0.5 U of S1 nuclease (gray line), as indicated. The emission and the excitation maxima were obtained as the means of Gaussian fits to the spectra.
TABLE 1.
Relative fluorescence intensities and anisotropies of free PC, ssRNA, and dsRNA
| Fluorescence intensity | ||||
| Buffer (%) | 50% ETOH (%) | 50% Formamide | Anisotropy | |
| Free PC | 100 ± 1 | 353 ± 1 | 216 ± 2 | 0.06 ± 0.02 |
| ssRNA | 48 ± 2 | 85 ± 2 | 162 ± 3 | 0.13 ± 0.01 |
| dsRNA | 31 ± 4 | 38 ± 2 | 98 ± 1 | 0.20 ± 0.02 |
Fluorescence anisotropy of PC reports on incorporation into RNA and duplex formation
Fluorescence anisotropy measurements sensitively report on the rotational diffusion of a fluorophore within a biomolecular complex; generally, the higher the anisotropy (or polarization), the less mobile the fluorophore and the larger the complex (Lakowicz 1999). This property should therefore allow us to monitor the incorporation of the small PC into the much larger ssRNA and dsRNA. We find that the free PC has a low anisotropy of 0.06 (Table 1 ▶), suggesting that it rotates relatively freely in solution. Upon incorporation into ssRNA, the anisotropy of PC increases by approximately twofold to 0.13, as expected. Addition of the complementary strand to form dsRNA further increases the anisotropy overall approximately threefold to 0.20 (Table 1 ▶). These data suggest that PC becomes increasingly immobile upon incorporation into ssRNA and dsRNA, presumably by a combination ofmore stable local stacking interactions and slower overall rotational diffusion of the complex.
Monovalent and divalent salts do not significantly affect PC fluorescence
Monovalent and divalent salts commonly supply the counter-ion charge necessary to promote secondary and tertiary structure folding as the basis for function of a polyanionic RNA in solution (Draper 2004; Woodson 2005). However, it is well known that salts also have the ability to quench fluorophores, for example, by promoting the formation of nonfluorescent states (Vamosi et al. 1996). Salts such as NaCl, KCl, and MgCl2, for example, have been observed to quench the fluorescence of 2-aminopurine and formycin by 15% (Ward et al. 1969). To evaluate PC as a probe for RNA folding and function, we therefore examined the effect of salt concentration on its fluorescence. Specifically, we titrated ssRNA and dsRNA, as well as the free PC, with NaCl and MgCl2 beginning from low-ionic strength buffer conditions (50 mM Tris, 25 mM MESNaOH, 25 mM acetic acid–NaOH at pH 7.5, containing ~12 mM Na+, at 25°C). Figure 3, A and B ▶, shows that, upon raising the MgCl2 and NaCl concentrations by 75 and 100 mM, respectively, the normalized fluorescence intensity of ssRNA decreases by ~40% and ~25%, respectively. In contrast, the normalized fluorescence intensities of both dsRNA and free PC remain virtually constant. These observations suggest that the quenching of fluorescence intensity observed for ssRNA with increasing ionic strength is not an intrinsic property of PC, but is most likely linked to properties of the incorporating ssRNA, for example, enhanced basestacking or secondary structure formation with increasing counter-ion concentration. (To control for the latter, mfold-based structure predictions were performed (Zuker 2003), which generally produced potential secondary structures of low thermodynamic stability [data not shown].) This relatively moderate intrinsic ionic strength dependence recommends PC as a fluorescent probe for a broad set of buffer conditions.
FIGURE 3.

