

Abstract
We have previously identified a phorbol ester-induced PKCϵ (protein kinase Cϵ) interaction with the (∼18 kDa) COIV [CO (cytochrome c oxidase) subunit IV] in NCMs (neonatal cardiac myocytes). Since PKCϵ has been implicated as a key mediator of cardiac PC (preconditioning), we examined whether hypoxic PC could induce PKCϵ–COIV interactions. Similar to our recent study with phorbol esters [Ogbi, Chew, Pohl, Stuchlik, Ogbi and Johnson (2004) Biochem. J. 382, 923–932], we observed a time-dependent increase in the in vitro phosphorylation of an approx. 18 kDa protein in particulate cell fractions isolated from NCMs subjected to 1–60 min of hypoxia. Introduction of a PKCϵ-selective translocation inhibitor into cells attenuated this in vitro phosphorylation. Furthermore, when mitochondria isolated from NCMs exposed to 30 min of hypoxia were subjected to immunoprecipitation analyses using PKCϵ-selective antisera, we observed an 11.1-fold increase in PKCϵ–COIV co-precipitation. In addition, we observed up to 4-fold increases in CO activity after brief NCM hypoxia exposures that were also attenuated by introducing a PKCϵ-selective translocation inhibitor into the cells. Finally, in Western-blot analyses, we observed a >2-fold PC-induced protection of COIV levels after 9 h index hypoxia. Our studies suggest that a PKCϵ–COIV interaction and an enhancement of CO activity occur in NCM hypoxic PC. We therefore propose novel mechanisms of PKCϵ-mediated PC involving enhanced energetics, decreased mitochondrial reactive oxygen species production and the preservation of COIV levels.
Keywords: cardiac hypoxia, cytochrome c oxidase subunit IV (COIV), electron-transport chain, mitochondria, preconditioning (PC), protein kinase Cϵ (PKCϵ)
Abbreviations: AMPK, AMP-dependent protein kinase; CO, cytochrome c oxidase; COIV, CO subunit IV; D-β-M, n-dodecyl-β-maltoside; DNP, 2,4-dinitrophenol; ETC, electron-transport chain; HMG-CoA reductase, 3-hydroxy-3-methylglutaryl-CoA reductase; IB, inhibitory buffer; IM, inner mitochondrial membrane; IP, immunoprecipitation; co-IP, co-immunoprecipitation; MAPK, mitogen-activated protein kinase; mKATP, mitochondrial ATP-dependent potassium; MPTP, mitochondrial permeability transition pore; NCM, neonatal cardiac myocyte; PC, preconditioning; PKA, protein kinase A; PKC, protein kinase C; ROS, reactive oxygen species
INTRODUCTION
Cardiac ischaemic/hypoxic PC (preconditioning) is a paradoxical response in which one or more brief cycles of oxygen deprivation and reperfusion lead to protection against injury induced by prolonged oxygen deprivation [1]. Experimentally, it is the most powerful form of cardiac protection known and it has been demonstrated in cardiac myocytes [2–5], intact hearts [6,7] and many animal species [8–11], including humans [12–14]. Downstream signalling molecules reported to be involved in PC include ATP-dependent potassium channels [4,12,15,16], PKC (protein kinase C) isoenzymes [2,3,12,15,17,18], MAPK (mitogen-activated protein kinase) enzymes [19–21], tyrosine kinases [22–24], PI3K (phosphoinositide 3-kinase) [22,25], heat-shock proteins [5,16,26], nitric oxide [27–29], free radicals [21,25,30] etc.
