

Abstract
Little is known about how substrates bind to CtXR (Candida tenuis xylose reductase; AKR2B5) and other members of the AKR (aldo–keto reductase) protein superfamily. Modelling of xylose into the active site of CtXR suggested that Trp23, Asp50 and Asn309 are the main components of pentose-specific substrate-binding recognition. Kinetic consequences of site-directed substitutions of these residues are reported. The mutants W23F and W23Y catalysed NADH-dependent reduction of xylose with only 4 and 1% of the wild-type efficiency (kcat/Km) respectively, but improved the wild-type selectivity for utilization of ketones, relative to xylose, by factors of 156 and 471 respectively. Comparison of multiple sequence alignment with reported specificities of AKR members emphasizes a conserved role of Trp23 in determining aldehyde-versus-ketone substrate selectivity. D50A showed 31 and 18% of the wild-type catalytic-centre activities for xylose reduction and xylitol oxidation respectively, consistent with a decrease in the rates of the chemical steps caused by the mutation, but no change in the apparent substrate binding constants and the pattern of substrate specificities. The 30-fold preference of the wild-type for D-galactose compared with 2-deoxy-D-galactose was lost completely in N309A and N309D mutants. Comparison of the 2.4 Å (1 Å=0.1 nm) X-ray crystal structure of mutant N309D bound to NAD+ with the previous structure of the wild-type holoenzyme reveals no major structural perturbations. The results suggest that replacement of Asn309 with alanine or aspartic acid disrupts the function of the original side chain in donating a hydrogen atom for bonding with the substrate C-2(R) hydroxy group, thus causing a loss of transition-state stabilization energy of 8–9 kJ/mol.
Keywords: aldo–keto reductase (AKR), Candida tenuis xylose reductase, hydrogen bonding, ketone reduction, structure–activity correlation, substrate selectivity
Abbreviations: AKR, aldo–keto reductase; CtXR, Candida tenuis xylose reductase (its amino acid numbering starts with the authentic N-terminal serine residue as ‘1’; the initiator methionine is processed off in the recombinant enzyme); QSAR, quantitative structure–activity relationship; XR, xylose reductase (EC 1.1.1.21)
INTRODUCTION
The AKRs (aldo–keto reductases) constitute a large protein superfamily of mainly NAD(P)H-dependent reductases that occur in a wide variety of organisms and cell types. The majority of the AKRs catalyse the reduction of aldehyde or ketone substrates and do so within diverse physiological contexts [1]. On the basis of sequence similarity, the (currently) 124 protein members are classified into 14 families [2]. More than half of the families (1–7 and 11) have been structurally characterized. Evidence from crystal structures and biochemical studies supports a canonical (β/α)8 barrel fold and a catalytic mechanism common to all AKRs [1]. Many AKRs show an unusually broad substrate specificity when assayed in vitro, converting with comparable catalytic efficiencies various carbonyl-group-containing compounds whose non-reacting parts represent a wide range of chemical structures. Therefore, this implies a low selectivity for the non-reacting part and has made it difficult to assign a clear physiological function to certain AKRs (for a discussion, see [3]). How the AKRs bind and achieve specificity for their purported in vivo substrate(s) has generally remained vague [4–6]. Little is also known about what determines the preference for reaction with aldehydes compared with ketones, and vice versa [7].
CtXR (Candida tenuis xylose reductase; AKR2B5) is a representative AKR of the energy-producing metabolism of pentose sugars in eukaryotic micro-organisms [8]. It belongs to family 2, the mannose and xylose reductases [2]. CtXR binds the open-chain form of D-xylose and reduces it to the corresponding polyol xylitol by utilizing NADPH or NADH [9,10]. The apparent affinity of CtXR for xylose (Km≈90 mM [9]) would seem rather low. However, it is roughly matched to reported pentose levels that are possible in a yeast cell [11], as expected for a catabolic enzyme that functions with good fluxional efficiency under the cellular boundary conditions [3]. In the present study we questioned how xylose-specific binding recognition is used by CtXR to selectively stabilize the transition state for the reduction of its natural substrate. In the light of its well-characterized structure–function relationships [12–15] and defined role in yeast physiology [8], CtXR was a particularly interesting candidate with which to examine molecular interactions leading to AKR substrate selectivity.
Although CtXR can reduce different aldehydes (R-CHO) in vitro whereby R may diverge from an uncharged polyhydroxylated side chain (Km range 10−1–102 mM) to hydrophobic aromatic or aliphatic side chains harbouring various kinds of substituents (Km range 100–102 mM), the best apparent binding and catalytic efficiency are found with xylose or a close structural analogue thereof [9,16]. After correction for the proportion of openchain free aldehyde in an aqueous solution of xylose which presents the true substrate of the enzyme [9], values of ≈18 μM (=90 mM/5000) and about 106 M−1·s−1 are obtained for Km and kcat/Km respectively. Consistent with results of earlier biochemical studies [9], the CtXR crystal structure revealed a substrate-binding pocket that is lined by hydrophobic residues and provides very few polar side chains that are candidate hydrogen-bonding partners with xylose hydroxy groups [12]. Contacts with the C-2(R) hydroxy group are thought to be a prime source of the observed stereochemical selectivity of the enzyme in the reaction with aldose substrates [9].
Energy-minimized docking of xylose into the active site of CtXR bound to NADP+, using ordered water molecules as guides, revealed a probable mode of substrate binding (Figure 1) [12]. When C-1 of the substrate is within hydride-transfer distance above the nicotinamide C-4 and the carbonyl oxygen hydrogen-bonded to the phenolic oxygen of Tyr51 and Nϵ2 of His113, the aldehyde proton points towards the indole ring of Trp23. The modelling predicts steric conflicts between the side chain of Trp23 and any substituents replacing the aldehyde hydrogen atom. This provides a hypothetical explanation for the large aldehyde compared with ketone substrate selectivity of the enzyme. Ketose sugars, for example, are not substrates of CtXR (R. Kratzer and B. Nidetzky, unpublished work). The sterically preferred orientation of the open-chain pentose brings the C-2(R) hydroxy group into hydrogen-bonding distance with the Nδ of Asn309. It suggests the possibility of additional hydrogen bonds between the side chains of Trp23, Asp50 and Asn309 and hydroxy groups on xylose C-3, C-4, and C-5 [12].