Relative fluorescence intensity of free pyrrolo-C ribonucleoside (gray triangles), ssRNA (filled circles), and dsRNA (open circles) as a function of NaCl (A) and MgCl2 (B) concentration in 50 mM Tris, 25 mM MES-NaOH, and 25 mM acetic acid–NaOH (pH 7.5; initially containing ~12 mM Na+) at 25°C. Errors are derived from at least two independent measurements.
pH titration of PC fluorescence yields apparent pKas
Many fluorophores are pH-dependent; however, an important characteristic of a fluorescent probe often is its ability to be used over a wide pH range. Therefore, we measured the fluorescence intensity of PC as a function of pH in the free PC, ssRNA, and dsRNA. Overall, PC fluorescence is pH-independent over a wide range, pH 4–9, and only significantly decreases below pH 4 in all three structural contexts (Fig. 4A ▶). This pH dependence allowed us to fit the data (see Materials and Methods) to derive apparent pKas for free PC and PC within ssRNA and dsRNA of 3.3, 2.7, and 3.6 (each ±0.3), respectively. The pKa of PC therefore appears to be <4, slightly lower than cytosine’s pKa of 4.2 (Saenger 1984). The fact that the three pKas in different structural contexts are similar suggests that the PC protonation equilibrium is not strongly influenced by stacking or hydrogen bonding interactions.
FIGURE 4.

The relative fluorescence intensity of free pyrrolo-C ribonucleoside (gray triangles), ssRNA (filled circles), and dsRNA (open circles) as a function of pH (A) and (v/v)% content (B) of formamide in standard buffer (50 mM Tris, 25 mM MES-NaOH, 25 mM acetic acid–NaOH at pH 7.5, 100 mM NaCl) at 25°C. Errors are derived from at least two independent measurements.
Organic cosolvents enhance the fluorescence of PC
Organic cosolvents such as formamide partially denature RNA, which can lead to escape from kinetic folding traps and functional enhancement (Pan and Sosnick 1997; Rook et al. 1998). To investigate the effects of organic cosolvents on PC fluorescence in the context of RNA, we performed a titration of ssRNA, dsRNA, and free PC with up to 50% formamide in standard buffer at 25°C. Figure 4B ▶ shows that in all three cases we observed a monotonous increase in fluorescence intensity by nearly twofold over this range of cosolvent, suggesting a direct effect on the fluorophore rather than the RNA structure.
To further analyze the effect of organic cosolvents on PC fluorescence, we tested the impact of 50% ethanol. Table 1 ▶ gives quantitative PC fluorescence signals for the free PC, ssRNA, and dsRNA in standard buffer with and without a supplement of 50% ethanol ethanol or 50% formamide (normalized to the signal of free PC in buffer). The free PC fluorescence increases 3.5-fold and 2.2-fold upon addition of 50% ethanol and formamide, respectively. By contrast, the ssRNA fluorescence increases by only twofold upon addition of 50% ethanol, while the dsRNA signal does not increase significantly. Taken together with our formamide titration data, these results suggest that, first, PC fluorescence is enhanced in a more hydrophobic solvent environment and, second, the presence of RNA secondary structure dampens this enhancement.
Application example: PC reports on the kinetics of formation and dissociation of an RNA/DNA duplex
To directly test the utility of PC in a dynamic RNA structure probing application, we designed a steady-state fluorescence assay in which we monitor binding and dissociation of a DNA oligonucleotide termed c1 to ssRNA. c1 is fully complementary to ssRNA and also contains a 10-nt 5′-overhang, which allows its removal by addition of the fully complementary DNA oligonucleotide c2. c2 forms an extended DNA duplex with c1 that displaces ssRNA (Fig. 5A ▶), in analogy to previous conformational shift assays (Yurke et al. 2000; Hoerter et al. 2004). Given that PC fluorescence in double-stranded nucleic acids is quenched, we expect duplex formation of ssRNA with c1 to cause a decrease in PC fluorescence that is reversed upon c2 addition.
FIGURE 5.