Many studies have also implicated PKCϵ in PC. Gray et al. [2] found that PKCϵ translocation was required for the induction of hypoxic PC in NCMs (neonatal cardiac myocytes). These authors used peptide-based inhibitors that block the translocation of PKCϵ to its isoenzyme-selective anchoring protein or RACK (receptor for activated C-kinase). Ping et al. [31] have established that induction of the early or late phases of PC in conscious rabbits triggered the translocation of the PKCϵ and PKCη isoenzymes to the cardiac particulate fraction. Qiu et al. [32] found that differential inhibition of PKCη, but not PKCϵ, translocation by low concentrations of the PKC inhibitor chelerythrine allowed the recovery of systolic ventricular wall thickening in rabbits, while elevated concentrations of chelerythrine blocked PKCϵ translocation and PC. This same group demonstrated that nitric oxide was responsible for PC-induced PKCϵ activation and protection in rabbits [28] by a mechanism involving nitric oxide-induced nitration of PKCϵ and enhanced PKCϵ interaction with its intracellular RACK [28,29]. Evidence has also emerged that one downstream mechanism of PKCϵ-mediated PC involves activation of the tyrosine kinases src and lck [23,24]. PKCϵ-induced PC also appears to involve enhanced activities of MAPKs, JNKs (c-Jun N-terminal kinases) and NF-κB (nuclear factor κB) and AP-1 (activator protein-1) transcription factors [20]. PC has been reported to involve the formation of signalling complexes between PKCϵ and ERKs (extracellular-signal-regulated kinases) inside cardiac mitochondria to promote the phosphorylation and inhibition of the apoptotic protein Bad [19]. In addition, overexpressed PKCϵ interacts with and inhibits the opening of the MPTP (mitochondrial permeability transition pore) [33], which, when open, is thought to play a major role in ischaemia/reperfusion injury. Low-level expression of a constitutively active PKCϵ in transgenic mice has been correlated with increased cardiac protection, whereas elevated levels of this mutated form of PKCϵ correlated with hypertrophy and heart failure [34]. Of interest, PKCϵ knockout mice are largely unable to precondition their myocardium [35,36]. In addition, expression of selective translocation inhibitors of PKCϵ blocks cardiac protection, while a peptide activator of PKCϵ enhances cardiac protection in transgenic mouse hearts [37].
CO (cytochrome c oxidase) is a 13-subunit enzyme complex positioned on the IM (inner mitochondrial membrane) of mammalian cells [38]. It is the last step in the mitochondrial ETC (electron-transport chain) and is intricately involved in mitochondrial ATP production and maintenance of the IM proton gradient [38,39]. In addition, when CO activity is inhibited or lost (as it is during prolonged myocardial ischaemia [40]), ATP production decreases and there are elevations of mitochondrial superoxide production due to disruption of electron flow (‘electron leak’) predominantly at ETC complexes I and III [39,40]. CO has also been shown to be modulated by protein kinases. For example, in vitro phosphorylation of COII (CO subunit II), COIII, COIV and COVb by PKA (protein kinase A) has been reported previously [41]. Bender and Kadenbach [42] also presented evidence that phosphorylation of COI on Ser441 by PKA switches on allosteric inhibition of complex IV by ATP. Miyazaki et al. [43] reported that phosphorylation of COII by c-Src in osteoclasts increases CO activity. Finally, our laboratory has previously shown, using an in vitro phosphorylation assay, that the PKCϵ isoenzyme phosphorylates COIV in particulate fractions isolated from phorbol ester-treated NCMs [44].
In the present study, we have demonstrated a stable interaction between PKCϵ and COIV, which correlates with a >2-fold enhancement of CO activity under conditions that precondition NMCs. We propose that a PKCϵ-mediated enhancement of CO activity during PC should improve the efficiency of electron flow from cytochrome c to molecular oxygen after ischaemic injury, which in turn would: (i) aid in maintenance of the IM proton gradient and improve myocardial aerobic respiration and (ii) reduce the production of ROS (reactive oxygen species) via complexes I and III of the mitochondrial ETC. To our knowledge, the present study is the first report of a PKCϵ modulation of CO in cardiac PC or any cell type.
EXPERIMENTAL
Primary NCMs
Primary cell cultures were isolated from 1-day-old neonatal Sprague–Dawley rats as described previously [45,46]. All work with animals was conducted in compliance with The Declaration of Helsinki, institutional, state and federal guidelines for the humane care and use of laboratory animals.
Induction of hypoxia in NCMs
For hypoxic PC and prolonged exposures to hypoxia, myocytes were placed in a PlasLabs Anaerobic chamber equilibrated at 37 °C with humidified atmosphere containing 1% CO2, <0.5% O2 and the balance N2. Hypoxic incubations were carried out in glucose-free M199 medium as described previously [2]. A Fyrite gas analyser was used to confirm O2 concentrations at or below 0.5%. Normoxic incubations of myocytes were carried out in a water-jacketed incubator gassed with 1% CO2 and 99% air at 37 °C in M199 medium containing 5 mM glucose. The PC method involves a 30 min period of hypoxia followed by a 30 min normoxic period. A similar method has been described previously [2].
