

Abstract
The reactions of the bis(cyclopentadienyl)(tert-butylimido)zirconium complex (Cp2Zr=N-t-Bu)(THF) (1) with epoxides, aziridines, and episulfides were investigated. Heterocycles without accessible β-hydrogens undergo insertion/protonation of the C–X bond to produce 1,2-amino alcohols (X = O) and 1,2-diamines (X = NR), whereas heterocycles with accessible β-hydrogens undergo elimination/protonation to produce allylic alcohols (X = O) and allylic sulfides (X = S). Mechanistic investigations support a stepwise pathway with zwitterionic intermediates for the first reaction class and a concerted pathway for the second reaction class. Additionally, the feasibility of chirality transfer from the planar-chiral ebthi (ebthi = ethylenebis(tetrahydroindenyl)) ligand was demonstrated with a chiral analogue, (ebthi)-Zr=NAr(THF) (Ar = 2,6-dimethylphenyl), 2, through the diastereoselective ring opening of meso epoxides.
Introduction
We have investigated the reactions of bis(cyclopentadienyl)(tert-butylimido)zirconium complex 11 and its chiral analogue (ebthi)Zr=NAr(THF) (ebthi = ethylenebis(tetrahydroindenyl), Ar = 2,6-dimethylphenyl), 2,2 with strained heterocycles. Two different modes of reactivity were observed, depending on the presence or absence of β-hydrogens, where the β-hydrogen is defined as being located two bonds away from the heterocyclic ring. Strained heterocycles without accessible β-hydrogens participate in insertion chemistry of the C–X bond, resulting in the formation of 1,2-aminoalkoxides (X = O) and 1,2-diamides (X = N) complexed to zirconium.3 Protonation of the 1,2-aminoalkoxide complexes releases the corresponding 1,2-amino alcohol. Use of early transition metals as Lewis acids to promote the opening of epoxides by amines and amine equivalents is an active area of research,4–6 due to the importance of the amino alcohol products in biologically active compounds and as ligands for other transition-metal-mediated reactions.7,8 Herein, we describe a mechanistically distinct pathway for the opening of epoxides by amine equivalents, in which imido complexes 1 and 2 act as both the Lewis acid and the nucleophilic amine source. This bifunctional reactivity allows for high levels of regio- and stereoselectivity; in most cases, only one of several possible products is obtained. The development of metal complexes that exhibit bifunctional reactivity is an active area of research for the design of new and highly chemo- and stereoselective reactions.9–12 In contrast, strained heterocycles with accessible β-hydrogens were found to participate in elimination reactions, resulting in allylic alkoxide complexes. The elimination reaction of 2-methyl-2-propene oxide with imido complex 1 was reported by our group previously.13 We now report the full details of our original reports, including mechanistic investigations and expansion of this reaction to include aziridines, episulfides, and additional epoxide substrates.
Results and Discussion
Reactions of Heterocycles without Accessible β-Hydrogens
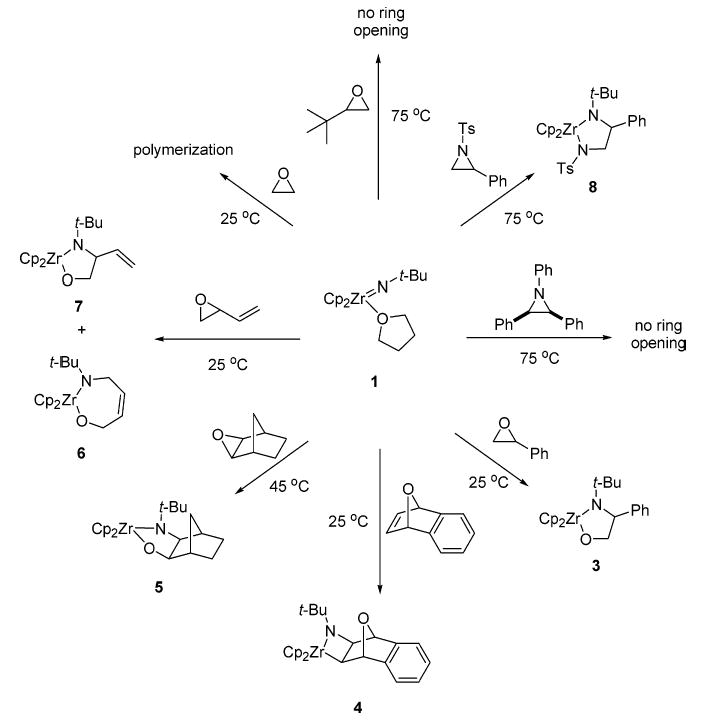
Imido complex 1 reacted cleanly with a variety of strained heterocycles without accessible β-hydrogens. Scheme 1 shows a summary of the reactivity of 1 with substrates of this class. In initial studies, we found that complex 1 reacted with styrene oxide through a highly regioselective ring opening, in 30 min at ambient temperature, to give the orange azaoxazircona-cyclopentane 3 (95% NMR yield, 44% isolated). Further insight into the pathway for reaction of the imido moiety with styrene oxide was gained by examining the reaction of the chiral analogue, (rac)-2, with (S)-styrene oxide. This ring opening proceeded with similar regio-chemistry, to give product (rac)-9 (83%) (eq 1). The structure of product 9 was confirmed by single-crystal X-ray diffraction (Figure 1). The Zr–O and Zr–N bond distances (1.966(3) and 2.112(3) Å, respectively) and the O–Zr–N bond angle (80.27(8)°) were similar to those of the other crystallographically characterized complexes synthesized in this study (vide infra). A comparison of selected bond lengths and angles can be found in Tables 2 and 3. The observed regio- and stereochemistries of 3 and 9 provided our first insight into the mechanism of this transformation. Formation of products containing a new N–C bond at the benzylic carbon was consistent with the reaction proceeding through a stepwise pathway that favored epoxide opening at the benzylic site. Furthermore, (rac)-9 was formed as a single diastereomer (R,R,R/S,S,S) (83% isolated yield), which implicated a pathway that allowed for scrambling of the original (S)-styrene oxide stereocenter. We considered both heterolytic and homolytic possibilities for this process: (a) ring opening to form a benzylic carbocation (10a), followed by trapping of the carbocation by the anionic imido ligand, and (b) ring opening to form a benzylic radical (10b), followed by trapping of the benzylic radical by the imido-based radical (Scheme 2). In our initial communication,3 we hypothesized that the reaction proceeded by pathway (a). However, since the neighboring phenyl group in intermediates 10a and 10b could help stabilize a charged or a radical species, further experimentation was needed to examine this issue.
Scheme 1.
Reactivity of 1 with Strained Heterocycles Lacking Accessible β-Hydrogens
Figure 1.

ORTEP diagram of 9.
Table 2.
Comparison of Selected Bond Distances (Å)
| complex | ||||
|---|---|---|---|---|
| 4 | Zr–N | 2.035(6) | ||
| 5 | Zr–N | 2.066(5) | Zr–O | 1.972(4) |
| 8 | Zr–N1 | 2.080(4) | Zr–N2 | 2.174(4) |
| 9 | Zr–N | 2.112(3) | Zr–O | 1.966(3) |
| 14 | Zr–N | 2.118(4) | Zr–O | 2.011(3) |
| 23 | Zr–N | 1.836(3) | Zr–O | 2.265(3) |
Table 3.
Comparison of Selected Bond Angles (deg)
| complex | ||
|---|---|---|
| 4 | C–Zr–N | 72.7(3) |
| 5 | O–Zr–N | 80.1(2) |
| 8 | N1–Zr–N2 | 78.2(2) |
| 9 | O–Zr–N | 80.27(8) |
| 14 | O–Zr–N | 78.4(1) |
| 23 | O–Zr–N | 96.8(1) |
Scheme 2.

Proposed Mechanism, Involving a Diradical or a Zwitterionic Intermediate
(1).

Substituent Effects
A Hammett study was conducted to determine the character of the intermediates (zwitterionic or diradical) in the stepwise process. The relative reaction rates of para-substituted styrene oxide derivatives were assessed via competition experiments in which a solution of imido complex 1 was added to a rapidly stirred solution of 10.0 equiv of the substituted styrene oxide and 10.0 equiv of the parent styrene oxide in C6D6. These reactions were monitored by 1H NMR spectroscopy and found to be complete in less than 10 min. The product ratios did not change upon standing. The relative reaction rates determined under these conditions are shown in Table 1, column 3.
Table 1.
Relative Rates of Reaction of para-Substituted Styrene Oxides, Compared to σ+ Constants
| p-X-Ar | σ+ | relative rate 10.0 equiv epoxide | relative rate 2.0 equiv epoxide |
|---|---|---|---|
| MeO– | −0.78 | 100a | |
| F– | −0.07 | 1.0 | 1.0 |
| H– | 0 | 1 | 1 |
| Cl– | 0.11 | 0.85 | 0.80 |
| Br– | 0.15 | 0.70 | 0.73 |
| CF3– | 0.61 | 0.15 | 0.15 |
Reaction with 20.0 equiv of styrene oxide and 2.0 equiv of p-methoxystyrene oxide.
A significantly better fit was obtained when the relative reaction rates were plotted against σ+14 than against σp, consistent with a direct resonance interaction between the para position on the aromatic ring and the reacting center. When electron-withdrawing (p-Cl, p-Br, p-CF3), neutral (p-F), and electron-donating substituents (p-OMe) were examined, a moderately good fit leading to a ρ+-value of −2.2 was obtained (Figure 2). This low absolute value of ρ+ contrasts with the higher absolute value of ρ+ observed for the Brønsted-acid-catalyzed aqueous solvolysis of para-substituted styrene oxides (−4.2).15 Therefore, the zirconocene imido-mediated opening of epoxides is significantly less sensitive to substituent effects than an epoxide-opening reaction known to proceed through a benzylic cation. However, a conceptually similar set of radical reactions do not give rise to viable Hammett correlations. Ingold and coworkers performed a computational study on a series of substituted toluene compounds (4-Y-C6H4CH2X, X = H, Br, Cl, F) and report the complete absence of a Hammett correlation between the bond dissociation enthalpies of C–X and the relevant substituent constants (either σ or σ+).16 An elegant review of this topic exists, which provides analysis and summary of both experimental and theoretical results in this area.16 For this reason, the results of our Hammett study are most consistent with the development of significant charge separation in the transition state leading to formation of the intermediates, and therefore with the intermediates themselves having zwitterionic character, despite the lower absolute value of ρ+ observed in this reaction than in the reported solvolysis reaction.
Figure 2.