Kinetics of formation and dissociation of an RNA/DNA duplex in standard buffer (50 mM Tris, 25 mM MES-NaOH, 25 mM acetic acid–NaOH at pH 7.5, 100 mM NaCl) at 25°C. This assay is analogous to previous conformational shift assays (Yurke et al. 2000; Hoerter et al. 2004) and demonstrates the reversibility of changes in pyrrolo-C fluorescence upon duplex formation. (A) Schematic of the assay. ssRNA binds DNA oligonucleotide c1 (cyan), which is removed by addition of DNA oligonucleotide c2 (green); gray indicates pyrrolo-C probe. (B) Upon addition of the DNA oligonucleotide c1 (cyan), the pyrrolo-C fluorescence decreases with a rate of 1.93 min−1. Upon addition of DNA oligonucleotide c2 (green), the fluorescence decrease is reversed, with a rate of 4.28 min−1. (C) Concentration dependence of the pseudo-first-order rate constant upon addition of excess c1 to ssRNA. The slope of the linear regression line yields the bimolecular rate constant kbind.
Indeed, upon addition of 1 μM c1 to 200 nM ssRNA in standard buffer at 25°C, we observed an exponential decrease in PC fluorescence over time with a rate constant of 1.93 min−1, indicative of duplex formation (Fig. 5B ▶). After the fluorescence signal equilibrated, we added 2 μM c2, which led to an exponential fluorescence increase at a rate constant of 4.28 min−1, signifying displacement of ssRNA from its hybrid with c1 (Fig. 5B ▶). These results provide direct evidence for PC’s capability to monitor in real time the formation and dissociation of a nucleic acid duplex. Furthermore, the pseudo-first-order rate constant of the fluorescence decrease upon c1 addition is linearly dependent on the c1 concentration. From the slope of this dependence, we were able to extract a bimolecular rate constant of 4.0 ± 0.2 × 106 M−1 min−1 for ssRNA binding to DNA oligonucleotide c1 (Fig. 5C ▶). This rate signifies that binding of c1 is not diffusion limited and is comparable to other bimolecular binding rate constants, such as those of substrate binding to the hairpin and HDV ribozymes (Walter et al. 1997; Harris et al. 2002; Pereira et al. 2002; Jeong et al. 2003).
Application example: PC reports on thermal melting of an RNA duplex
As a second application example, we sought to demonstrate PC’s ability to report on thermodynamic parameters of a dsRNA such as its equilibrium melting. Optical melting curves derived from the hyperchromic shift upon denaturation are frequently used to provide such information indiscriminately of position in a nucleic acid. By contrast, a site-specifically incorporated fluorescent reporter is expected to monitor nucleic acid melting locally, which can be used to probe specific regions of interest. To demonstrate the validity of using PC to report on local melting in RNA, we obtained first a standard optical melting profile by following the absorption at 260 nm, and second, a melting profile monitored by fluorescence at 347 nm, employing our dsRNA duplex under standard buffer conditions. The equilibrium melting profile monitored by absorption shows two transitions represented by melting temperatures (Tm) of 70°C and 80°C (Fig. 6A ▶). This may represent two different regions of the dsRNA structure melting sequentially. The corresponding melting transition monitored by PC fluorescence at 347 nm exhibits only a single melting transition at 81°C, within error (±1°C) of the major transition in the absorption melting profile (Fig. 6B ▶). Under the same buffer conditions, the fluorescence of the free PC shows only a very slight intrinsic temperature dependence, with a minimum near ~75°C (Fig. 6C ▶); this may explain the slight drift in baseline <70°C observed in our fluorescence melting profile (Fig. 6B ▶). As a further control, we obtained optical melting profiles at 260 nm for ssRNA and cssRNA alone under the same buffer conditions. The profiles each show a very minor transition represented by melting temperatures of 29°C and 39°C, respectively (Fig. 6D ▶), consistent with mfold calculations (Zuker 2003) that predict only potential secondary structures of low thermodynamic stability (data not shown). Therefore, the minor melting transition we observe at 70°C in the absorptive melting profile of the ssRNA/cssRNA duplex cannot be attributed to melting of an excess of one of the two RNA strands. Taken together, our results demonstrate that PC incorporated into the middle of an RNA duplex accurately reports on the main melting transition, independent of minor transitions in the RNA as a whole.