Figure 1. Enzyme–substrate interactions in the active site of CtXR, as predicted by modelling experiments [12].

The open-chain xylose bound to the enzyme is shown.
In the present study we individually mutated Trp23, Asp50 and Asn309 into different residues and determined the functional consequences in purified point mutants to characterize the predicted enzyme–substrate interactions. The crystal structure of the N309D mutant was determined at a resolution of 2.4 Å (1 Å=0.1 nm). The results provide distinct structure–function assignments for each side chain and, to the best of our knowledge, present the first comprehensive molecular-level analysis of an AKR substrate-binding pocket. They may find application in biocatalysis for the stereoselective transformation of prochiral carbonyl-group-containing compounds [17–19].
EXPERIMENTAL
Materials
All carbonyl substrates were of the highest purity available from Sigma–Aldrich or Fluka. Other materials were reported elsewhere [20].
Site-directed mutagenesis, enzyme production and purification
Site-directed mutagenesis was carried out by using procedures described elsewhere [21,22]. The mutagenic oligonucleotide primers are listed below with the mismatched bases underlined:
N309D forward: 5′ TTGAGATTCGATGATCCGTGGG 3′
N309A forward: 5′ TTGAGATTCGCAGATCCTTGGG 3′
N309 reverse: 5′ GCCGATGTCCAACTTAGCGATT 3′
D50A forward: 5′ GAGGCCTACGGTAACGAAAAG 3′
D50 reverse: 5′ GGCACCGTCGAACAATCTGTA 3′
W23F forward: 5′ GGTTTCGGCTGTTTCAAACTCG 3′
W23Y forward: 5′ GGTTTCGGCTGTTATAAACTCG 3′
W23 reverse: 5′ GATGGATGGCATTAAGTGGCCG 3′
To facilitate colony screening by restriction-site analysis, the NcoI restriction site in the N309 forward primers was deleted and a FokI restriction site in the W23 reverse primer was inserted by introducing silent mutations (marked in bold). Introduction of additional silent mutations avoided dimerization of oligonucleotides. The authenticity of each mutagenized gene was confirmed by dideoxy sequencing. Recombinant wild-type CtXR and the mutants thereof were produced in Escherichia coli and purified to apparent homogeneity using reported protocols [23]. Purified N309D, N309A, W23F, W23Y and D50A mutants migrated in SDS/PAGE as single protein bands to exactly the same position as the wild-type (results not shown).
Crystallization of N309D holoenzyme and data collection
The purified N309D mutant was crystallized by using the hanging-drop vapour-diffusion method at 25 °C. The drop consisted of 1 μl of well solution and 1 μl of protein solution (15 mg/ml) with 2.5 mM NAD+ added. The best-diffracting crystals were grown using a well solution consisting of 2.1 M (NH4)2SO4, 100 mM sodium acetate and 100 mM sodium citrate, pH 6.4. After 2.5 months the crystals had dimensions of 1.5 mm×0.2 mm×0.1 mm and were harvested into a cryoprotectant containing 75% (v/v) well solution and 25% (v/v) ethylene glycol (with 5 mM NAD+ added) prior to data collection at 100 K. Diffraction data were collected on an R-AXIS IV imaging plate detector with a rotating anode. The programs DENZO and SCALEPACK [24] were used to integrate and reduce data, yielding a 2.4 Å data set with an Rmerge of 8.4%. Other relevant data-collection statistics are given in Table 1. The structure of the mutant was determined using the isomorphous model of the CtXR K273R/N275D mutant (PDB accession code 1SM9; [22]), followed by rigid body refinement. The model was improved by alternating cycles of manual refitting and refinement using the software suites O [25] and CNS (Crystallography & NMR System; [26]). Final statistics describing the refinement and model quality are given in Table 1.
Table 1. Summary of data collection, refinement and models for the N309D mutant bound to NAD+.
| Parameter | Value |
|---|---|
| Data collection | |
| Space group | C2 |
| Unit cell | a=180.64 Å, b=128.40 Å, c=80.00 Å, β=90.71° |
| Monomers per asymmetric unit | 4 |
| Resolution range (Å) | 30–2.4 |
| Number of observations/unique reflections | 138182/64303 |
| Rmerge (overall/high-resolution shell*) (%) | 0.084/0.196 |
| Completeness (overall/high-resolution shell*) | 90.2/82.9 |
| Mosaicity (°) | 0.615 |
| I/σ(I) (overall/high-resolution shell*) | 8.17/3.20 |
| Model | |
| Protein atoms | 10128 |
| NAD+ atoms | 176 |
| Water molecules | 789 |
| Overall temperature factors (Å2) | |
| Monomer A | 26.3 |
| Monomer B | 20.7 |
| Monomer C | 24.3 |
| Monomer D | 16.6 |
| Refinement | |
| Reflections used (I>0) | 64105 |
| Rcryst | 0.176 |
| Rfree | 0.231 |
| R.m.s.† deviation from ideal bond length (Å) | 0.007 |
* The high-resolution shell is 2.49–2.40.
† Root-mean-square.
Steady-state assays
Unless mentioned otherwise, all experiments were performed at 25 °C in 50 mM potassium phosphate buffer, pH 7.0. Initial-rate measurements were carried out as described recently [22], whereby 2 or 5% ethanol was added as required to enhance the solubility of hydrophobic carbonyl compounds. Kinetic parameters for enzymatic xylose reduction by NADH were not affected by the added ethanol. The initial rates were recorded immediately after preparation of the substrates to avoid their non-enzymatic decomposition in aqueous solution [27]. Unless indicated, initial rates were obtained under conditions in which the substrate concentration was varied and the coenzyme concentration was constant and saturating [carbonyl reduction, 230 μM NAD(P)H; alcohol oxidation, 600 μM NAD(P)+]. The enzyme concentration in the assays was in the range 0.03–10.0 μM, depending on the activity towards the respective substrate. Appropriate controls containing enzyme and coenzyme, or the substrate and coenzyme, were determined under conditions otherwise exactly identical with those used in the enzymatic assay. If required, the initial rates were corrected for blank readings. Steady-state enzyme binding to NAD+ was recorded on a Hitachi F-2000 spectrofluorimeter as described previously [14]. Corrections for inner-filter effects were not necessary.