Hammett study showing correlation between log(k/ko) and σ+.
We considered the possibility that, due to the presence of a large excess of epoxide, the THF-bound complex might no longer be the ground state of the system. Instead, an epoxide-bound complex might be the ground state. The competition experiments using 20.0 equiv of epoxide were too rapid to observe starting materials at room temperature. Therefore, the nature of the ground state was investigated by low-temperature 1H NMR spectroscopy. When 20.0 equiv of styrene oxide was added to imido complex 1 at −20 °C, complete displacement of THF by the epoxide was observed. In contrast, monitoring the reaction of imido complex 1 with 2.0 equiv of p-trifluoromethylstyrene oxide and 2.0 equiv of styrene oxide at partial conversion showed that, under these conditions, the ground state remained the THF adduct. We next examined whether this difference affected the outcome of the competition experiments. The competition experiment was repeated for the series of substrates using 2.0 equiv of each epoxide (Table 1, column 4). Relative rates were found to be identical in the experiments that employed 2.0 equiv of each epoxide and 10.0 equiv of each epoxide, within the error of the measurement (±10%). These identical relative rates imply that the reaction operates under Curtin–Hammett kinetics; that is, our measurement reflects ΔΔG‡, the difference in energy between the transition states for the opening of styrene oxide and substituted styrene oxide and does not reflect the energy difference between ground states. This case would be expected for rapid and reversible binding of each epoxide, prior to rate-determining ring opening. Activation parameters supporting the rapid and reversible binding of Lewis bases to imido complex 1 at ambient temperature were recently reported by our group.17
Stereochemistry of Metallacycle Formation
Further support for the proposed stepwise mechanism was obtained through examination of the reactivity of 1 with cis- and trans-stilbene oxides in separate reactions. In this case, 1H NMR analysis of the reaction mixture showed formation of the same metallacycle, independent of the stereochemistry of the starting material, consistent with ring opening to form cis (11) and trans (12) zwitterions, which interconverted rapidly relative to trapping by the imido group (Scheme 3). Furthermore, performing the reaction in the presence of excess stilbene oxide showed that the unreacted epoxide did not isomerize under the reaction conditions. This supports a mechanism that proceeds through rate-limiting ring opening, since once the epoxide opens, it does not revert to a bound epoxide, which would be expected to rapidly dissociate from the metal. We initially assigned product 13 as the trans isomer, since we theorized that the lower steric interaction between the adjacent phenyl groups would make the trans transition state more favorable. This stereochemical assignment was later confirmed for the ebthi system (vide infra).
Scheme 3.

Mechanism of Reaction of cis- and trans-Stilbene Oxides with 1, Showing Curtin–Hammett Kinetics
The reaction of chiral ebthi complex (rac)-2 with cis-or trans-stilbene oxide was also studied (eq 2). In theory, this reaction could lead to the generation of four different diastereomers, arising from both the planar chirality of the ebthi ligand and from the benzylic stereocenters. A 1H NMR spectrum of the crude reaction mixture showed that product (rac)-14 was formed as the same single diastereomer independent of the starting stereochemistry of the epoxide (eq 2). An X-ray diffraction study revealed that (rac)-14 was the R,R,R,R/S,S,S,S racemate, confirming the trans stereochemistry (Figure 3). This offered support for our initial hypotheses that 13 was the trans isomer (vide supra). The high diastereoselectivity of this reaction suggested the possibility of a kinetic resolution of the racemic imido compound (rac)-2 by readily available enantiopure trans-stilbene oxide.18 Addition of 0.55 equiv of (R,R)-trans-stilbene oxide (>98% ee) to a solution of 1.0 equiv of (rac)-2 in C6H6 at ambient temperature resulted in a modest product diastereoselectivity of 3:1, corresponding to 50% ee of unreacted 2.19
Figure 3.

ORTEP diagram of 14 (obtained as the RRRR/SSSS racemate).
(2).

Vinylcyclopropane Oxide
In an effort to gain further support for the presence of a zwitterionic intermediate, we investigated the ring opening of vinylcyclopropane oxide20 (15) with imido complex 1. We reasoned that if the reaction proceeds via a radical pathway, we should observe a product corresponding to ring opening of the cyclopropylcarbinyl radical.21 Unfortunately, the reaction of 1 with 15 led to a complex mixture of products as determined by multiple Cp resonances in the 1H NMR spectrum. Treatment of N-TBS imido complex 1613 (TBS = tert-butyldimethylsilyl), however, with epoxide 15 led to formation of a single product, 17, that corresponded to simple ring opening of the epoxide (Scheme 4). Given the fast rate of cyclopropylcarbinyl radical ring opening,22 failure to observe the corresponding cyclopropane ring-opened product 18 suggests again that the reaction proceeds via a zwitterionic mechanism (path a) as opposed to a radical mechanism (path b).23
Scheme 4.

Reaction of N-TBS Imido Complex 16 with Vinylcyclopropane Oxide
Reactions with Other Strained Heterocyclic Systems
We next examined the reaction of butadiene monoepoxide with 1 (Scheme 5). Stepwise ring opening would be expected to form allylic intermediate 19, in which cationic character would be distributed between the two terminal carbon atoms of the allylic system. Trapping to form a five-membered ring or to form a seven-membered ring would then produce metallacycle 6 or 7, respectively. When 1 was added to a solution of butadiene monoepoxide in C6D6 at ambient temperature, the solution turned bright orange immediately. 1H NMR spectroscopy showed formation of two metallacycle isomers, assigned as 6 and 7 by proton coupling patterns (43% and 41% NMR yields, respectively), and the absence (6) or presence (7) of diastereotopic cyclopentadienyl ligands. Metallacycle 6 exhibited fluxional behavior that resulted in line broadening in the 1H NMR spectrum at ambient temperature, possibly originating from different, equilibrating ring conformations. Warming the sample to 70 °C resulted in a sharpening of the resonances. Protonolysis by benzoic acid produced zirconocene dibenzoate 20 as well as the two previously unknown amino alcohols 21 and 22, in a ratio identical to the ratio of metallacycle 6 to 7. The structures of 21 and 22 were assigned by HRMS and 1H NMR spectroscopy.
Scheme 5.

Imidozirconium-Mediated Opening of Butadiene Monoepoxide
Although epoxide complexes with styrene oxide, stilbene oxide, or butadiene monoepoxide bound to zirconium were not observed by 1H NMR spectroscopy in the reactions with 1 or 2, we presumed that these complexes were intermediates formed along the reaction pathway. Therefore, we sought a system with a higher barrier for ring opening that would allow for observation of an epoxide complex. A C6D6 solution of 1 and exo-norbornene oxide exhibited a broad 1H NMR spectrum, consistent with exchange of the THF and exo-norbornene oxide ligands at ambient temperature (Scheme 6). Removal of the THF in vacuo from this solution produced exo-norbornene oxide complex 23 quantitatively, as observed by 1H NMR spectroscopy. Crystals of 23 were obtained at low temperature and characterized by X-ray diffraction (Figure 4). The long Zr–O bond (2.265(3) Å) is consistent with the presence of a dative interaction between the epoxide and the zirconium center. The short Zr–N bond (1.836(3) Å) indicates that the double-bond character of the imido moiety remains and is similar to the Zr–N bond length in 1 (1.826(4) Å).1 A comparison of selected bond lengths and angles for the complexes reported in this study can be found in Tables 2 and 3. This is one of the few examples of proposed intermediate epoxide complexes that have been isolated and structurally characterized.24,25
Scheme 6.

Proposed Mechanism for the Opening of exo-Norbornene Oxide by 1
Figure 4.