FIGURE 6.

Temperature-dependent UV absorbance at 260 nm and fluorescence emission at 347 nm of dsRNA. (A) UV melting profile of dsRNA. The relative absorbance of the RNA at 260 nm (open circles, left axis) and a bimodal Gaussian distribution fit to the smoothed first derivative (solid and dashed/dotted lines, right axis) are shown. Two different melting temperatures Tm1 and Tm2 are derived (see Materials and Methods). (B) Fluorescence melting profile of ssRNA (open circles), analyzed as described in A. (C) Temperature dependence of the free PC fluorescence (gray triangles). (D) UV melting profile of ssRNA (closed circles) and cssRNA (closed diamonds). The relative absorbance of the RNA at 260 nm (left axis) and Gaussian distribution fits to the smoothed first derivatives (ssRNA, solid line; cssRNA, dotted line) (right axis) are shown.
Conclusions
In summary, we have incorporated the fluorescent cytosine analog 3-[β-D-2-ribofuranosyl]-6-methylpyrrolo[2,3-d]pyrimidin-2(3H)-one, or PC, into an RNA and shown that it serves as effective probe of kinetic and thermodynamic parameters associated with nucleic acid structure formation. PC is fluorescent over a broad range of solution conditions and becomes reversibly quenched upon basepairing and -stacking in an RNA (or RNA/DNA) duplex. Our results therefore highlight the potential of PC in further applications such as those where the 2′ deoxy modified PC has previously been applied. For example, the local structure in an RNA or RNA/DNA duplex may be probed (Dash et al. 2004; Johnson et al. 2005) or a change in basepairing and/or -stacking upon protein binding be followed (Liu and Martin 2001, 2002; Chen and He 2004). Its relatively small size and full hydrogen bond complementarity to G promise to make PC a valuable addition to the uridine- paired 2-aminopurine ribonucleoside (Berry et al. 2004).
MATERIALS AND METHODS
Preparation of RNA oligonucleotides
PC cyanoethyl phosphoramidite with 2′-O-triisopropylsilyloxymethyl (TOM) protection group and the free ribonucleoside were a gift from Berry and Associates (Dexter, MI). The TOM protected phosphoramidite is now commercially available from Glen Research Corporation; a TBS-protected version is available from Berry and Associates. RNA oligonucleotides (sequences given in Fig. 1 ▶) were synthesized commercially by the Howard Hughes Medical Institute Biopolymer/Keck Foundation Biotechnology Resource RNA Laboratory at the Yale University School of Medicine. The RNA contained 2′-protection groups and was deprotected as suggested by the manufacturer by using standard protocols based on triethylamine trihydrofluoride (http://info.med.yale.edu/wmkeck/) (Walter 2001, 2002). Deprotected RNA was purified by denaturing 20% polyacrylamide and 8 M urea; gel electrophoresis; diffusion elution into 0.5 M NH4OAc, 0.1% SDS, and 0.1 mM EDTA overnight at 4°C; chloroform extraction; ethanol precipitation; and C8 reverse-phase HPLC with a linear acetonitrile gradient in triethylammonium acetate as described previously (Walter 2001, 2002). The PC ribonucleoside concentration was calculated from its absorbance at 347 nm (ɛ347 = 3700 M−1 cm−1), and RNA concentrations were calculated from their absorption at 260 nm.
DNA sequences used in this study were as follows: c1 (5′-GA GCTGTCTCATAGTCCATTGGCCATCGAAT-3′), c2 (5′-ATTCGAT GGCCAATGGACTATGAGACAGCTC-3′). Oligodeoxynucleotides were synthesized and desalted by Invitrogen. Synthetic DNA was purified by denaturing 20% polyacrylamide and 8 M urea, gel electrophoresis, and ethanol precipitation as described above. Stock concentrations were determined from their absorption at 260 nm.