Data processing and statistical analysis were carried out as reported elsewhere [22]. The limited solubility of certain compounds or a very low apparent binding affinity prevented saturation of the enzymes with some substrates. In these cases, the catalytic efficiency was obtained from the part of the Michaelis–Menten plot where under conditions of [substrate]≪Km, the reaction rate is linearly dependent on [substrate] with a slope that equals (kcat/Km) divided by the molar concentration of enzyme.
Enzymatic reduction of carbonyl substrates
Ethyl 4-chloroacetoacetate (50 mM; 100 mM Tris/HCl buffer, pH 7.0 [28]) or ethyl benzoylformate (5 mM; 50 mM potassium phosphate buffer, pH 7.0) was incubated at 25 °C in the presence of equimolar NADH and enzyme (wild-type, W23F or W23Y). The reaction mixture (2 ml) contained about 2 units of enzyme activity, measured with the substrate to be reduced. After complete (>98%) ketone reduction, measured as the depletion of NADH, the alcohol product was extracted with dichloromethane (2 ml). HPLC analysis of the dried organic phase, along with authentic standards of (R) and (S)-configured alcohols, was performed with a CHIRALPAK AD-H column (Daicel; purchased from VWR) on an Agilent 1100 Series instrument equipped with a G1365B MWD UV detector (210 nm). n-Heptane/propane (9:1, v/v) was the eluent.
QSAR (quantitative structure–activity relationship) analysis
The following equation:
 |
(1) |
was used to correlate observed substituent effects on kcat/Km for enzymatic ketone reduction with effects of the substituent on the electronic properties, expressed by the Taft factor, σ* [29] (a polar substituent parameter in aliphatic systems first defined by R. W. Taft), as well as the hydrophobic (logP) and molecular (logMol) characteristics of the parent compound, here acetophenone. logP values (the logarithm of the compound's partition coefficient between n-octanol and water) were derived from SciFinder Scholar (edition 2004), and logMol values (logarithm of the molecular volume measured in Å3) were calculated using the program Sybyl 6.9.1 (edition 2003). In eqn (1), ρ is the Taft factor coefficient, A and B are molecular volume and hydrophobicity correction coefficients respectively, and C is a constant term.
Multivariate regression analysis was performed as described elsewhere [14].
RESULTS AND DISCUSSION
Purification and characterization of site-directed mutants
Table 2 compares apparent kinetic parameters for NADH-dependent reduction of xylose by the wild-type and the five purified mutants. Catalytic efficiencies of the mutants were decreased to 3.7 and 1.0% of the wild-type value, indicating that the substituted side chains are important for xylose reduction by CtXR.
Table 2. Apparent kinetic parameters of wild-type and mutants for NADH-dependent reduction of xylose.
| Enzyme | Km (mM) | kcat (s−1) | 103×(kcat/Km)′* (M−1·s−1) |
|---|---|---|---|
| Wild-type | 91 | 12.4 | 680 |
| W23F | 1475 | 7.4 | 25 |
| W23Y | – | – | 6.5 |
| D50A | 243 | 3.4 | 14 |
| N309D | 1349 | 2.4 | 9 |
| N309A | 1787 | 6.8 | 19 |
* kcat/Km′ values are corrected for the 0.02% of open-chain free aldehyde in aqueous solution of xylose [34].
Kinetic consequences of replacing Asp50 with alanine
The multiple sequence alignment in Figure 2 shows that, except for family 2 AKRs, the majority of AKR families have a non-ionizable and mostly non-polar residue at the position equivalent to Asp50 in CtXR. Asp50 contributes the most to the relative polarity of the substrate binding site of CtXR and is close in structure to the catalytic residues Tyr51 and His113 [12]. Therefore, in addition to its predicted interaction with C-2(R) and C-3(S) hydroxy groups of xylose (see Figure 1), Asp50 was a generally interesting candidate for site-directed mutagenesis.
Figure 2. Conservation pattern for Trp23 and Asp50 revealed by partial protein sequence alignment of representative members of AKR subfamilies 1A–5E.