ORTEP diagram of epoxide complex 23.
Heating either a solution of 1 and exo-norbornene oxide (45 °C for 24 h) or a solution of 23 (45 °C for 5 h) yielded cis-metallacycle 5 (94% NMR yield, 46% isolated). The stereochemistry of 5 was confirmed by X-ray diffraction (Figure 5). The cis stereochemistry of 5 cannot result from “back side” SN2 displacement of the epoxide oxygen and therefore is consistent with the initial conversion of 23 to an intermediate that allows for subsequent “front side” attack. This intermediate may be the carbon-bridged (“nonclassical”) zwitterion 24 or the nonbridged (“classical”) diradical.26 Interestingly, no trapping product 25, which would result from the rearranged norbornyl framework, is observed. This lack of rearrangement seems somewhat inconsistent with the zwitterionic hypothesis. However, the possibility that closure to form 5 from zwitterion 24 proceeds preferentially through a kinetically favored five-membered ring transition state instead of trapping at the adjacent carbon atom, which would require a six-membered ring transition state, cannot be ruled out.
Figure 5.
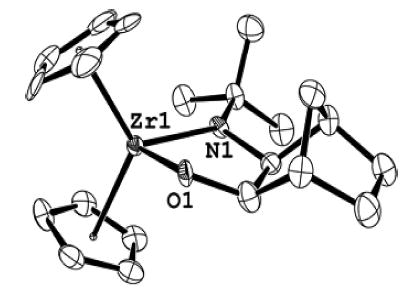
ORTEP diagram of cis-metallacycle 5.
Kinetic studies of the conversion of 23 to 5 were performed, and the reaction was found to exhibit first-order kinetics with respect to 23 (k = 0.017 min−1 (59.8 °C), k = 0.051 min−1 (69.8 °C), k = 0.084 min−1 (79.8 °C), k = 0.22 min−1 (89.9 °C)).27 The rate was zero-order in concentration of excess epoxide, consistent with unimolecular conversion of 23 to 5. Rates measured over this temperature range gave activation parameters ΔH‡ = 19 ± 2 kcal mol−1 and ΔS‡ = −18 ± 4 cal mol−1 K−1.
When 1 was treated with 1,2-epoxy-3,3-dimethylbutane (Scheme 1), 1H NMR spectroscopy showed line broadening at ambient temperature, consistent with establishment of an equilibrium between bound THF and bound epoxide complexes, similar to the equilibrium between 1 and 23. Heating this mixture to 75 °C, however, did not produce ring-opened product. This lack of reactivity corroborated the requirement for substantial resonance stabilization from a group at the α position to facilitate ring opening and therefore provided indirect support for resonance-stabilized (“nonclassical”) zwitterion 24 over the (“classical”) diradical structure.
Addition of ethylene oxide to 1 in C6D6 resulted in rapid precipitation of a white solid, consistent with polymerization of the epoxide. 1H NMR spectroscopy performed on the remaining solution showed the formation of an intractable mixture of products, implying that ethylene oxide accessed different reactivity than the other epoxides in this study.
The reaction of imido complex 1 with 7-oxabenzonor-bornadiene, which had been shown previously to undergo metal-mediated ring opening and subsequent trapping by nucleophiles,28 was investigated next (Scheme 7). This substrate, however, reacted with imido complex 1 to form the [2 + 2] cycloaddition product, 4, in quantitative NMR yield. Removal of the solvent in vacuo produced analytically pure material (87%). Complex 4 was characterized by X-ray crystallography (Figure 6). The X-ray crystal structure verified that cycloaddition favors the exo face of the olefin exclusively, which was observed previously in the reaction of 1 with norbornene.13 Formation of 4 was found to be irreversible up to 75 °C, as evidenced by the stability of 4 in the presence of styrene oxide. The stability of 4 contrasted sharply with the previously reported observation that imido complex 1 formed highly labile [2 + 2] cyclcoaddition products with ethylene and norbornene.13 The replacement of the methylene bridge of norbornene with an oxygen therefore caused a marked shift in reactivity, perhaps through the increase in strain of the bicyclic ring system, which would destabilize the π-bond of the starting heterocycle.
Scheme 7.

Reaction of 1 with 7-Oxabenzonorbornadiene
Figure 6.

ORTEP diagram of 4.
The reactivity of imido complex 1 with aziridines was highly dependent on the nature of the substituent on the nitrogen. For example, N-tosyl-2-phenylaziridine reacted with 1 over 2 days at 75 °C to give the magenta diazazirconacyclopentane 8 (>95% NMR yield, 84% isolated; eq 3), while cis-1,2,3-triphenylaziridine did not undergo ring opening. This discrepant behavior can be explained by the better leaving group ability of tosylamides over phenylamides. The structure of 8 was confirmed by single-crystal X-ray diffraction (Figure 7). The bond between the zirconium atom and the tert-butylamide group was found to be shorter (2.080 Å) than the bond between the zirconium and the tosylamide (2.174 Å). This is consistent with partial donation of the lone pair on the tert-butylamide to the electron-deficient zirconium center, creating multiple bond character. The lone pair on the tosylamide is less able to donate to the zirconium center, due to interaction with the sulfonyl group. Importantly, the observed regiochemistry of 8 supports a zwitterionic ring-opening mechanism analogous to the one we have proposed for the reactions of 1 with epoxides.
Figure 7.
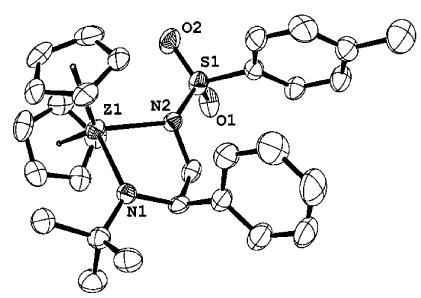
ORTEP diagram of 8.
(3).

Reactions of Strained Heterocycles with Accessible β-Hydrogens
Imido complex 1 exhibited clean elimination reactivity with a variety of epoxides with accessible β-hydrogens. Scheme 8 shows a summary of the reactivity of 1 with substrates of this class; the individual reactions will be described in detail in the subsequent paragraphs. Propylene oxide underwent clean ring opening with 1 in C6D6 at ambient temperature to give the corresponding allylic alkoxy amide 26. Removal of the solvent produced analytically pure 26 (>95%). Similarly, cyclohexene oxide reacted with 1 to give complex 27 (90% NMR yield, 37% isolated), and cyclooctene oxide reacted with 1 to give complex 28 (> 95% NMR yield) as a mixture of cis and trans isomers (88:12). Removal of the solvent produced analytically pure 28 (>95%). Due to the complexity of the 1H NMR spectrum of the substrate, the isomer assignment was not determined. Instead, the diastereoselectivity of the elimination reaction was investigated in detail through the reaction of 1 with the simpler substrate, 1,2-butene oxide.
Scheme 8.

Reactivity of 1 with Strained Heterocycles with Accessible β-Hydrogens
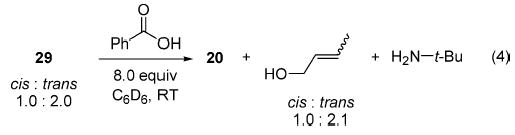
When a solution of 1,2-butene oxide in C6D6 was added to imido complex 1 at ambient temperature, 1H NMR spectroscopy showed the formation of two isomeric zirconocene products in the ratio 2.0:1.0 (29). Identification of these products was facilitated by cleavage of the organic fragments from the metal center with 8 equiv of benzoic acid. This produced the zirconocene dibenzoate complex 20, tert-butylamine, and cis- and trans-crotyl alcohol (>95% NMR yield; eq 4). Protic cleavage did not change the ratio of isomers significantly (2.1: 1.0). Comparison with a commercially available mixture of cis- and trans-crotyl alcohol by 1H NMR spectroscopy and by 2-D TOCSY NMR spectroscopy showed that the major isomer corresponded to trans-crotyl alcohol and the minor isomer to cis-crotyl alcohol. Therefore, imido complex 1 exhibited a modest preference for removal of the proton corresponding to formation of the trans olefin.
(4).
The substrate trans-phenylpropene oxide provided an opportunity to examine the preference of imido complex 1 to participate in insertion (vide supra) or elimination reactions, via an intramolecular competition experiment (Scheme 9). The addition of trans-phenylpropylene oxide to 1 in C6D6 at ambient temperature resulted in the rapid (<30 min) formation of a single product 30 (82% isolated) corresponding to an elimination reaction. Product 32, which would form via insertion, was not observed. Therefore, the elimination pathway has a significantly lower barrier than the insertion pathway. This has important consequences for the mechanism of the elimination reaction. Since a reaction that proceeds via significant charge buildup or radical character would be expected to favor reactivity at the benzylic position, the fact that elimination at a saturated center proceeds more rapidly suggests a concerted process for this reaction (Scheme 10).
Scheme 9.
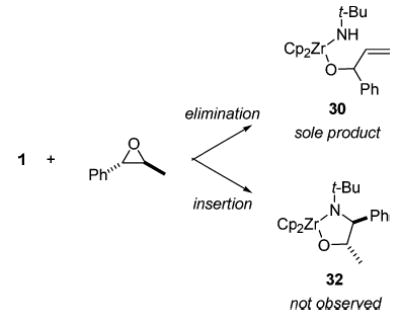
Intramolecular Competition Experiment, Showing Preference for Elimination Reactivity
Scheme 10.

Proposed Concerted Pathway for Elimination Reaction of Propene Oxide
The potential for chirality transfer from the planar-chiral ebthi ligand set of (rac)-2 was investigated in the ring-opening desymmetrization reaction of the meso epoxide cis-2,3-butene oxide (eq 5). This ring-opening reaction could theoretically result in the formation of two possible diastereomers due to the chirality of the ligand and that of the newly formed carbon-based stereocenter. This is of interest due to the limited number of methods currently available to enantioselectively deprotonate meso epoxides.29 When 1 equiv of cis-2,3-butene oxide was added to 1 equiv of (rac)-2, a 1H NMR spectrum showed that the reaction proceeded with a high degree of diastereoselectivity (>95:5). This established the feasibility of using the ebthi ligand for effective chirality transfer in the elimination reactions of zirconium imido complexes.
(5).