Fluorescence experiments
All steady-state fluorescence measurements were conducted on an Aminco-Bowman Series 2 (AB2) spectrofluorometer (Thermo Spectronic) using 3-mm path length and 225-μL quartz cuvettes. Emission spectra were obtained by exciting at 337 nm (4-nm bandwidth) and monitoring fluorescence between 400 and 600 nm (8-nm bandwidth), and excitation spectra were obtained by monitoring fluorescence at 450 nm (8-nm bandwidth) while scanning excitation wavelengths between 300 and 400 nm (4-nm bandwidth). Fluorescence anisotropies were measured using the Autopolarizer equipment of the AB2 spectrofluorometer with excitation at 337 nm (4-nm bandwidth) and emission detection at 450 nm (8-nm bandwidth). Excitation and emission polarizers were sequentially set to all four possible combinations of vertical (v, 0°) and horizontal (h, 90°) alignment—Ivv, Ivh, Ihv, and Ihh— to derive the instrument-factor corrected anisotropy A from the expression (Lakowicz 1999):
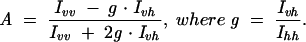 |
Experiments were generally carried out at concentrations of 1 μM free PC, ssRNA, and dsRNA in a standard buffer of 50 mM Tris-HCl, 25 mM MES-NaOH, and 25 mM acetic acid–NaOH (pH 7.5), 100 mM NaCl, at 25°C (to produce dsRNA, ssRNA was annealed with a twofold excess of the complementary strand cssRNA, by heating to 70°C for 2 min and cooling to room temperature over 5 min). Nuclease digests were performed by addition of 0.5 U of S1 nuclease to a fluorescence cuvette with 1 μM ssRNA in standard buffer and incubation for 1 h at 25°C, at which point the fluorescence signal had stabilized.
All titrations with the exception of pH were performed in standard buffer by slowly increasing the amount of titrant. This was accomplished by adding small aliquots of an identical solution but supplemented with 200 mM MgCl2, 300 mM NaCl, or 75% formamide, followed by mixing and removal of a solution volume equivalent to that of the added aliquot. This procedure ensures that neither the concentrations of RNA and buffer nor the total solution volume changed over the course of the titration. After each titration step, the solution was equilibrated for 5–10 min until the fluorescence signal had stabilized, and an emission spectrum was recorded as described above. The fluorescence intensity of the peak was extracted by averaging 10 adjacent data points. pH titrations were performed by preparing a single solution of 1 μM RNA or free PC, supplemented with 100 mMNaCl, which was separated into aliquots and buffered to the required pH using final concentrations of 25 mM sodium citrate (pH 2.0–4.0) or 50 mM Tris, 25 mM MES, and 25 mM acetic acid (pH 4.0–9.0). Emission spectra were recorded and analyzed as described above. To derive pKa values, the PC fluorescence dependence on pH was fit with the equation F = F0 + ΔFmax/(1 + 10(pKa − pH)), where F0 is the fluorescence intensity at low pH and ΔFmax is the maximum increase in fluorescence intensity at high pH.
Kinetic binding and dissociation assays were performed by preparing 200 nM ssRNA in standard buffer, which was heated to 70°C for 2 min and cooled to room temperature over 5 min. Oligodeoxynucleotide c1 (5 μL) was manually added to a final concentration of 1 μM. After the fluorescence signal equilibrated, oligodeoxynucleotide c2 (5 μL) was added to a final concentration of 2 μM. The resulting time traces were fit with single-exponential decay and increase functions of the form y = y0 + A(e−t/τ) and y = y0 + A(1 − e−t/τ), respectively, employing Marquardt-Levenberg nonlinear least-squares regression (Microcal Origin 7.0), where A is the amplitude and 1/τ an estimated pseudo-first-order rate constant. To obtain a binding rate constant for c1, its concentration was varied from 1 to 2.5 μM, and the resulting concentration dependence was fit by linear regression to yield the bimolecular rate constant kbind as the slope.