The protein sequences were compared with the program Vector NTI Suite 7.0 (2001) using the scoring matrix PAM250 [37]. All sequences start with the initiator methionine residue and are shown up to the active-site tyrosine residue. Residues at position 23 are framed. Residues at position 50 are shown second to the right in the alignment, preceding the conserved tyrosine residue. For each subfamily (in bold below), the statistics of the occurrence of Trp23 homologues are shown in parentheses as the number of homologous residues per total number of protein members. 1A (4W/4), Homo sapiens (human) liver aldehyde reductase [1A1, acc. (accession number on the NCBI protein sequence database) NP_006057, EC 1.1.1.2]; 1B 12W/12, Homo sapiens placenta aldose reductase (1B1, acc. NP_001619, EC 1.1.1.21); 1E 1W/1, Mus musculus (house mouse) AKR (1E, acc. AAB37274); 2A 2W/2, Malus domestica (apple) sorbitol-6-phosphate reductase (2A1, acc. P28475, EC 1.1.1.200); 2B 8W/8, CtXR (2B5, acc. AAC25601, EC 1.1.1.21); 2D 1W/1, Aspergillus niger (onion black mould) xylose reductase (2D, acc. AAF61912, EC 1.1.1.21); 3A 2W/2, Saccharomyces cerevisiae (baker's yeast) Gcy1p, putative glycerol dehydrogenase (3A1, acc. P14065, EC 1.1.1.72); 3B 1W/1, Sporidiobolus salmonicolor (red yeast) aldehyde reductase (3B, acc. AAB17362, EC 1.1.1.2); 3C 1A/1, Saccharomyces cerevisiae Ara1p, D-arabinose dehydrogenase (3C, acc. CAA85107, EC 1.1.1.117); 3D 1W/1, Magnaporthe grisea (rice-blastdisease fungus) aldose reductase (3D, acc. AAK55762, EC 1.1.1.21); 4C 6W/6, Hordeum vulgare (barley) aldehyde reductase (4C1, acc. P23901, EC 1.1.1.21); 1C 15Y/20, 2T/20, 2F/20, 1S/20, Homo sapiens liver 20α-hydroxysteroid dehydrogenase (1C1, acc. NP_1344, EC 1.3.1.20); 1D 3Y/3, Homo sapiens liver 3-oxo-5β-steroid 4-dehydrogenase (1D1, acc. NP_005980, EC 1.3.99.6); 2C 1W/1, 4-dihydromethyltrisporate dehydrogenase of the pin mould Mucor mucedo (2C, acc. CAA98021); 2E 1Y/2, 1F/2, Spodoptera littoralis (Egyptian cotton leafworm) 3-dehydroecdysone 3β-reductase (2E1, acc. CAB41997); 4A 4A/4, Glycine max (soya bean) chalcone polyketide reductase (4A1, acc. P26690, EC 2.3.1.170); 4B 1S/3, 2A/3, chalcone polyketide reductase of the tropical leguminous plant Sesbania rostrata (4B1, acc. P26690); 5B 1F/1, morphine dehydrogenase of the bacterium Pseudomonas putida (5B, acc. Q02198, EC 1.1.1.218); 5C 1F/1 Corynebacterium sp. 2,5-diketo-D-gluconic acid reductase (5C, acc. AAA83535, EC 1.1.1.125); 5D 1Y/1, Corynebacterium sp. 2,5-diketo-D-gluconic acid reductase (5D, acc. P15339, EC 1.1.1.274); 5E 1Y/1, 2,5-diketo-D-gluconic acid reductase of the bacterium Zymomonas mobilis (5E1 acc. 5354195, EC 1.1.1.125).
A full steady-state kinetic analysis for xylose reduction and xylitol oxidation by the D50A mutant was carried out and kinetic parameters are summarized in Table 3, along with the corresponding wild-type parameters. Using NAD(H) as the coenzyme, the catalytic-centre activities in the reduction and oxidation directions of the mutant (indicated by kcat) were decreased to 31 and 18% of the respective wild-type parameters. By contrast, the Michaelis constants (Km) for substrates and coenzymes as well as the apparent dissociation constants (Kd) for NADH and NAD+ were not affected in the mutant or showed less than a two-fold increase in comparison with the corresponding wild-type values. The kcat for NADPH-dependent reduction of xylose by the mutant was three times that for the corresponding NADH-dependent reaction, whereas the wild-type kcat increased by a factor of 1.73 when NADPH replaced NADH. Interpretation of changes in kcat for the reduction direction caused by the mutation are complicated, because the wild-type kcat is largely governed by the rate of release of NAD+ [30] or NADP+ (W. Neuhauser and B. Nidetzky, unpublished work). By contrast, the wild-type kcat for xylitol oxidation is limited by the chemical reaction rate [30]. Its decrease in D50A is thus attributed to a slowing down of catalysis by about the same factor.
Table 3. Steady-state kinetic parameters for xylose reduction and xylitol oxidation catalysed by wild-type and D50A mutant.
Parameters had standard errors <15%.
| Value | |||
|---|---|---|---|
| Parameter | Enzyme… | Wild-type | D50A |
| NADH-dependent xylose reduction* | |||
| kcat (s−1) | 11 | 3.4 | |
| Km,xylose (mM) | 142 | 158 | |
| Km,NADH (mM) | 38 | 40 | |
| Ki,NADH (μM) | 19 | 20 | |
| NADPH-dependent xylose reduction† | |||
| kcat (s−1) | 19 | 11 | |
| Km,NADPH (mM) | 3.2 | 2.3 | |
| NAD+-dependent xylitol oxidation‡ | |||
| kcat (s−1) | 1.1 | 0.2 | |
| Km,xylitol (mM) | 334 | 537 | |
| Kd,NAD+ (μM)§ | 234 | 373 | |
* From initial rates (v) recorded at varied [NADH] and [xylose], and fits of data to the equation v=kcat[E][NADH][xylose]/(Ki,NADHKm,xylose+Km,xylose[NADH]+Km,NADH[xylose]+[NADH] [xylose]); Ki,NADH is an apparent dissociation constant for NADH.
† From initial rates recorded with varied [NADPH] at [xylose]=1 M.
‡ From initial rates of xylitol oxidation recorded at [NAD+]=600 μM.
§ From fluorescence titration data. Kd is an apparent dissociation constant for NAD+.
Table 4 compares kinetic parameters of D50A and wild-type for the reduction of a series of aldohexoses differing in content and stereochemistry of their hydroxy groups. Mutation of Asp50 caused no change in the specificity pattern of the wild-type with these substrates. Therefore, the wild-type efficiency for reduction of the polyhydroxylated compounds tested is not clearly dependent on interactions between Asp50 and substrate hydroxy groups.
Table 4. Apparent kinetic parameters of wild-type CtXR and mutants thereof for the reduction of polyhydroxylated aldehyde substrates.