Propene sulfide also underwent an ambient-temperature elimination reaction with imido complex 1, producing allylic sulfide complex 31 (72% NMR yield; Scheme 11). Heating complex 1 in the presence of excess propylene sulfide at 75 °C for 8 h resulted in catalytic desulfurization of the substrate, producing propene (510% NMR yield, relative to 1) and multiple other products, both soluble and insoluble in the reaction medium. Mass spectral analysis of the soluble products indicated the presence of mass fragments corresponding to oligomerization of the starting material. Previous reports in the literature suggest that episulfides are readily polymerized when treated with a variety of Lewis acids and readily desulfurized to yield the corresponding olefin.30 Therefore, it seems unlikely that catalysis specifically requires the presence of complex 1 or 31, but rather may involve minor amounts of uncharacterized zirconocene products. Characterization of 31 was facilitated by protic cleavage with benzoic acid, which produced 20, tert-butylamine, and allyl sufide (>95% NMR yield). In contrast to the insertion chemistry observed with 1 (vide supra), N-tosylaziridines bearing accessible β-hydrogens did not participate in ring-opening elimination reactions.
Scheme 11.
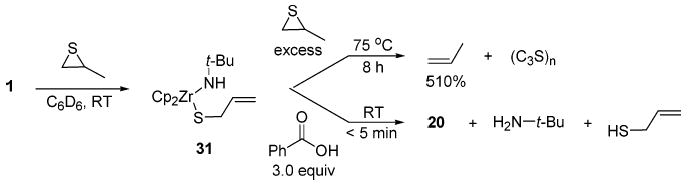
Ring Opening of Propene Sulfide and Subsequent Reactions
Conclusion
Imidozirconocene complexes 1 and 2 underwent highly regio- and stereoselective insertion and elimination reactions with a variety of epoxides. This reactivity can be extended to properly substituted aziridines and episulfides. Appropriately substituted substrate probes were used to investigate the mechanisms of these reactions and have provided strong evidence for a stepwise reaction pathway for the ring-opening reaction of heterocycles without β-hydrogens. This mechanism was shown to involve rate-determining ring opening, leading to stereochemically labile intermediates. A good correlation between reaction rate and the Hammett substituent constants (σ+) suggested that the intermediates in this process possessed substantial zwitterionic character. The absence of cyclopropyl ring opening when vinylcyclopropane oxide reacted with imido complex 16 further supported the action of a zwitterionic mechanism. In contrast, the results of substrate probe experiments were most consistent with the ring-opening reaction of heterocycles with accessible β-hydrogens proceeding through a concerted pathway. The results demonstrate the bifunctional importance of the Lewis-acidity of the zirconium center and the nucleophilicity and basicity of the imido moiety in promoting these ring-opening reactions.
Experimental Section
General Methods
All manipulations were carried out under inert atmosphere, using a nitrogen-filled recirculating glovebox or standard Schlenk line techniques. Solvents were degassed using three freeze–pump–thaw cycles and dried over 3 Å activated molecular sieves (benzene-d6, toluene-d8), passed through an activated alumina column and sparged with nitrogen (benzene, toluene, pentane, THF, ether),31 or distilled from purple Na/benzophenone ketyl (THF, ether), prior to use. Bis(cyclopentadienyl)(tert-butylimido) zirconium complex 1,1 (ebthi)Zr=NAr(THF) (ebthi = ethylenebis(tetrahydroindenyl, Ar = 2,6-dimethylphenyl), 2,2 bis(cyclopentadienyl)(tert-butyldimethylsilylimido) zirconium complex 16,13 and vinylcyclopropane oxide20 were synthesized according to previously published procedures. Liquid and low-melting-solid substrates were degassed using three freeze–pump–thaw cycles and vacuum transferred from CaH2 or distilled from CaH2 prior to use. p-Methoxystyrene oxide was synthesized by dimethyldioxirane (DMDO) epoxidation of p-methoxystyrene.32
All 1H NMR and 13C NMR spectra were recorded on Bruker AV300, AV400, and DRX500 spectrometers. 1H NMR and 13C-{1H} NMR chemical shifts (δ) are reported in parts per million (ppm) downfield of tetramethysilane and referenced to the residual protiated solvent peak. When listed, 13C{1H} NMR peak assignments were determined by DEPT 135 experiments. NMR yields were measured by integration of product resonances in single-scan 1H NMR spectra, relative to integration of THF resonances. X-ray structural analyses were performed by Dr. Fred Hollander and Dr. Allen Oliver in the University of California, Berkeley CHEXRAY facility.
Metallacycle 3
A solution of styrene oxide (28 mg, 0.23 mmol) in benzene (0.2 mL) was added to a solution of imido complex 1 (75 mg, 0.21 mmol) in benzene (0.3 mL), producing an orange solution. To remove the benzene, the volatile materials were removed in vacuo to give a viscous orange oil, which was dissolved in ether (0.2 mL) to form a clear orange solution, and again the volatile materials were removed in vacuo to give an orange oil. The oil was again taken up in ether (0.75 mL) to form a cloudy solution and cooled to −35 °C. After 2 days, a red solid had precipitated. The mother liquor was decanted, the solid was washed quickly with cold pentane, and the volatile materials were removed in vacuo to leave the red solid (38 mg, 44%). 1H NMR (300 MHz, C6D6): δ 7.63 (d, J = 7.0 Hz, 2H), 7.30 (t, J = 7.8 Hz, 2H), 7.16 (m, 1H), 6.24 (s, 5H), 6.10 (s, 5H), 4.80 (dd, J = 10.2, 5.3 Hz, 1H), 4.46 (d, J = 5.3 Hz, 1H) 3.99 (d, J = 10.2 Hz, 1H), 0.80 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 151.4, 127.6, 127.0, 125.9, 113.6, 112.1, 78.6, 75.4, 55.8, 30.3. IR (Nujol): 1352, 1196, 1071, 1059, 1043, 1012, 965, 768, 761, 722, 707, 598, 552 (s), 529 (s), 508 (s) cm−1. Anal. Calcd for C22H27NOZr: C, 64.03; H, 6.59; N, 3.39. Found: C, 64.43; H, 6.67; N, 3.40.
p-Trifluoromethylstyrene Oxide Metallacycle
A solution of p-trifluoromethylstyrene oxide (16 mg, 0.082 mmol) in benzene (0.3 mL) was added to imido complex 1 (30 mg, 0.082 mmol). After 4 days, the solution had turned bright orange, and the volatile materials were removed in vacuo, producing an orange gum. The gum was taken up in pentane (6 drops) to form a cloudy orange solution, which was cooled to −35 °C. After 2 days, clusters of pale orange crystals had formed. The mother liquor was removed by pipet, and the residual solvent was removed from the crystals in vacuo, yielding 32 mg (81%). 1H NMR (500 MHz, C6D6): δ 7.51 (m, 4H), 6.18 (s, 5H), 6.08 (s, 5H), 4.73 (dd, J = 10, 5.0 Hz, 1H), 4.31 (d, J = 5.5 Hz, 1H), 3.79 (d, J = 10 Hz, 1H), 0.71 (s, 9H). 13C{1H} NMR (125 MHz, C6D6): δ 156.1, 126.2, 124.6 (CF3), 113.7, 112.3, 78.0, 75.1, 55.8, 30.2. Anal. Calcd for C23H26F3NOZr: C, 57.47; H, 5.45; N, 2.91. Found: C, 57.47; H, 5.27; N, 2.88.
p-Bromostyrene Oxide Metallacycle
A solution of p-bromostyrene oxide (16 mg, 0.082 mmol) in benzene (0.3 mL) was added to imido complex 1 (30 mg, 0.082 mmol). The solution turned bright orange over 10 min. After 2 days, the volatile materials were removed in vacuo, producing an orange gum. The gum was taken up in ether (0.5 mL), producing a cloudy orange solution. Pentane vapor diffusion was initiated at −35 °C. After 6 days, orange crystals were present above the solvent line and in the pale yellow mother liquor. The mother liquor was removed by pipet, and the crystals were washed quickly with cold pentane. The residual solvent was removed in vacuo, yielding 18 mg (45%). 1H NMR (300 MHz, C6D6): δ 7.35 (m, 4H), 6.17 (s, 5H), 6.09 (s, 5H), 4.72 (dd, J = 9.5, 4.8 Hz, 1H), 4.26 (d, J = 5.1 Hz), 3.81 (d, J = 10.2 Hz, 1H), 0.72 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 150.7, 130.7, 128.8, 119.5, 113.7, 112.3, 77.8, 75.3, 55.8, 34.1, 25.5. Anal. Calcd for C22H26BrNOZr: C, 53.75; H, 5.33; N, 2.85. Found: C, 53.92; H, 5.18; N, 2.77.