Thermal denaturation
Thermal denaturation of 1 μM dsRNA at 260 nm was carried out using a Beckman DU-640B UV-Vis spectrophotometer with High Performance Temperature Controller and Micro Auto 6 Tm cell holder. RNA samples (~300 μL) were prepared as described above in standard buffer, degassed for 5 min prior to obtaining UV melting curves. Absorbance readings at 260 nm were collected every 1.0°C as the sample was heated from 40°C to 90°C at a rate of 0.2°C/min. Melting curves in Figure 6 ▶ represent averaged data collected from two melts of freshly prepared dsRNA. After normalization, the first derivative was determined and smoothed by adjacent averaging. The derivative was fit best with two Gaussian distributions using MicroCal Origin 7.0, yielding two melting temperatures.
Thermal denaturation of 1 μMdsRNA at 347 nm was carried out using a Cary Eclipse spectrophotometer (Varian) with a multicell Peltier temperature control. RNA samples (~1 mL) were prepared as described above in standard buffer and degassed for 5 min prior to obtaining fluorescence melting curves. Fluorescence readings at 347 nm were collected every 0.2°C, as the sample was heated from 40°C to 90°C at a rate of 0.2°C/min. Fluorescence melting curves represent data collected from two melts of freshly prepared dsRNA. After normalization, the first derivative was determined and smoothed by adjacent averaging. The derivative was best fit with a single Gaussian function using MicroCal Origin 7.0.
Acknowledgments
We thank Will Pearson and Berry and Associates for supplying the PC phosphoramidite before it became commercially available. We also thank Carol Fierke for use of her Cary Eclipse spectrophotometer. This work was supported in part by NIH grant GM62357 (N.G.W.) and supplement GM62357-S1 (R.A.T.).
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2165806.
REFERENCES
- Berry, D.A., Jung, K.Y., Wise, D.S., Sercel, A.D., Pearson, W.H., Mackie, H., Randolph, J.B., and Somers, R.L. 2004. Pyrrolo-dC and pyrrolo-C: Fluorescent analogs of cytidine and 2′-deoxycytidine for the study of oligonucleotides. Tetrahedron Lett. 45: 2457– 2461. [Google Scholar]
- Bradrick, T.D. and Marino, J.P. 2004. Ligand-induced changes in 2- aminopurine fluorescence as a probe for small molecule binding to HIV-1 TAR RNA. RNA 10: 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P. and He, C. 2004. A general strategy to convert the MerR family proteins into highly sensitive and selective fluorescent biosensors for metal ions. J. Am. Chem. Soc. 126: 728–729. [DOI] [PubMed] [Google Scholar]
- Clerte, C. and Hall, K.B. 2004. Global and local dynamics of the U1A polyadenylation inhibition element (PIE) RNA and PIE RNA-U1A complexes. Biochemistry 43: 13404–13415. [DOI] [PubMed] [Google Scholar]
- Dash, C., Rausch, J.W., and Le Grice, S.F. 2004. Using pyrrolo-deoxy-cytosine to probe RNA/DNA hybrids containing the human immunodeficiency virus type-1 3′ polypurine tract. Nucleic Acids Res. 32: 1539–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna, J.A., Szostak, J.W., Rich, A., and Usman, N. 1990. Chemical synthesis of oligoribonucleotides containing 2-aminopurine: Substrates for the investigation of ribozyme function. J. Org. Chem. 55: 5547–5549. [DOI] [PubMed] [Google Scholar]
- Draper, D.E. 2004. A guide to ions and RNA structure. RNA 10: 335– 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto, J., Nuesca, Z., Mazurek, M., and Sowers, L.C. 1996. Synthesis and hydrolysis of oligodeoxyribonucleotides containing 2-aminopurine. Nucleic Acids Res. 24: 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godde, F., Toulme, J.J., and Moreau, S. 1998. Benzoquinazoline derivatives as substitutes for thymine in nucleic acid complexes: Use of fluorescence emission of benzo[g]quinazoline-2,4-(1H,3H)-dione in probing duplex and triplex formation. Biochemistry 37: 13765– 13775. [DOI] [PubMed] [Google Scholar]