Abbreviation: nd, not determined.
| Value | |||||
|---|---|---|---|---|---|
| Parameter | Enzyme… | Wild-type | N309D | N309A | D50A |
| DL-Glyceraldehyde (25%†) | |||||
| Km (mM) | 3.5 | 35 | 54 | nd | |
| kcat (s−1) | 7.8 | 2.3 | 4.2 | nd | |
| 103×(kcat/Km)′* (M−1·s−1) | 8.9 | 0.3 | 0.3 | nd | |
| D-Glucose (0.0024%†) | |||||
| Km (mM) | 236 | – | – | 486 | |
| kcat (s−1) | 4.8 | – | – | 2.2 | |
| 103×(kcat/Km)′* (M−1·s−1) | 833 | 8.3 | 8.3 | 188 | |
| D-Galactose (0.02%†) | |||||
| Km (mM) | 208 | 264 | – | 244 | |
| kcat (s−1) | 11 | 0.3 | – | 3.3 | |
| 103×(kcat/Km)′* (M−1·s−1) | 265 | 5.5 | 9.0 | 70 | |
| 2-Deoxy-D-galactose (0.03%†) | |||||
| Km (mM) | 742 | 187 | 178 | – | |
| kcat (s−1) | 2.1 | 0.2 | 0.9 | – | |
| 103×(kcat/Km)′* (M−1·s−1) | 9.3 | 3.7 | 17 | 6.3 | |
* kcat/Km′ values are corrected for the proportion of open-chain free aldehyde in aqueous solution (shown in parentheses and indicated by
† [34]).
Kinetic consequences of replacing Asn309 with alanine or aspartic acid
Table 4 shows kinetic parameters of N309A and N309D for reductions of polyhydroxylated aldehydes that were chosen as particular probes of hydrogen bonding between the mutated side chain and hydroxy groups at substrate C-2 and C-5 (see Figure 1). Comparison of the kcat/Km values for D-galactose and 2-deoxy-D-galactose revealed that the 30-fold preference of the wild-type for reaction with D-galactose was completely lost in the two mutants, each reducing the two substrates with almost the same catalytic efficiencies. Values of kcat/Km for the reduction of 2-deoxy-D-galactose were similar for wild-type and the two mutants, supporting the suggestion that Asn309 forms a hydrogen bond with the C-2(R) hydroxy group of the substrate (Figure 1; [12]). The contributed net binding energy of this interaction to a stabilization of the transition state can be estimated in either of two ways, using the relationships:
 |
(2) |
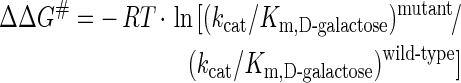 |
(3) |
Where ΔΔG# is Gibbs free-energy change, T is temperature and R is the gas constant. These equations estimate that ΔΔG# is between 8.4 kJ/mol (eqn 2) and 9.6 kJ/mol (eqn 3; N309A).
Prior studies in which the natural enzyme isolated from C. tenuis was used yielded a greater ΔΔG# value of >17 kJ/mol [9]. Given the high purity of the commercial substrates, we do not know the basis of this significant difference in differential binding energy. The modified N-terminal amino acid in the natural enzyme [10] is not found in recombinant CtXR [12], but is unlikely to have significant functional consequences.
Mutations of Asn309 and substrate deoxygenation appear to be fully complementary in locally disrupting the proposed hydrogen bond (Figure 1; [12]), giving rise to the consequent loss of binding energy. However, the quantitatively similar disruptive effect of the substitutions N309A and N309D warrants discussion. Unless water was bound in the space vacated by the replacement Asn309→Ala and restored partly the original hydrogen bond formed in the wild-type, the N309A mutant would be expected not to interact with the hydroxy group at C-2 of D-galactose. This scenario is consistent with the experimental findings. However, considerations of side-chain polarity and steric bulk implicate Asp309 as a valid alternative partner for hydrogen-bonding with the substrate 2-hydroxy. Its obvious lack of performance in N309D suggests that the directionality of the original hydrogen bond is E-H····O-H; hence, the C-2(R) hydroxy group functions as acceptor of a hydrogen for bonding, in excellent agreement with the findings of Neuhauser et al. [9].
X-ray structure of N309D mutant bound to NAD+
The mutant enzyme folds into the canonical (β/α)8-barrel. Four XR molecules (two dimers) are within the crystallographic asymmetric unit, each of them showing clear electron density for NAD+. Every monomer consists of residues 3–321 out of 321 residues predicted from the sequence. The asymmetric unit contains 789 water atoms. The structure of N309D was determined at 2.4 Å with the refinement statistics (Table 1) indicating a well-defined structure. A Ramachandran plot was generated using the program PROCHECK [31] and showed that 89.9% of the residues are in the most favoured regions, 10.0% are in additional allowed regions and 0.1% in generously allowed regions. The root-mean-square deviation between Cα atoms of mutant and wild-type was calculated to be 0.11 Å. The electron density for Asp309 was well defined. The structural analysis of the substrate-binding site and the active site, as well as of the coenzyme-binding site, revealed that the mutation was highly conservative. The only structural change induced by the site-directed replacement was a rotation of the mutant carboxylate relative to the wild-type amide, changing the χ2 angle from 28.6° in the wild-type to −39.4° in N309D (Figure 3).
Figure 3. Stereo view of an overlay of the substrate binding sites of CtXR wild-type enzyme (green) with that of the N309D mutant (light grey).

The bound NAD+ is coloured orange for the wild-type and yellow for N309D (however, since the cofactor shows the same conformation in both of the structures, the NAD+ which is bound to the N309D mutant covers the NAD+ bound to the wild-type almost entirely). The Figure was generated with Molscript [35] and Raster3D [36].
Taken together, the structural and functional studies of the N309D mutant indicate that the negatively charged side chain of Asp309 has lost the ability to donate a hydrogen atom for bonding with the C-2(R)-OH group of D-galactose. As shown in Tables 2 and 4, the decrease in kcat/Km for reactions catalysed by N309A and N309D mutants, compared with the corresponding wild-type kcat/Km values, was not dependent on the number of carbon atoms of the aldoses tested: three carbon atoms, 3.4% of wild-type; five carbon atoms, 2.8 and 1.3% of wild-type; six carbon atoms, 1% of wild-type (D-glucose); 3.4 and 2.1% of wild-type (D-galactose). Asn309-mediated substrate-binding recognition by CtXR is therefore clearly regioselective for the C-2 hydroxy group. These results provide the first molecular-level foundation of carbohydrate substrate specificity in a member of the AKR superfamily. Asn309 is conserved in all currently known members of family 2B, emphasizing that its proposed function in CtXR is likely to be general.