p-Chlorostyrene Oxide Metallacycle
A solution of p-chlorostyrene oxide (23.4 mg, 0.151 mmol) in benzene (1.5 mL) was added to imido complex 1 (50.0 mg, 0.137 mmol). The solution turned bright orange over 10 min. After 1 day, the volatile materials were removed in vacuo, producing an orange gum. The gum was taken up in ether (1.5 mL), producing a cloudy orange solution. Pentane vapor diffusion was initiated at −35 °C. After 9 days, orange crystals were present above the solvent line and in the pale yellow mother liquor. Some fine white powder (presumably decomposition products) had settled to the bottom of the solution. The mother liquor was removed by pipet, concurrently removing the fine white powder as a suspension. The crystals were washed quickly with cold pentane and the residual solvent was removed in vacuo, yielding 18 mg (30%). 1H NMR (500 MHz, C6D6): δ 7.37 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 6.17 (s, 5H), 6.08 (s, 5H), 4.73 (dd, J = 10.5, 5.0 Hz, 1H), 4.29 (d, J = 5.5 Hz, 1H), 3.83 (d, J = 10 Hz, 1H), 0.74 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 150.4 (C), 131.8 (C), 128.4 (CH), 127.8 (CH), 113.7 (CH), 112.3 (CH), 78.0 (CH), 75.6 (CH2), 56.1 (CH), 30.6 (CH3). Anal. Calcd for C22H26ClNOZr: C, 59.10; H, 5.86; N, 3.13. Found: C, 58.99; H, 5.93; N, 2.97.
p-Fluorostyrene Oxide Metallacycle
A solution of p-fluorostyrene oxide (20.8 mg, 0.151 mmol) in benzene (1.5 mL) was added to imido complex 1 (50.0 mg, 0.137 mmol). The solution turned bright orange over 10 min. After 1 day, the volatile materials were removed in vacuo, producing an orange gum. The gum was taken up in ether (1.5 mL), producing a cloudy orange solution. Pentane vapor diffusion was initiated at −35 °C. After 6 days, red crystals were present above the solvent line and in the mother liquor. Some fine white powder (presumably decomposition products) had settled to the bottom of the solution. The mother liquor was removed by pipet, concurrently removing the fine white powder as a suspension. The crystals were washed quickly with cold pentane, and the residual solvent was removed in vacuo, yielding 36.5 mg (62%). 1H NMR (500 MHz, C6D6): δ 7.42 (dd, J = 8.0, 2.5 Hz, 2H), 6.93 (t, J = 9 Hz, 2H), 6.19 (s, 5H), 6.09 (s, 5H), 4.75 (dd, J = 10.0, 5.0 Hz, 1H), 4.35 (d, J = 5.0 Hz, 1H), 3.87 (d, J = 10.0 Hz, 1H), 0.75 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 162.1 (d, JC–F = 220 Hz, C), 147.4 (d, JC–F = 3.8 Hz, C), 128.6 (d, JC–F = 7.5, CH), 114.6 (d, JC–F = 20 Hz, CH), 113.9 (CH), 112.5 (CH), 78.0 (CH), 75.8 (CH2), 56.1 (C), 30.6 (CH3). Anal. Calcd for C22H26FNOZr: C, 61.35; H, 6.09; N, 3.25. Found: C, 61.27; H, 6.29; N, 3.45.
p-Methoxystyrene Oxide Metallacycle
A solution of p-methoxystyrene oxide (28.2 mg, 0.188 mmol) in benzene (3 mL) was added to imido complex 1 (65.4 mg, 0.188 mmol). The solution turned bright orange with dissolution of 1. After 15 min, the volatile materials were removed in vacuo, producing an orange gum. The complex decomposed upon attempted purification. 1H NMR (500 MHz, C6D6): δ 7.54 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 6.26 (s, 5H), 6.09 (s, 5H), 4.81 (dd, J = 10.0, 5.0 Hz, 1H), 4.46 (d, J = 5.0 Hz, 1H), 4.02 (d, J = 10 Hz, 1H), 0.84 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 158.8 (C), 143.6 (C), 128.3 (CH), 113.9 (CH), 112.5 (CH), 78.3 (CH), 76.1 (CH2), 56.2 (C), 54.8 (CH3), 30.7 (CH3).
Hammett Study General Procedures. 10.0 Equiv of Epoxide
A solution of imido complex 1 (10.0 mg, 0.0274 mmol) in benzene-d6 (0.5 mL) was added dropwise to a rapidly stirred solution of substituted styrene oxide (0.274 mmol, 10.0 equiv) and styrene oxide (0.274 mmol, 10.0 equiv) in benzene-d6 (0.7 mL). The solution turned bright orange upon addition. The reaction was monitored by 1H NMR spectroscopy and found to be complete in <30 min. The product ratios did not change upon standing.
2.0 Equiv of Epoxide
A solution of imido complex 1 (10 mg, 0.027 mmol) in benzene-d6 (0.5 mL) was added dropwise to a rapidly stirred solution of substituted styrene oxide (0.055 mmol, 2.0 equiv) and styrene oxide (0.055 mmol, 2.0 equiv) in benzene-d6 (0.7 mL). The solution turned bright orange over 5 min. The reaction was monitored by 1H NMR spectroscopy and found to be complete in 2–8 h. The product ratios did not change upon standing.
Low-Temperature 1H NMR Study of THF Displacement by Excess Epoxide
In the glovebox, a NMR tube was charged with a solution of imido complex 1 (15 mg, 0.041 mmol) in toluene-d8 and capped with a rubber septum. A 250 μL gastight syringe was charged with styrene oxide (94 μL, 0.82 mmol, 20 equiv), and the tip was protected from air by insertion into a plastic stopper. Both items were removed from the box.
The NMR tube was placed in a −78 °C bath for 10 min, at which time styrene oxide was injected into the solution through the septum. After 2 min, the NMR tube was removed from the bath, the viscous solution was quickly shaken, and the tube was dropped into a precooled (−20 °C) spectrometer probe. A 1H NMR spectrum obtained at this temperature showed complete displacement of THF by styrene oxide (no formation of metallacycle product 3 was observed).
Metallacycle 4
7-Oxabenzonorbornadiene (19.7 mg, 0.137 mmol) was dissolved in benzene (1.0 mL) and added to a solution of imido complex 1 (50.0 mg, 0.137 mmol) in benzene (1.0 mL), producing a dark orange solution. The solvent was removed in vacuo, to give an orange glass (52 mg, 87%). 1H NMR (500 MHz, C6D6): δ 7.20 (d, J = 6.0 Hz, 1H), 7.15 (m, 1H), 7.12 (dt, J = 7.0, 1.0 Hz), 7.04 (dt, J = 7.0, 1.0 Hz, 1H), 6.04 (s, 5H), 5.56 (s, 5H), 5.36 (s, 1H), 5.25 (s, 1H), 2.67 (d, J = 7.5 Hz, 1H), 2.17 (d, J = 7.0 Hz, 1H). 13C{1H} NMR (125 MHz C6D6): δ 150.7, 144.4, 126.3, 125.0, 111.2, 110.2, 84.6, 83.2, 67.8, 55.9, 42.9, 31.8. Satisfactory elemental analysis could not be obtained on this compound.
Procedure for Obtaining Crystals Suitable for X-ray Diffraction Analysis
7-Oxabenzonorbornadiene (19.7 mg, 0.137 mmol) was dissolved in benzene (1.0 mL) and added to a solution of imido complex 1 (50.0 mg, 0.137 mmol) in benzene (1.0 mL), producing a dark orange solution. The solvent was removed in vacuo, to give an orange powder. The powder was taken up in ether (3 mL), and pentane vapor diffusion was started at −35 °C. After 3 days, orange crystals were present along the solvent front and on the bottom of the solution. The mother liquor was removed by pipet. Satisfactory elemental analysis could not be obtained on this compound.
Metallacycle 5
A solution of exo-norbornene oxide (19 mg, 0.17 mmol) in benzene (1 mL) was added to a solution of imido complex 1 (58 mg, 0.16 mmol), in benzene (1 mL), producing a tan solution. The solution was transferred to a resealable J. Young NMR tube, sealed, and removed from the box. The NMR tube was heated at 45 °C for 3 days, and the solvent was then removed in vacuo to produce a red gum. The gum was dissolved in pentane (0.2 mL), and orange crystals suitable for X-ray diffraction analysis formed upon standing at ambient temperature for 5 days. The mother liquor was removed by pipet, and the residual solvent was removed in vacuo (30 mg, 46%). 1H NMR (300 MHz, C6D6): δ 6.13 (d, J = 16.2 Hz, 10H), 4.25 (d, J = 3.3 Hz, 1H), 3.38 (d, J = 3.3 Hz, 1H), 2.39 (d, J = 5.7 Hz, 1H), 2.24 (s, 1H), 2.14 (s, 1H), 1.30–1.11 (m, 6H), 0.96 (s, 9H). 13C{1H} NMR (125 MHz, C7D8): δ 113.2, 111.8, 83.8, 81.3, 55.2, 44.1, 42.8, 34.0, 27.0, 23.6, 30.9. Anal. Calcd for C21H29NOZr: C, 62.64; H, 7.26; N, 3.48. Found: C, 62.53; H, 7.38; N, 3.45.