- Ha, T. 2004. Structural dynamics and processing of nucleic acids revealed by single-molecule spectroscopy. Biochemistry 43: 4055– 4063. [DOI] [PubMed] [Google Scholar]
- Harris, D.A., Rueda, D., and Walter, N.G. 2002. Local conformational changes in the catalytic core of the trans-acting hepatitis delta virus ribozyme accompany catalysis. Biochemistry 41: 12051–12061. [DOI] [PubMed] [Google Scholar]
- Hoerter, J.A., Lambert, M.N., Pereira, M.J., and Walter, N.G. 2004. Dynamics inherent in helix 27 from Escherichia coli 16S ribosomal RNA. Biochemistry 43: 14624–14636. [DOI] [PubMed] [Google Scholar]
- Inoue, H., Imura, A., and Ohtsuka, E. 1985. Synthesis and hybridization of dodecadeoxyribonucleotides containing a fluorescent pyridopyrimidine deoxynucleoside. Nucleic Acids Res. 13: 7119–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson, D.M. and Eccleston, J.F. 1997. Fluorescent nucleotide analogs: Synthesis and applications. Methods Enzymol. 278: 363–390. [DOI] [PubMed] [Google Scholar]
- Jean, J.M. and Hall, K.B. 2001. 2-Aminopurine fluorescence quenching and lifetimes: Role of base stacking. Proc. Natl. Acad. Sci. 98: 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, S., Sefcikova, J., Tinsley, R.A., Rueda, D., and Walter, N.G. 2003. Trans-acting hepatitis delta virus ribozyme: Catalytic core and global structure are dependent on the 5′ substrate sequence. Biochemistry 42: 7727–7740. [DOI] [PubMed] [Google Scholar]
- Johnson, N.P., Baase, W.A., and von Hippel, P.H. 2005. Investigating local conformations of double-stranded DNA by low-energy circular dichroism of pyrrolo-cytosine. Proc. Natl. Acad. Sci. 102: 7169–7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klostermeier, D. and Millar, D.P. 2001. RNA conformation and folding studied with fluorescence resonance energy transfer. Methods 23: 240–254. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2d ed. Springer Publishers, Dordrecht, Netherlands.
- Liu, C. and Martin, C.T. 2001. Fluorescence characterization of the transcription bubble in elongation complexes of T7 RNA polymerase. J. Mol. Biol. 308: 465–475. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Promoter clearance by T7 RNA polymerase: Initial bubble collapse and transcript dissociation monitored by base analog fluorescence. J. Biol. Chem. 277: 2725–2731. [DOI] [PubMed] [Google Scholar]
- Millar, D.P. 1996. Fluorescence studies of DNA and RNA structure and dynamics. Curr. Opin. Struct. Biol. 6: 322–326. [DOI] [PubMed] [Google Scholar]
- O’Neill, M.A. and Barton, J.K. 2002. 2-Aminopurine: A probe of structural dynamics and charge transfer in DNA and DNA:RNA hybrids. J. Am. Chem. Soc. 124: 13053–13066. [DOI] [PubMed] [Google Scholar]
- Pan, T. and Sosnick, T.R. 1997. Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat. Struct. Biol. 4: 931–938. [DOI] [PubMed] [Google Scholar]
- Patel, S.S. and Bandwar, R.P. 2003. Fluorescence methods for studying the kinetics and thermodynamics of transcription initiation. Methods Enzymol. 370: 668–686. [DOI] [PubMed] [Google Scholar]
- Pereira, M.J., Harris, D.A., Rueda, D., and Walter, N.G. 2002. Reaction pathway of the trans-acting hepatitis delta virus ribozyme: A conformational change accompanies catalysis. Biochemistry 41: 730–740. [DOI] [PubMed] [Google Scholar]
- Rist, M.J. and Marino, J.P. 2002. Fluorescent nucleotide base analogs as probes of nucleic acid structure, dynamics and interactions. Curr. Org. Chem. 6: 775–793. [Google Scholar]
- Rook, M.S., Treiber, D.K., and Williamson, J.R. 1998. Fast folding mutants of the Tetrahymena group I ribozyme reveal a rugged folding energy landscape. J. Mol. Biol. 281: 609–620. [DOI] [PubMed] [Google Scholar]
- Roy, S. 2003. On the use of 2-aminopurine as a probe for base pair opening during transcription initiation. Methods Enzymol. 370: 568–576. [DOI] [PubMed] [Google Scholar]
- Saenger, W. 1984. Principles of nucleic acid structure. Springer-Verlag, New York.