Trp23 determines ketone-versus-aldehyde substrate selectivity of CtXR
The kcat/Km values for xylose reduction by W23F and W23Y mutants were lowered to 3.7 and 1.0% of the wild-type value respectively (Table 2). A similar loss of catalytic efficiency, to 8% of the wild-type value, was seen with a W20Y mutant of human aldose reductase [32], whereby Trp20 is the positional analogue of Trp23 of CtXR (see Figure 2).
A series of oxo-group-containing compounds were tested as substrates of wild-type CtXR and the two Trp23 mutants in initial-rate experiments. The ketones span a wide range of intrinsic reactivities of the carbonyl group undergoing enzymatic reduction, reflected by their substituent Taft factors (σ*). Kinetic parameters are summarized in Table 5. The formation of alcohol product(s) coupled to the enzymatic oxidation of NADH was confirmed using selected substrates.
Table 5. Apparent kinetic parameters of wild-type and W23F and W23Y mutants for NADH-dependent reduction of a series of ketones.
| Value | ||||
|---|---|---|---|---|
| Substrate/parameter | Enzyme… | Wild-type | W23F | W23Y |
| Acetophenone† (Taft σ*=0.60; molecular volume=108.1 A3) | ||||
| kcat/Km (M−1·s−1) | 0.5 | 0.2 | 0.2 | |
| Acetoin (Taft σ*=0.46; molecular volume=82.9 A3) | ||||
| Km (mM) | 451 | 851 | 519 | |
| kcat (s−1) | 0.8 | 0.7 | 0.4 | |
| kcat/Km (M−1·s−1) | 1.8 | 0.8 | 0.8 | |
| 4-Hydroxybutan-2-one (Taft σ*=0.21; molecular volume=98.1 A3) | ||||
| Km (mM) | 392 | 373 | 289 | |
| kcat (s−1) | 0.02 | 0.02 | 0.01 | |
| kcat/Km (M−1·s−1) | 0.05 | 0.05 | 0.03 | |
| Diacetyl (Taft σ*=1.65; molecular volume=74.2 A3) | ||||
| Km (mM) | 15 | 34 | 107 | |
| kcat (s−1) | 12 | 9.6 | 13 | |
| kcat/Km (M−1·s−1) | 800 | 282 | 121 | |
| Acetylacetone† (Taft σ*=0.60; molecular volume=91.3 A3) | ||||
| kcat/Km (M−1·s−1) | 0.07 | 0.03 | 0.03 | |
| Oxopantoyl-lactone (Taft σ*=1.81; molecular volume=104.7 A3) | ||||
| kcat/Km (M−1·s−1) | 8.0 | 46 | 36 | |
| Ethyl pyruvate (Taft σ*=2.26; molecular volume=97.7 A3) | ||||
| Km (mM) | 12 | 15 | 14 | |
| kcat (s−1) | 6.5 | 2.4 | 0.8 | |
| kcat/Km (M−1·s−1) | 542 | 160 | 57 | |
| Ethyl benzoylformate‡ (Taft σ*=2.86; molecular volume=47.9 A3) | ||||
| kcat/Km (M−1·s−1) | 269 | 911 | 636 | |
| Ethyl acetoacetate† (Taft σ*=0.82; molecular volume=114.6 A3) | ||||
| kcat/Km (M−1·s−1) | 0.7 | 0.3 | 0.2 | |
| Ethyl 4-chloroacetoacetate†§ (Taft σ*=1.83; molecular volume=127.0 A3) | ||||
| kcat/Km (M−1·s−1) | 34 | 68 | 62 | |
| Ethyl 4,4,4-trifluoroacetoacetate† (Taft σ*=3.38; molecular volume=122.9 A3) | ||||
| kcat/Km (M−1·s−1) | 0.3 | 0.6 | 0.3 | |
† 2% (v/v) Ethanol was added.
‡ 5% (v/v) Ethanol was added.
§ 100 mM Tris/HCl buffer, pH 7.0.
The chosen ketones were approx. 103–107-fold poorer substrates of the wild-type enzyme than was the open-chain xylose. Mutation of Trp23 caused a decrease in this large xylose compared with ketone substrate selectivity of the wild-type by factors of up to 156 and 471 respectively. The relatively better utilization by the mutants of ketones than of xylose, compared with the wild-type, reflects a strong decrease in kcat/Km for xylose reduction (Table 2) and a modest increase in kcat/Km values for ketone reduction (Table 5). A role of Trp23 in selectively binding and achieving specificity for aldehydes is thus supported.
Comparison of the conservation pattern of Trp23 with the reported substrate specificities of other AKRs (families 1–5; Figure 2) suggests that the proposed structure–function relationship for this residue is of a general importance. Aldehyde-preferring AKRs often (though not exclusively) have a tryptophan residue at a position homologous with Trp23. By contrast, ketone-preferring AKRs often display a tyrosine or phenylalanine residue, but also other residues, as the positional analogue of Trp23. Members of AKR families 7–14 show divergent N-terminal sequences such that no firm conclusion regarding the conservation of Trp23 can be made (see the AKR superfamily homepage at http://www.med.upenn.edu/akr/).
QSAR analysis
Values of kcat/Km,ketone for the wild-type and the Trp23 mutants in Table 5 displayed a 16000-fold variation in the dependence of the side-chain structure of the substrate. Eqn (1) was fitted to the set of data for each enzyme, excluding in all cases the kcat/Km,ketone for ethyl 4,4,4-trifluoroacetoacetate, which was inexplicably low. The results indicated that only electronic and molecular-volume factors of the substituent were important parameters affecting kcat/Km. Parameter estimates along with the coefficient of determination (r2) and the F-test value were obtained from two-parameter correlations, as shown in eqns. 4(a)–4(c).