Metallacycles 6 and 7
A solution of butadiene monoepoxide (42 mg, 0.60 mmol) in benzene-d6 (0.5 mL) was added to a solution of imido complex 1 (200 mg, 0.548 mmol) in benzene-d6 (1 mL), producing a bright orange solution. A 1H NMR spectrum showed formation of two metallacycle isomers, assigned as 6 and 7 via proton coupling patterns (43% and 41% NMR yields, respectively), and via the distinctive absence (6) or presence (7) of diastereotopic cyclopentadienyl ligands. Spectral information for 6: 1H NMR (300 MHz, C6D6): δ 6.09 (s, 10H), 5.37 (m, 2H), 4.69 (br, 2H), 3.45 (br, 2H), 1.00 (s, 9H). Spectral information for 7: 1H NMR (300 MHz, C6D6): δ 6.10 (s, 5H), 6.01 (s, 5H), 4.36 (dd, J = 6.0, 4.8 Hz, 1H), 3.83 (dd, J = 6.0, 4.8 Hz, 1H), 0.99 (s, 9H).
The high solubility of these complexes hampered purification; therefore, protic cleavage of the alkoxide moiety from the metal was used to corroborate the product assignments. To the crude reaction mixture containing 6 and 7 was added benzoic acid (669 mg, 5.5 mmol) in benzene (3 mL). The reaction mixture became colorless immediately. The solution was diluted with ether (3 mL) and extracted with aqueous 1 M HCl (10 mL × 3). The combined aqueous layers were back extracted with ether (7 mL × 3), and then aqueous NaOH was added until pH = 9, producing a white precipitate. The aqueous layer was extracted with ether (20 mL × 3). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo to a cloudy oil. The oil was extracted with benzene-d6 (1 mL). A 1H NMR spectrum showed amino alcohols 21 and 22 and tert-butylamine (>90% purity). Spectral information for 21: 1H NMR (300 MHz, C6D6): δ 5.82 (m, 2H), 4.21 (d, J = 5.7 Hz, 1H), 4.08 (d, J = 4.5 Hz, 1H), 3.16 (d, J = 4.5 Hz, 1H), 2.93 (d, J = 5.7 Hz, 1H), 0.95 (s, 9H). Spectral information for 22: 1H NMR (300 MHz, C6D6): δ 5.49 (m, 2H), 4.90 (m, 1H), 3.48 (m, 1H), 0.97 (s, 9H). HRMS (EI): m/z calcd (C8H17NO) 144.1388, found 144.1393 [M+].
Metallacycle 8
N-Tosyl-2-phenylaziridine (24 mg, 0.086 mmol) was dissolved in toluene-d8 (0.8 mL) and added to solid imido complex 1 (100 mg, 0.27 mmol), producing a pale tan solution. The solution was transferred to a resealable J. Young NMR tube, sealed, and removed from the box. The NMR tube was heated at 75 °C for 1 day, producing a deep purple-brown solution. A 1H NMR spectrum obtained at this time showed product 8 (98% NMR yield). The volatile materials were removed in vacuo to produce a magenta gum. The gum was taken up in ether (0.5 mL), and pentane vapor diffusion was started at −35 °C. After 3 days, deep purple crystals suitable for X-ray diffraction analysis had formed. The mother liquor was removed by pipet, and the residual solvent was removed in vacuo, yielding 42 mg of crystals. A 1H NMR spectrum showed 1 equiv of ether, and a X-ray diffraction study showed inclusion of ca. 0.5 equiv ether into the crystal lattice. An adjusted yield taking this ether into account was calculated: 38 mg of 8 (82%). 1H NMR (500 MHz, C6D6): δ 7.55 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 7.0 Hz, 2H), 6.99 (m, 3H), 6.63 (d, J = 8.0 Hz, 2H), 6.44 (s, 5H), 6.40 (s, 5H), 3.86 (d, J = 5.0 Hz, 1H), 3.78 (dd, J = 12.5. 5.0 Hz, 1H), 3.22 (d, J = 12.5 Hz, 1H), 1.91 (s, 3H), 0.63 (s. 9H). 13C{1H} NMR (125 MHz, C6D6): δ 149.9 (C), 140.2 (C), 140.1 (C), 128.7 (CH), 127.3 (CH), 126.9 (CH), 126.5 (CH), 125.5 (CH), 115.1 (CH), 114.0 (CH), 65.6 (C), 57.3 (C), 54.8 (CH2), 30.2 (CH3), 20.8 (CH3). The elemental composition is not altered significantly upon inclusion of 0.5 equiv of ether. Anal. Calcd for C29H34N2O2SZr: C, 61.55; H, 6.06; N, 4.95. Found: C, 61.57; H, 6.30; N, 4.83.
Metallacycle 9
A solution of styrene oxide (13 mg, 0.11 mmol) in toluene (0.3 mL) was added to solid orange imido complex (rac)-2 (50 mg, 0.091 mmol), producing a deep red-brown solution. After 10 min, the solution was concentrated in vacuo to a viscous red oil. Ether (3 drops) was added to dissolve the oil, and pentane vapor diffusion was started at −35 °C. After 24 h, orange crystals had formed. The mother liquor was removed, the crystals were washed with cold pentane, and the residual solvent was removed in vacuo (45 mg, 83%). 1H NMR (500 MHz, C6D6): δ 7.01 (d, J = 5.0 Hz, 1H), 7.00–6.92 (m, 1H), 6.88 (t, J = 7.5 Hz, 1H), 6.28 (d, J = 3.0 Hz, 1H), 6.21 (d, J = 2.5 Hz, 1H), 6.03 (dd, J = 5.0, 4.5 Hz, 1H), 5.51 (t, J = 9.5 Hz, 1H), 5.43 (d, J = 3.0 Hz, 1H), 5.24 (d, J = 3.0 Hz, 1H), 5.16 (dd, J = 5.0, 4.5 Hz, 1H), 3.14 (dt, J = 16 Hz, 1H), 2.98–2.94 (m, 1H), 2.91–2.84 (m, 1H), 2.70–2.63 (m, 2H), 2.57 (m, 2H), 2.38 (s, 3H), 2.34 (s, 3H), 2.2–2.0 (m, 1H), 1.94–1.90 (m, 2H), 1.64–1.54 (m, 3H), 1.40–1.37 (m, 2H), 0.90–0.85 (m, 2H). 13C{1H} NMR (125 MHz, C6D6): δ 153.6, 140.3, 135.1, 133.8, 133.1, 131.0, 130.5, 128.3, 128.1, 127.0, 122.1, 121.2, 119.6, 112.2, 110.5, 110.3, 106.4, 83.5, 72.9, 29.0, 27.8, 25.3, 24.5, 23.6, 23.4, 23.0, 22.4, 22.1, 21.9. Anal. Calcd for C36H41NOZr: C, 72.68; H, 6.95; N, 2.35. Found: C, 72.42; H, 6.95; N, 2.46.
Procedure for Obtaining Crystals Suitable for X-ray Diffraction Analysis
A solution of styrene oxide (13 mg, 0.11 mmol) in toluene (6 drops) was added to solid (rac)-2 (50 mg, 0.091 mmol), producing a cloudy orange solution. After 20 min, vapor diffusion of pentane into the solution was started and the system was cooled to −35 °C. After 5 days, nonsuitable yellow crystals had formed. The mother liquor was removed, and a new vapor diffusion of pentane into this mother liquor was started. After 4 days, yellow crystals suitable for X-ray diffraction analysis had formed. The mother liquor was removed by pipet, and the residual solvent was removed in vacuo.
Metallacycle 13
trans-Stilbene oxide (22 mg, 0.11 mmol) and imido complex 1 (38 mg, 0.10 mmol) were dissolved in toluene (4 drops), producing an orange solution after 15 min. Vapor diffusion of ether into the solution at −35 °C was started. After 1 day, clusters of red-orange crystals had formed. The mother liquor was removed by pipet, and the residual solvent was removed in vacuo (18 mg, 37%). 1H NMR (300 MHz, C6D6): δ 7.13–7.02 (m, 10H), 6.22 (d, J = 3.3 Hz, 10H), 5.67 (d, J = 9.0 Hz, 1H), 4.80 (d, J = 9.0 Hz, 1H), 0.76 (s, 9H). 13C{1H} NMR (125 MHz, C6D6): δ 146.1, 144.4, 129.8, 127.9, 127.2, 126.9, 126.8, 126.4, 113.5, 113.1, 88.6, 86.7, 55.9, 30.4. Anal. Calcd for C28H31NOZr: C, 68.80; H, 6.39; N, 2.87. Found: C, 69.04; H, 6.53; N, 2.95.
Metallacycle 14
A solution of cis-stilbene oxide (40 mg, 0.20 mmol) in benzene (4 mL) was added to an orange suspension of (rac)-2 (100 mg, 0.18 mmol) in benzene (1 mL), to produce a dark red solution after 5 min. The solution was layered with pentane (15 mL) and cooled to −35 °C. After 4 weeks the pale orange mother liquor was decanted to reveal clusters of orange crystals, suitable for X-ray diffraction analysis, of a 1:1 mixture of the desired product and benzene (65 mg, 48%). Purified material therefore contained 1 equiv of benzene. 1H NMR (500 MHz, C6D6): δ 7.51 (d, J = 7.5 Hz, 1H), 7.27 (d, J = 7 Hz, 1H), 7.19–6.90 (m, 16H), 6.76 (t, J = 5 Hz, 1H), 6.61 (d, J = 10.5 Hz, 1H), 6.45 (d, J = 3 Hz, 1H), 5.72 (d, J = 6 Hz, 1H), 5.72 (s, 1H), 5.41 (d, J = 3 Hz, 1H), 4.39 (d, J = 3 Hz, 1H), 3.53 (m, 1H), 2.90 (m, 1H), 2.95–2.25 (m, 1H), 2.52 (s), 2.07–2.04 (m, 1H), 2.04 (s), 1.94–1.90 (m, 1H), 1.60–1.50 (m, 2H), 1.45–1.32 (m, 1H), 1.30–1.19 (m, 1H). 13C{1H} NMR (125 MHz, C6D6): δ 156.7, 146.6, 144.5, 132.7, 131.5, 131.4, 131.0, 129.5, 126.4, 126.3, 126.2, 122.6, 120.6, 119.6, 115.7, 111.9, 110.5, 108.6, 28.4, 27.6, 25.2, 25.1, 24.7, 23.4, 23.0, 22.9, 22.6, 22.4, 22.3. Anal. Calcd for C48H49NOZr: C, 77.16; H, 6.61; N, 1.87. Found: C, 76.78; H, 6.96; N, 1.83.