LIVE/DEAD cell viability assays
Cell viability was confirmed using the LIVE/DEAD kit (Molecular Probes). Cells were grown on glass chamber slides as described previously [2]. Culture medium was replaced with medium containing 2 μM calcein acetoxymethyl ester and 4 μM propidium iodide. Cells were viewed using a Zeiss Axiophot microscope equipped with a ×40 water immersion objective. All viable cells were stained green by the calcein ester, but only cells with compromised plasma membranes were stained red by the propidium dye. A total of 15 random fields of cells were selected and the numbers of green (live) and red (dead) cells were counted. Results were expressed as percentage viability (see Supplementary Figure 1 at http://www.BiochemJ.org/bj/393/bj3930191add.htm).
Cell lysis and isolation of particulate cell fractions
The culture medium was removed and the cells were washed twice in chilled Ca2+/Mg2+-free PBS. Unless otherwise indicated, all other operations were conducted with chilled reagents on ice or at 4 °C. Care was taken to siphon off all excess PBS. Cells were scrapped from the dish in 350 μl of IB (inhibitory buffer; 50 mM KH2PO4, 5 mM EDTA, 0.5 mM EGTA, 10 nM calyculin A, 5 mM Na4P2O7 and 25 μg/ml each of PMSF, leupeptin, aprotinin and soya-bean trypsin inhibitor). Cells were triturated three times in IB using a tuberculin syringe and a 22-gauge needle. Next, cells were centrifuged at 100000 g for 30 min at 4 °C. The resulting supernatants were assayed by adding 20 μl of the fraction to an in vitro phosphorylation assay (see below). Particulate fractions were then resuspended in IB plus 0.2% Triton X-100 using a tuberculin syringe and a 22-gauge needle. The solubilized particulate fractions were then incubated on ice for 5 min. Two more cycles of resuspension were carried out prior to transferring 20 μl aliquots of the particulate cell fraction into microfuge tubes for the in vitro 32P incorporation (phosphorylation) assay.
In vitro phosphorylation assays
Particulate cell fractions isolated from NCMs as described above were subjected to an in vitro assay for 5 min at room temperature (22 °C). The final phosphorylation assay buffer consisted of 50 mM Tris/HCl (pH 7.4), 5 mM KH2PO4, 1 mM EDTA, 0.1 mM EGTA, 2 nM calyculin A, 1 mM Na4P2O7, 5 μg/ml each of PMSF, leupeptin, aprotinin and soya-bean trypsin inhibitor, 0.4 mM free Mg2+ (calculated as described previously [47]), 10 μM [γ-32P]ATP (3000 c.p.m./pmol), 1–3 of μg 1,2-sn-diacylglycerol and 5 μg of phosphatidylserine in the presence and absence of 0.5 mM CaCl2. Reactions were terminated by the addition of SDS Laemmli sample buffer at 90 °C. Samples were then heated for 5 min and subjected to SDS/PAGE.
Peptide synthesis and delivery into cells
PKC translocation inhibitor peptides [2,36,37,48–50] containing the following sequences were synthesized at the Emory University Microchemical Facility: (i) ϵV1-2 [EAVSLKPT; PKCϵ (amino acids 14–21)], (ii) βC2-4 [SLNPEWNET; PKCβ (amino acids 218–226)], (iii) δV1-1 [SFNSYELGSL; PKCδ (amino acids 8–17)] These peptides were then conjugated to the protein transducing domain of the HIV-Tat protein (amino acids 47–57) as described previously [37]. An HIV-Tat carrier–carrier peptide (YGRKKRRQRRR-YGRKKRRQRRR) was synthesized as a control.
Electrophoresis, gel drying and autoradiography
Samples obtained from in vitro incubations or other analyses were subjected to SDS/PAGE on 1 mm thick 13.5% (w/v) polyacrylamide gels by the method of Laemmli. It was essential to de-gas our gel solutions for 45 min prior to casting gels for polymerization. Gels were run overnight at approx. 3 mA per gel. When in vitro 32P-labelling experiments were carried out, gel bottoms (containing the dye front) were removed and discarded. Gels were dried in a Bio-Rad gel drying apparatus at 80 °C for 1 h, and subjected to autoradiography and densitometry. All autoradiographs used for densitometry were scanned in the linear range of detection.