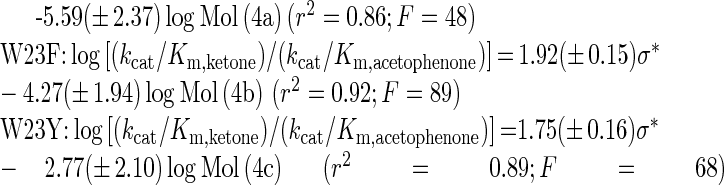 |
The r2 values of ≥0.86, together with high F values (≥48), point out a useful fit of each equation to the corresponding data. Values of log(kcat/Km,ketone) for the W23F mutant adjusted for the logMol effect according to eqn 4(b) are plotted against the substituent Taft factor in Figure 4.
Figure 4. Dependence on Taft factors (σ*) of the second-order rate constants for NADH-dependent reduction of ketones catalysed by W23F.

The continuous line shows bivariate regression analysis of data.
The results in Figure 4 and eqns 4(a)–4(c) underline the strong dependence of log(kcat/Km,ketone) on the electronic characteristics of the substrate. This conclusion is reinforced by taking into account that carbonyl-group hydration in aqueous solution will increase in response to an increasing electron-withdrawing character of the substituent [33]. The full electronic substituent effect on kcat/Km,ketone is therefore partly masked in the observable parameter coefficient. The positive value of ρ(kcat/Km,ketone) implies that electron-withdrawing substituents speed the enzymatic conversion of ketones or, in other words, a higher partial positive charge on the reactive carbon leads to a more rapid enzymatic reaction. Structure–reactivity correlations for CtXR-catalysed conversions of ketones (the present study) and aromatic aldehydes [14,20] yield consistent mechanistic interpretations, suggesting that there will probably be a decrease in positive charge at the reaction centre upon moving along the reaction co-ordinate from the free substrate and the binary complex of enzyme and NADH to the transition state. Values of ρ(kcat/Km,ketone) for W23F and W23Y are similar to the wild-type value, indicating that the site-directed replacements of Trp23 do not perturb the electronic substituent effect on the free-energy profile of ketone reduction by CtXR.
Conclusions
How do broadly specific AKRs bring about the requisite selectivity for their physiological carbonyl substrate? A rigorous quantitative analysis relies on strictly local disruption of non-covalent enzyme–substrate interactions, using ideally both point mutants and complementary substrate analogues, and determination of resulting kinetic consequences. The situation is complicated for plastic binding sites such as those seen in AKRs. Here we assign clear structure–function relationships to Asn309 and Trp23 of the substrate-binding pocket of CtXR. The results explain the high aldehyde compared with ketone substrate selectivity of CtXR and the requirements for non-covalent contacts with the C-2(R) hydroxy group of xylose to attain optimum catalytic efficiency. The linear free-energy relationships for ketone reduction are novel and provide a practically useful reactivity scale for biocatalytic conversions of prochiral carbonyl substrates [17–19] by CtXR and mutants thereof. The third residue examined, Asp50, probably functions in fine tuning of the catalytic rate.
Acknowledgments
Dr Mario Klimacek and Franz Wührer, both of the Institute of Biotechnology and Biochemical Engineering, Graz University of Technology, Graz, Austria, are thanked for their helpful contributions. Financial support is acknowledged from the Austrian Science Funds (grant P-15208-MOB to B.N.), the National Institutes of Health (grant to D.K.W.) and the Keck Foundation (grant to D.K.W.).
References
- 1.Jez J. M., Bennett M. J., Schlegel B. P., Lewis M., Penning T. M. Comparative anatomy of the aldo–keto reductase superfamily. Biochem. J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyndman D., Baumann D. R., Heredia V. V., Penning T. M. The aldo–keto reductase superfamily homepage. Chem.–Biol. Interact. 2003;143–144:621–631. doi: 10.1016/s0009-2797(02)00193-x. [DOI] [PubMed] [Google Scholar]
- 3.Grimshaw C. E. Aldose reductase: model for a new paradigm of enzymic perfection in detoxification catalysts. Biochemistry. 1992;31:10139–10145. doi: 10.1021/bi00157a001. [DOI] [PubMed] [Google Scholar]
- 4.Heredia V. V., Cooper W. C., Kruger R. G., Jin Y., Penning T. Alanine scanning mutagenesis of the testosterone binding site of rat 3α-hydroxysteroid dehydrogenase demonstrates contact residues influence the rate-determining step. Biochemistry. 2004;43:5832–5841. doi: 10.1021/bi0499563. [DOI] [PubMed] [Google Scholar]
- 5.Ma H., Penning T. M. Conversion of mammalian 3α-hydroxysteroid dehydrogenase to 20α-hydroxysteroid dehydrogenase using loop chimeras: changing specificity from androgens to progestins. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11161–11166. doi: 10.1073/pnas.96.20.11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barski O. A., Bohren K. M., Gabbay K. H. The C-terminal loop of aldehyde reductase determines the substrate and inhibitor specificity. Biochemistry. 1996;35:14276–14280. doi: 10.1021/bi9619740. [DOI] [PubMed] [Google Scholar]
- 7.Tarle I., Borhani D. W., Wilson D. K., Quiocho F. A., Petrash J. M. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J. Biol. Chem. 1993;268:25687–25693. [PubMed] [Google Scholar]
- 8.Ellis E. M. Microbial aldo–keto reductases. FEMS Microbiol Lett. 2002;216:123–131. doi: 10.1111/j.1574-6968.2002.tb11425.x. [DOI] [PubMed] [Google Scholar]
- 9.Neuhauser W., Haltrich D., Kulbe K. D., Nidetzky B. Noncovalent enzyme–substrate interactions in the catalytic mechanism of yeast aldose reductase. Biochemistry. 1998;37:1116–1123. doi: 10.1021/bi9717800. [DOI] [PubMed] [Google Scholar]