- Vamosi, G., Gohlke, C., and Clegg, R.M. 1996. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: Multiple conformers of singlestranded and double-stranded dye-DNA complexes. Biophys. J. 71: 972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, N.G. 2001. Structural dynamics of catalytic RNA highlighted by fluorescence resonance energy transfer. Methods 25: 19–30. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Probing RNA structural dynamics and function by fluorescence resonance energy transfer (FRET). Curr. Protocols Nucleic Acid Chem. 11.10: 11.10.11–11.10.23. [DOI] [PubMed] [Google Scholar]
- Walter, N.G. and Burke, J.M. 1997. Real-time monitoring of hairpin ribozyme kinetics through base-specific quenching of fluoresceinlabeled substrates. RNA 3: 392–404. [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. Fluorescence assays to study structure, dynamics, and function of RNA and RNA-ligand complexes. Methods Enzymol. 317: 409–440. [DOI] [PubMed] [Google Scholar]
- Walter, N.G., Chan, P.A., Hampel, K.J., Millar, D.P., and Burke, J.M. 2001. A base change in the catalytic core of the hairpin ribozyme perturbs function but not domain docking. Biochemistry 40: 2580– 2587. [DOI] [PubMed] [Google Scholar]
- Walter, N.G., Harris, D.A., Pereira, M.J., and Rueda, D. 2002. In the fluorescent spotlight: Global and local conformational changes of small catalytic RNAs. Biopolymers 61: 224–242. [DOI] [PubMed] [Google Scholar]
- Ward, D.C., Reich, E., and Stryer, L. 1969. Fluorescence studies of nucleotides and polynucleotides, I: Formycin, 2- aminopurine riboside, 2,6-diaminopurine riboside, and their derivatives. J. Biol. Chem. 244: 1228–1237. [PubMed] [Google Scholar]
- Wilhelmsson, L.M., Holmen, A., Lincoln, P., Nielsen, P.E., and Norden, B. 2001. A highly fluorescent DNA base analogue that forms Watson-Crick base pairs with guanine. J. Am. Chem. Soc. 123: 2434–2435. [DOI] [PubMed] [Google Scholar]
- Woodson, S.A. 2005. Metal ions and RNA folding: A highly charged topic with a dynamic future. Curr. Opin. Chem. Biol. 9: 104–109. [DOI] [PubMed] [Google Scholar]
- Wu, P.G., Nordlund, T.M., Gildea, B., and McLaughlin, L.W. 1990. Base stacking and unstacking as determined from a DNA decamer containing a fluorescent base. Biochemistry 29: 6508–6514. [DOI] [PubMed] [Google Scholar]
- Yurke, B., Turberfield, A.J., Mills Jr., A.P., Simmel, F.C., and Neumann, J.L. 2000. A DNA-fuelled molecular machine made of DNA. Nature 406: 605–608. [DOI] [PubMed] [Google Scholar]
- Zhuang, X. 2005. Single-molecule RNA science. Annu. Rev. Biophys. Biomol. Struct. 34: 399–414. [DOI] [PubMed] [Google Scholar]
- Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]