Isolation of mitochondria from NCMs
Ten 100 mm dishes of NCMs were used for each treatment group. Cells were washed twice in chilled MSE buffer (10 mM Tris/HCl, pH 7.4, 220 mM mannitol, 70 mM sucrose, 1 mM EGTA, 2 mM taurine, 1 mM carnitine, 1 mM Na2P4O7, 10 nM calyculin A and 20 μg/ml each of PMSF, soya-bean trypsin inhibitor, aprotinin and leupeptin). Cells were then gently scraped from each dish and collected in a total volume of 1 ml of MSE buffer and incubated on ice with 0.025 unit of trypsin (Sigma Chemical) for 20 min. Cells were then subjected to Dounce homogenization. The resulting homogenate was sequentially centrifuged twice at 500 g for 5 min to pellet nuclei and cell debris. The resulting pellets were discarded and the 500 g (post-nuclear) supernatant was then layered over Percoll/Optiprep gradients (see below).
Preparation of Percoll/Optiprep density gradients
The 500 g post-nuclear supernatant (prepared as described above) was layered over a combination Percoll/Optiprep (Accurate Chemical and Scientific, Westbury, NY, U.S.A.) gradient prepared on the same day as follows. Each gradient was prepared in Beckman-Ultraclear 14 mm×89 mm centrifuge tubes. The first step in gradient formation involved overlaying 1.74 ml of a 17% (v/v) Optiprep solution on a 1.74 ml cushion of 35% Optiprep solution. Next, 4.35 ml of a 6% (v/v) Percoll solution was layered on top of the 17% Optiprep solution. All Optiprep and Percoll solutions were prepared using MSE buffer as the diluent. Gradients were stored on ice until use. Next, 4.2 ml of post-nuclear supernatant was gently layered on top of the 6% Percoll portion of the gradient. All tubes were centrifuged in a Beckman SW.41 swinging bucket rotor at 50000 g for 30 min using the lowest acceleration and deceleration speeds. Using this procedure, mitochondria partition at the 17%/35% Optiprep interface. Mitochondria were removed from each gradient using a Pasteur pipette and placed on ice until use in co-IP (co-immunoprecipitation) and CO assays.
CO activity assays
Intact mitochondria were solubilized and assayed for CO activity using a commercially available kit (Sigma Chemical) as described previously [43]. Assays were conducted for 1 min (initial rates) at 25 °C in duplicate. The kit measures the oxidation of ferrocytochrome c to ferricytochrome c via the activity of CO. CO activity is therefore monitored as a decrease in absorbance at 550 nM. Equivalent amounts of protein for each mitochondrial extract were assayed.
IP (immunoprecipitation) method
Anti-PKCϵ or anti-COIV antiserum (5 μg/ml each) was chemically coupled with 500 μl of Bio-Rad Affi-gel according to the manufacturer's instructions. Bound antiserum was pelleted at 300 g for 10 min and resuspended in IP buffer (150 mM NaCl, 20 mM Tris/HCl, pH 7.4, 10 mM EDTA, 1% Triton X-100, 1 mM Na2P4O7 and 10 nM calyculin A). Intact mitochondria were added to the IP buffer such that the final volume of the immunoprecipitate was 1 ml. Solubilized mitochondria were incubated with 150 μl of antibody-coupled Affi-gel with agitation at 4 °C for 4 h overnight. Affi-gel immunoprecipitates were pelleted at 300 g for 10 min and washed three times in IP buffer. After the final wash, the resulting Affi-gel pellets were solubilized in SDS/PAGE sample buffer with 90 °C heating. The solubilized proteins were liberated by pelleting the Affi-gel at 1000 g for 5 min. The supernatants from each IP were subjected to SDS/PAGE (15% polyacrylamide).
Western-blot analyses
Western blotting was carried out as described previously [46].
Statistical analysis
Results were expressed as means±S.E.M. Differences from control values were considered statistically significant at P≤0.05 using the unpaired Student's t test.
RESULTS AND DISCUSSION
Hypoxia induces PKCϵ-mediated in vitro phosphorylation of an approx. 18 kDa protein in NCMs
It has previously been demonstrated that a 30 min hypoxia exposure induces a PKCϵ-mediated PC of NCMs against prolonged hypoxia-induced cell death [2]. In the present study, we have confirmed this result in each of our experiments (results not shown). In addition, we have previously demonstrated that preferential activation (translocation) of the PKCϵ isoenzyme by 3 nM 4β-PMA [46] induces the in vitro phosphorylation of COIV in NCM particulate fractions [44]. As PKCϵ is activated during PC, we hypothesized that a brief PC (hypoxia exposure) would also induce the in vitro phosphorylation of COIV by PKCϵ in NCMs. Figure 1 demonstrates a time-dependent in vitro phosphorylation of an approx. 18 kDa protein in particulate fractions isolated from NCMs exposed to hypoxia. The in vitro phosphorylation of the approx. 18 kDa protein reached a peak at approx. 5 min of hypoxia and remained elevated by >2-fold above normoxic control levels for 1 h. Given the similarity between this PC-induced response and that observed following 3 nM 4β-PMA treatment [44], we hypothesized that the approx. 18 kDa protein was COIV and that PKCϵ and COIV may interact to play a role in cardiac PC.