- 10.Neuhauser W., Haltrich D., Kulbe K. D., Nidetzky B. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis. Isolation, characterization and biochemical properties of the enzyme. Biochem. J. 1997;326:683–692. doi: 10.1042/bj3260683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gárdonyi M., Jeppsson M., Lidén G., Gorwa-Grauslund M. F., Hahn-Hägerdal B. Control of xylose consumption by xylose transport in recombinant Saccharomyces cerevisiae. Biotechnol. Bioeng. 2003;82:818–824. doi: 10.1002/bit.10631. [DOI] [PubMed] [Google Scholar]
- 12.Kavanagh K. L., Klimacek M., Nidetzky B., Wilson D. K. The structure of apo and holo forms of xylose reductase, a dimeric aldo–keto reductase from Candida tenuis. Biochemistry. 2002;41:8785–8795. doi: 10.1021/bi025786n. [DOI] [PubMed] [Google Scholar]
- 13.Klimacek M., Szekely M., Grießler R., Nidetzky B. Exploring the active site of yeast xylose reductase by site-directed mutagenesis of sequence motifs characteristic of two dehydrogenase/reductase family types. FEBS Lett. 2001;500:149–152. doi: 10.1016/s0014-5793(01)02609-6. [DOI] [PubMed] [Google Scholar]
- 14.Kratzer R., Kavanagh K. L., Wilson D. K., Nidetzky B. Studies of the enzymic mechanism of Candida tenuis xylose reductase (AKR 2B5): X-ray structure and catalytic reaction profile for the H113A mutant. Biochemistry. 2004;43:4944–4954. doi: 10.1021/bi035833r. [DOI] [PubMed] [Google Scholar]
- 15.Kratzer R., Nidetzky B. Electrostatic stabilization in a pre-organized polar active site: the catalytic role of Lys-80 in Candida tenuis xylose reductase (AKR2B5) probed by site-directed mutagenesis and functional complementation studies. Biochem. J. 2005;389:507–515. doi: 10.1042/BJ20050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nidetzky B., Mayr P., Hadwiger P., Stütz A. E. Binding energy and specificity in the catalytic mechanism of yeast aldose reductases. Biochem. J. 1999;344:101–107. [PMC free article] [PubMed] [Google Scholar]
- 17.Kataoka M., Kita K., Wada M., Yasohara Y., Hasegawa J., Shimizu S. Novel bioreduction system for the production of chiral alcohols. Appl. Microbiol. Biotechnol. 2003;62:437–445. doi: 10.1007/s00253-003-1347-y. [DOI] [PubMed] [Google Scholar]
- 18.Zhou B., Gopalan A. S., VanMiddlesworth F., Shieh W.-R., Sih C. J. Stereochemical control of yeast reductions. 1. Asymmetric synthesis of L-carnitine. J. Am. Chem. Soc. 1983;107:5925–5926. [Google Scholar]
- 19.Müller M. Chemoenzymatic synthesis of building blocks for statin side chains. Angew. Chem. Int. Ed. Engl. 2005;44:362–365. doi: 10.1002/anie.200460852. [DOI] [PubMed] [Google Scholar]
- 20.Mayr P., Nidetzky B. Catalytic reaction profile for NADH-dependent reduction of aromatic aldehydes by xylose reductase from Candida tenuis. Biochem. J. 2002;366:889–899. doi: 10.1042/BJ20020080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemsley A., Arnheim N., Toney M. D., Cortopassi G., Galas D. J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petschacher B., Leitgeb S., Kavanagh K. L., Wilson D. K., Nidetzky B. The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem. J. 2005;385:75–83. doi: 10.1042/BJ20040363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayr P., Brüggler K., Kulbe K. D., Nidetzky B. D-Xylose metabolism by Candida intermedia: isolation and characterisation of two forms of aldose reductase with different coenzyme specificities. J. Chromatogr. B. 2000;737:195–202. doi: 10.1016/s0378-4347(99)00380-1. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation model. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 26.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Kaluzna I. A., Matsuda T., Sewell A. K., Stewart J. D. Systematic investigation of Saccharomyces cerevisiae enzymes catalyzing carbonyl reductions. J. Am. Chem. Soc. 2004;126:12827–12832. doi: 10.1021/ja0469479. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu S., Kataoka M., Katoh M., Morikawa T., Miyoshi T., Yamada H. Stereoselective reduction of ethyl 4-chloro-3-oxobutanoate by a microbial aldehyde reductase in an organic solvent–water diphasic system. Appl. Environ. Microbiol. 1990;56:2374–2377. doi: 10.1128/aem.56.8.2374-2377.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansch C., Leo A., Hoekman D. H. Washington, DC: American Chemical Society; 1995. Exploring QSAR – Hydrophobic, Electronic, and Steric Constants. [Google Scholar]
- 30.Nidetzky B., Klimacek M., Mayr P. Transient-state and steady-state kinetic studies of the mechanism of NADH-dependent aldehyde reduction catalyzed by xylose reductase from the yeast Candida tenuis. Biochemistry. 2001;40:10371–10381. doi: 10.1021/bi010148a. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;23:283–291. [Google Scholar]
- 32.Hohman T. C., El-Kabbani O., Malamas M. S., Lai K., Putilina T., McGowan M. H., Wane Y.-Q., Carper D. A. Probing the inhibitor-binding site of aldose reductase with site-directed mutagenesis. Eur. J. Biochem. 1998;256:310–316. doi: 10.1046/j.1432-1327.1998.2560310.x. [DOI] [PubMed] [Google Scholar]
- 33.Guthrie J. P., Pitchko V. Hydration of carbonyl compounds, an analysis in terms of no barrier theory: prediction of rates from equilibrium constants and distortion energies. J. Am. Chem. Soc. 2000;122:5520–5528. [Google Scholar]
- 34.Angyal S. J. The composition of reducing sugars in solution: current aspects. Adv. Carbohydr. Chem. Biochem. 1991;49:19–35. [Google Scholar]
- 35.Kraulis P. J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 36.Merritt E. A., Bacon D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 37.Altschul S. F. Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 1991;219:555–565. doi: 10.1016/0022-2836(91)90193-A. [DOI] [PMC free article] [PubMed] [Google Scholar]