Metallacycle 17
Vinylcyclopropane oxide (20 mg, 0.24 mmol) was added to a solution of imido complex 16 (100 mg, 0.24 mmol) in benzene (2.5 mL), producing a yellow solution. The reaction mixture was allowed to sit for 24 h, at which time pentane (2.5 mL) was added to the solution and the reaction mixture was filtered to remove a white insoluble byproduct. The solvent was removed in vacuo, and the resultant yellow solid was taken up in ether (1.0 mL), layered with pentane (1.0 mL), and cooled to −35 °C. After 1 day, a yellow crystalline solid had precipitated. The mother liquor was decanted, and the volatile materials were removed in vacuo to leave the yellow solid (52 mg, 43%). 1H NMR (400 MHz, C6D6): δ 6.21 (s, 5H), 6.14 (s, 5H), 4.71 (dd, 1H, J = 4.0, 9.6 Hz), 3.97 (d, 1H, J = 9.6 Hz), 3.17 (dd, 1H, J = 4.0, 7.6 Hz), 1.51 (m, 1H), 1.02 (s, 9H), 0.62 (m, 1H), 0.48 (m, 1H), 0.32 (m, 1H), 0.26 (s, 3H), 0.16 (m, 1H), −0.27 (s, 3H). 13C{1H} NMR (100 MHz, C6D6): δ 113.9, 113.4, 80.0, 76.1, 28.4, 20.3, 18.5, 7.8, 3.7, 0.2, −0.5. Anal. Calcd for C21H33NOSiZr: C, 58.01; H, 7.65; N, 3.22. Found: C, 58.12; H, 7.70; N, 3.14.
Complex 23
A solution of exo-norbornene oxide (22 mg, 0.20 mmol) in toluene (1.2 mL) was added to imido complex 1 (50 mg, 0.14 mmol), producing a tan solution. The volatile materials were removed in vacuo, yielding tan crystals. An additional portion of toluene (0.5 mL) was added, and the volatile materials were again removed in vacuo, to remove residual THF. The resulting tan crystals were redissolved in minimal toluene (1.2 mL), and the solution was filtered. The tan solution was then layered with pentane (3.5 mL) and cooled to −35 °C. After 9 days the mother liquor was decanted to reveal tan crystals suitable for X-ray diffraction analysis (28 mg, 38%). 1H NMR (300 MHz, C6D6): δ 6.15 (s, 10 H), 3.35 (s, br, 2H), 1.96 (s, 2H), 0.95 (br, 2H), 0.88 (m, br, 4 H), 0.58 (m, br, 4 H), 0.25 (d, J = 9.6 Hz, 2H). 13C{1H} NMR (125 MHz, C7D8): δ 137.0, 110.0, 61.4, 36.0, 34.1, 26.1, 23.6. Anal. Calcd for C21H29NOZr: C, 62.64; H, 7.26; N, 3.48. Found: C, 62.48; H, 7.25; N, 3.38.
Kinetic Analysis of Formation of Metallacycle 25 from Complex 23
Epoxide complex 23 (7.0 mg, 0.017 mmol), exo-norbornene oxide (3.7 mg, 0.034 mmol), and 1,3,5-trimethoxy-benzene internal standard (8.0 mg, 0.048 mmol) were dissolved in toluene-d8 and transferred to a resealable J. Young NMR tube (total solution volume 0.380 mL). Product formation was monitored by 1H NMR spectroscopy, and the probe temperature was calibrated with an ethylene glycol standard. The reaction exhibited clean first-order kinetics: k = 0.017 ± 0.000086 min−1 (59.8 °C), k = 0.051 ± 0.00033 min−1 (69.8 °C), k = 0.084 ± 0.00021 min−1 (79.8 °C), k = 0.22 ± 0.0016 min−1 (89.9 °C). Error limits are reported at one standard deviation for each k and were calculated by comparison between the actual and simulated data. The rate constants were independent of the initial concentration of complex 23 and of excess epoxide: k = 0.048 ± 0.00034 min−1 (3.0 mg 23, 69.5 °C), k = 0.048 ± 0.0059 min−1 (10 mg exo-norbornene oxide, 0.265 mL total solvent volume, 69.8 °C). The reproducibility was conservatively estimated by doubling the difference in the k values for a triplicate run at 69.5 °C (i.e., error = 2 × [0.051 min−1 –0.048 min−1] = 0.006 min−1). Errors in ΔH‡ and ΔS‡ were calculated from this estimation.
Complex 26
Propylene oxide (8.7 mg, 0.15 mmol) was dissolved in benzene (1.0 mL) and added to a solution of imido complex 1 (50 mg, 0.14 mmol) in benzene (1.0 mL), producing a pale yellow solution. After 5 min, the solution was concentrated in vacuo to a cloudy oil that solidified upon standing (40 mg, 82%). 1H NMR (500 MHz, C6D6): δ 5.97–5.90 (m, 1H), 5.89 (s, 10H), 5.31 (dq, J = 17, 2.5 Hz, 1H), 5.08 (dt, J = 11, 2.0 Hz, 1H), 3.92 (s, br, 1H), 1.22 (s, 1H). 13C{1H} NMR (125 MHz C6D6): δ 140.2, 111.8, 110.7, 73.7, 55.1, 34.2. Anal. Calcd for C17H25NOZr: C, 58.24; H, 7.19; N, 3.99. Found: C, 58.15; H, 6.99; N, 4.00.
Complex 27
Cyclohexene oxide (29 mg, 0.30 mmol) was dissolved in benzene (0.3 mL), and the resulting solution was added to solid imido complex 1 (100 mg, 0.27 mmol), producing a pale tan solution. After 10 min, pentane was layered on top, and the sample was cooled to −35 °C. After 14 days, a scant amount of powder had precipitated on the bottom of the vial. The colorless mother liquor was removed carefully by pipet so as not to disturb the fine precipitate. The mother liquor was concentrated in vacuo, yielding a white powder (39 mg, 37%). 1H NMR (500 MHz, C6D6): δ 5.95 (s, 5H), 5.94 (s, 5H), 5.76 (m, 1H), 5.67 (m, 1H), 4.37 (m, 1H), 3.62 (s, 1H), 1.9–1.3 (m, 6 H), 1.25 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 133.1, 126.9, 110.7, 75.3, 54.8, 34.3, 33.3, 25.3, 19.8. Anal. Calcd for C20H29NOZr: C, 61.49; H, 7.48; N, 3.59. Found: C, 61.43; H, 7.43; N, 3.86.
Complex 28
Cyclooctene oxide (19 mg, 0.15 mmol) was dissolved in benzene (1.0 mL), and the resulting solution was added to a solution of imido complex 1 (50 mg, 0.14 mmol) in benzene (1.0 mL), producing a pale yellow solution. After 5 min, the solvent was removed in vacuo to give a clear gel (58 mg, 99%). 1H NMR analysis showed a 88:12 mixture of isomers. Major isomer: 1H NMR (500 MHz, C6D6): δ 5.97 (s, 10H), 5.63 (ddd, J = 10.5, 6.5, 1.0 Hz, 1H), 5.51 (m, 1H), 4.81 (quintet, J = 5.5, 1H), 3.68 (s, br, 1H), 2.20 (m, 1H), 1.92 (m, 2H), 1.6–1.4 (m, 6H), 1.31 (m 1H), 1.27 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 138.5, 125.6, 110.6, 78.8, 54.8, 39.9, 34.2, 29.3, 26.1, 23.8. Anal. Calcd for C22H33NOZr: C, 63.10; H, 7.94; N, 3.35. Found: C, 63.36; H, 7.71; N, 3.18.
Complex 29
1,2-Butene oxide (9.9 mg, 0.14 mmol) was dissolved in benzene (1.0 mL) and added to a solution of imido complex 1 (50 mg, 0.14 mmol) in benzene (1.0 mL). After 30 min, the solvent was removed in vacuo, to give a light yellow oil (14 mg, 28%). 1H NMR analysis showed a 2.0:1.0 mixture of isomers. Trans isomer (major): 1H NMR (500 MHz, C6D6): δ 6.19 (s, 10H), 5.62 (m, 2H), 4.39 (br, 1H), 1.63 (m, 3H), 1.21 (s, 9H). Cis isomer (minor): 1H NMR (500 MHz, C6D6): δ 5.92 (s, 10H), 5.69 (m, 1H), 5.43 (m, 1H), 4.52 (d, J = 6.3), 3.79 (br, 1H), 1.53 (d, J = 6.6 Hz), 1.21 (s, 9H). Anal. Calcd for C18H27NOZr: C, 59.29; H, 7.46; N, 3.84. Found: C, 58.61; H, 7.29; N, 3.39.
Assignment of Isomers Was Facilitated by Protic Cleavage of the Alkoxide Moiety from the Complex
A solution of benzoic acid (26 mg, 0.22 mmol) in benzene-d6 (0.5 mL) was added to a solution of complex 29 (10 mg, 0.027 mmol) in benzene-d6 (0.5 mL). After 20 min, 1H NMR and TOCSY spectra showed the formation of trans- and cis-crotyl alcohol (2.1:1.0), which were compared to a commercially available sample of 90:10 trans:cis-crotyl alcohol to confirm isomer assignment.
Complex 30