Figure 1. Brief hypoxia induces in vitro 32P labelling of an approx. 18 kDa protein.

NCMs were exposed to 0–60 min of hypoxia, and particulate cell fractions were then subjected to an in vitro 32P-labelling reaction with [γ-32P]ATP as described in the Experimental section. The reaction mixture was then subjected to SDS/PAGE and autoradiography. Molecular-mass sizes are given in kDa. The arrow points to a protein of approx. 18 kDa. (A) A typical autoradiograph from these analyses is shown. (B) Means±S.E.M. densitometry values for four independent experiments each taken from a separate myocyte preparation. All hypoxia treatment groups were significantly different from the normoxic control value at or below the P<0.05 level.
Introduction of a PKCϵ-selective translocation inhibitor into NCMs attenuates the hypoxia-induced in vitro phosphorylation of the approx. 18 kDa protein
To confirm that the hypoxia-induced in vitro phosphorylation of COIV involved PKCϵ, we introduced a PKCϵ-selective translocation inhibitor coupled with the protein transduction domain of the HIV-Tat protein into cells prior to a 30 min hypoxia exposure (Figure 2). A Tat-carrier peptide control and PKC isoenzyme-selective translocation inhibitors directed against the classical PKC isoenzymes [2,37,48,49] were also tested. Following hypoxia, particulate cell fractions were isolated and subjected to the in vitro phosphorylation reactions. Introduction of the PKCδ-selective translocation inhibitor (δV1-1) or the classical PKC inhibitor βC2-4 into NCMs prior to hypoxic exposure did not attenuate the in vitro phosphorylation of COIV (Figure 2). Of interest, we observed a 1.9±0.1-fold increase (n=3, P<0.01) in the in vitro 32P labelling of the approx. 18 kDa protein in particulate fractions isolated from cells receiving the δV1-1 (PKCδ)-inhibitory peptide (Figure 2). We are currently investigating the mechanism (direct or indirect) by which inhibition of the PKCδ isoenzyme enhances the in vitro phosphorylation of the approx. 18 kDa protein. In contrast, the PKCϵ translocation inhibitor inhibited the in vitro 32P labelling of the approx. 18 kDa protein by 79±15% (n=3, P<0.05). Our results are consistent with hypoxic PC of NCMs being associated with the translocation of the PKCϵ isoenzyme to the particulate cell fraction and the subsequent in vitro 32P labelling of the approx. 18 kDa protein by PKCϵ (Figure 2).
Figure 2. A PKCϵ-selective translocation inhibitor attenuates the in vitro 32P labelling of the approx. 18 kDa protein.
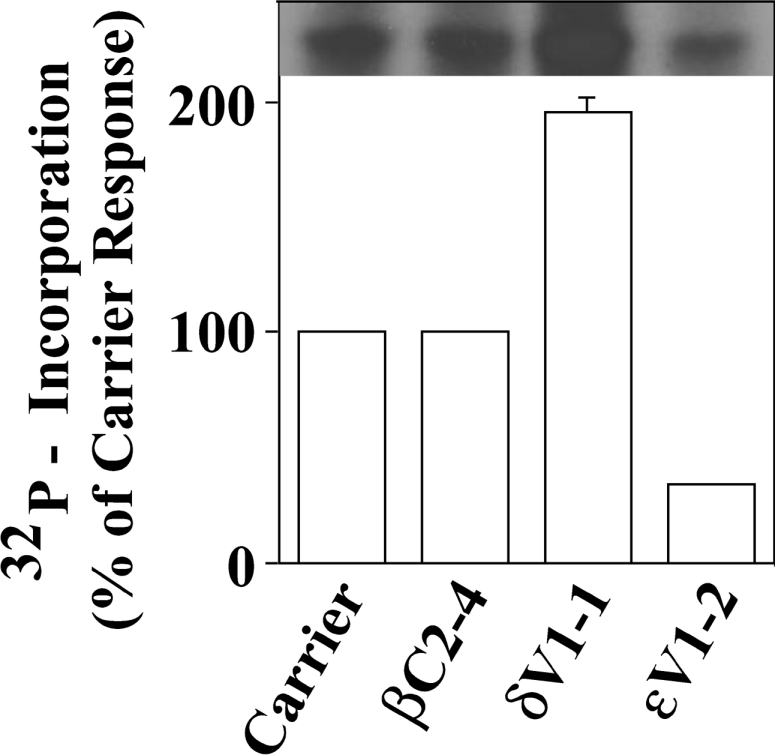
NCMs were preincubated for 30 min in the presence of control (HIV-Tat carrier) peptide, the classical PKC inhibitor βC2-4, the PKCδ translocation inhibitor δV1-1 or the PKCϵ-selective translocation inhibitory peptide (ϵV1-2) as described in the Experimental section. Next, cells were given a 30 min hypoxic incubation. Cell particulate fractions were then harvested and subjected to in vitro 32P-labelling reaction, SDS/PAGE, gel drying and autoradiography (see the Experimental section). The autoradiograph shown in the upper part of the Figure is from a single representative experiment. The histograms in the lower part of the Figure are the means±S.E.M. for three independent experiments, each conducted on a separate myocyte preparation.