Phenylpropene oxide (19 mg, 0.15 mmol) was dissolved in benzene (1.0 mL) and added to a solution of imido complex (50 mg, 0.14 mmol) in benzene (1.0 mL), producing a pale yellow solution. After 20 min, the solvent was removed in vacuo, to give a brown oil (48 mg, 82%). 1H NMR (500 MHz, C6D6): δ 7.38 (d, J = 7.0 Hz, 2H), 7.26 (t, J = 7.5 Hz, 2H), 7.12 (t, J = 7.5 Hz, 1H), 5.96 (m, 1H), 5.92 (s, 5H), 5.91 (s, 1H), 5.26 (m, 2H), 5.01 (d, J = 10 Hz, 1H), 4.10 (s, br, 1H), 1.20 (s, 9H). 13C{1H} NMR (125 MHz C6D6): δ 145.8 (C), 143.7 (CH), 126.6 (CH), 126.3 (CH), 111.7 (CH2), 110.7 (CH), 85.1 (CH), 55.3 (C), 34.2 (CH3). One of the Cp resonances was not found, presumably due to overlap with the solvent resonance. Anal. Calcd for C23H29NOZr: C, 64.74; H, 6.85; N, 3.28. Found: C, 64.52; H, 6.75; N, 3.21.
Complex 31
Propene sulfide (6.1 mg, 0.082 mmol) was dissolved in benzene-d6 (0.3 mL) and added to a solution of imido complex (30 mg, 0.082 mmol) in benzene-d6 (0.2 mL), producing a bright yellow solution. Analysis of the crude reaction mixture by 1H NMR spectroscopy showed 69% yield of complex 31. The complex decomposed upon attempted purification. 1H NMR (300 MHz, C6D6): δ 6.20 (m, 1H), 5.78 (s, 10H), 5.48 (s, br, 1H), 5.29 (d, J = 12.9 Hz, 1H), 5.02 (d, J = 7.5 Hz, 1H), 3.57 (d, J = 3.3 Hz, 2H), 1.14 (s, 9H).
Assignment of the Product Was Facilitated by Protic Cleavage of the Sulfide Moiety from the Complex
A solution of benzoic acid (30 mg, 0.25 mmol) in benzene-d6 (0.5 mL) was added to the crude solution of complex 31. The solution became colorless immediately. Analysis of the solution by 1H NMR spectroscopy showed formation of allyl sulfide (>95% from 31). Comparison with commercially available allyl sulfide confirmed the assignment.
NMR Desymmetrization Experiment: Formation of Complex 34
To a solution of imido complex (rac)-2 (5.5 mg, 0.010 mmol) in benzene-d6 (0.4 mL) was added 2,3-butene oxide (0.97 μL, 0.011 mmol), producing a yellow solution. A 1H NMR spectrum obtained on this solution after 1 h showed that all (rac)-2 had been consumed, to produce complex 34 as one diastereomer (>95:5). A 1H NMR spectrum obtained after 1 day showed no change. Structural assignment of 34 was made by analogy to complexes 26–31. 1H NMR (300 MHz, C6D6): δ 7.16 (m, 2H), 6.91 (t, J = 7.6 Hz, 1H), 6.16 (d, J = 3.0 Hz, 1H), 6.01 (ddd, J = 17.2, 10.4, 7.2 Hz, 1H), 5.52 (d, J = 3.0 Hz, 1H), 5.39 (d, J = 3.0 Hz, 1H), 5.27 (d, J = 3.0 Hz, 1H), 5.06 (dm, J = 17.4 Hz, 1H), 4.96 (dm, J = 11.1 Hz, 1H), 4.86 (s, br, 1H), 4.65 (p, J = 6.4 Hz, 1H), 2.38 (s, 1H), 1.32 (d, J = 6.3 Hz, 3H).
Catalytic Desulfurization of Propene Sulfide
Propene sulfide (50 mg, 0.68 mmol) was dissolved in benzene-d6 (0.3 mL) and added to a solution of imido complex 1 (10 mg, 0.027 mmol) in benzene-d6 (0.2 mL), producing a bright yellow solution. The solution was transferred to a NMR tube, which was fitted with a Cajon adaptor, sealed, and removed from the glovebox. The NMR tube was flame sealed under vacuum and heated at 75 °C for 8 h. The dark brown, heterogeneous reaction mixture was then analyzed by 1H NMR spectroscopy, which showed the formation of propene (5.1 equiv, relative to 1), multiple products giving rise to baseline resonances, and unreacted propene sulfide (3.6 equiv). The NMR tube was opened to air, and the solution was flushed through a silica plug and diluted with THF. GC-MS analysis of the resultant solution indicated the presence of mass fragments corresponding to oligomerization of the starting material.
Supplementary Material
Acknowledgments
This work was supported by the NSF though a graduate fellowship to S.A.B, and by the NIH through grant no. GM-25459 to R.G.B. and postdoctoral fellowships to R.T.R. and F.E.M. We thank Dr. Fred Hollander and Dr. Allen Oliver of the UC Berkeley CHEXRAY facility for the X-ray structure determinations, Dr. Herman van Halbeek for help with NMR experiments, and Mr. Jordan Markham for the gift of N-tosyl-2-phenylaziridine.
Footnotes
Supporting Information Available: Kinetics data for the conversion of 23 to 5; X-ray structural data for compounds 5, 7, 8, 9, 14, and 23. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Walsh PJ, Hollander FJ, Bergman RG. J Am Chem Soc. 1988;110:8729. [Google Scholar]
- 2.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blum SA, Walsh PJ, Bergman RG. J Am Chem Soc. 2003;125:14276. doi: 10.1021/ja037267t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartoli G, Bosco M, Carlone A, Locatelli M, Massaccesi M, Melchiorre P, Sambri L. Org Lett. 2004;6:2173. doi: 10.1021/ol049372t. [DOI] [PubMed] [Google Scholar]
- 5.Larrow JF, Schaus SE, Jacobsen EN. J Am Chem Soc. 1996;118:7420. [Google Scholar]
- 6.Caron M, Sharpless KB. J Org Chem. 1985;50:1557. [Google Scholar]
- 7.Bergmeier SC. Tetrahedron. 2000;56:2561. [Google Scholar]
- 8.Ager DJ, Prakash I, Schaad DR. Chem Rev. 1996;96:835. doi: 10.1021/cr9500038. [DOI] [PubMed] [Google Scholar]
- 9.Hill M, Krker G, Kehr G, Frohlich R, Kataeva O. J Am Chem Soc. 2004;126:11046. doi: 10.1021/ja048447l. [DOI] [PubMed] [Google Scholar]
- 10.Ichikawa E, Suzuki M, Yabu K, Albert M, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:11808. doi: 10.1021/ja045966f. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy-Smith JJ, Nolin KA, Gunterman HP, Toste FD. J Am Chem Soc. 2003;125:4056. doi: 10.1021/ja029498q. [DOI] [PubMed] [Google Scholar]
- 12.Eldred SE, Stone DA, Gellman SH, Stahl SS. J Am Chem Soc. 2003;125:3422. doi: 10.1021/ja028242h. [DOI] [PubMed] [Google Scholar]
- 13.Walsh PJ, Hollander FJ, Bergman RG. Organometallics. 1993;12:3705. [Google Scholar]
- 14.Takeuchi K, Takasuka M, Ohga Y, Okazaki T. J Org Chem. 1999:64. [Google Scholar]
- 15.Blumenstein JJ, Ukachukwu VC, Mohan RS, Whalen DL. J Org Chem. 1993;58:924. [Google Scholar]
- 16.Pratt DA, Dilabio GA, Mulder P, Ingold KU. Acc Chem Res. 2004;37:334. doi: 10.1021/ar010010k. [DOI] [PubMed] [Google Scholar]
- 17.Blum SA, Bergman RG. Organometallics. 2004;23:4003. doi: 10.1021/om0495776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tu Y, Wang ZX, Shi Y. J Am Chem Soc. 1996;118:9806. [Google Scholar]
- 19.Priego J, Carratero JC. Synlett. 1999:1603. [Google Scholar]
- 20.Kirmse W, Kornrumpf B. Angew Chem, Int Ed Engl. 1969;8:75. [Google Scholar]
- 21.Nonhebel DC. Chem Soc Rev. 1993;22:347. [Google Scholar]
- 22.Bowry VW, Lusztyk J, Ingold KU. J Am Chem Soc. 1991;113:5687. [Google Scholar]
- 23.There was no evidence for the presence of olefinic resonances in the 1H NMR spectrum.
- 24.Darensbourg DJ, Wildeson JR, Lewis SJ, Yarbrough JC. J Am Chem Soc. 2002;124:7075. doi: 10.1021/ja020184c. [DOI] [PubMed] [Google Scholar]
- 25.Groves JT, Han Y, Engen D. J Chem Soc, Chem Commun. 1990:436. [Google Scholar]
- 26.Warner CR, Strunk RJ, Kuivila HG. J Org Chem. 1966;31:3381. [Google Scholar]
- 27.We estimate the error to be ±0.006 min−1 at 69.5 °C, based on a triplicate run.
- 28.Lautens M, Fagnou K, Hiebert S. Acc Chem Res. 2003;36:48. doi: 10.1021/ar010112a. [DOI] [PubMed] [Google Scholar]
- 29.For leading references, see: Eames J.Eur J Inorg Chem 2002393 [Google Scholar]
- 30.Adams RD, Queisser JA, Yamamoto JH. J Am Chem Soc. 1996;118:10674. and references therein. [Google Scholar]
- 31.Alaimo PJ, Peters DW, Arnold J, Bergman RG. J Chem Educ. 2001;78:64. [Google Scholar]
- 32.Murray, R. W.; Singh, M. Organic Syntheses; Wiley: New York; Vol. IX.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.