The PKCϵ isoenzyme co-precipitates with COIV following exposure of NCMs to hypoxia
COIV exists in the IM and has exposed regions on the matrix side of the IM. Previous Western-blot studies, however, have indicated that little PKCϵ exists inside mitochondria in the basal state, suggesting that PKCϵ must translocate to mitochondria following PC in order to interact with COIV. Following activation, PKCϵ has been reported to exist inside mitochondria isolated from mouse hearts [33], NCMs [51] and guinea-pig cardiac myocytes [52]. For example, Baines et al. [19] found that PKCϵ forms intramitochondrial signalling complexes with MAPK following PC of mouse myocardium. Lawrence et al. [51] reported enhanced PKCϵ levels in mitochondria isolated from NCMs exposed to the cardioprotective drug urocortin. Ohnuma et al. [53] reported PC/diazoxide-induced translocation of PKCϵ to mitochondria in rabbit hearts. There has also been a report of Gqα receptor-induced PKCϵ translocation to mitochondria isolated from human atrial appendage tissue exposed to PC [54]. These reports of PKCϵ localization in mitochondria, the hypoxia-induced in vitro labelling of the approx. 18 kDa protein (Figures 1 and 2), and our previous phorbol ester studies [44] suggested to us that PKCϵ may interact with COIV in cardiac PC. We therefore administered normoxia (control) or 30 min of hypoxia to NCMs. Cells were then gently scraped from dishes and lysed in MSE buffer, and intact mitochondria were isolated using Percoll/Optiprep density gradients as described in the Experimental section. We next conducted IPs using anti-PKCϵ antisera and then probed for COIV in Western blots (Figure 3A). We observed a 11.1±2.9-fold (n=6, P<0.03) increase in PKCϵ–COIV co-precipitation in cells subjected to hypoxic PC. We then repeated the IPs using anti-COIV antisera and probed in Western blots using anti-PKCϵ antisera (Figure 3B). Following 30 min of hypoxia, we observed means±S.E.M. increases in PKCϵ co-IP with COIV-selective antisera above normoxic control levels of 8.2±2.7-fold (n=3, P<0.03). These results were consistent with a 30 min NCM hypoxia exposure triggering an interaction between PKCϵ and COIV inside NCM mitochondria. We propose that the PKCϵ–COIV interaction occurs on the matrix side of the IM, but future studies employing mitochondrial subfractionation and/or immunoelectron microscopy techniques will be required to confirm this hypothesis. We are currently conducting these studies. We are also currently pursuing studies to determine the nature of the PKCϵ–COIV interaction (e.g. substrate–enzyme interactions and direct binding partner). Finally, despite the fact that we used density- gradient-purified mitochondria, we cannot completely rule out the possibility that contaminants from non-mitochondrial fractions contributed to the PKCϵ–COIV interaction.
Figure 3. PKCϵ and the COIV subunit co-immunoprecipitate following exposure of NCMs to hypoxic PC.

NCMs were exposed to normoxic (Con) or 30 min hypoxia (Hx 30′) as shown. Mitochondria isolated from each treatment group were then subjected to IPs using antisera selective for the PKCϵ (A) isoenzyme or COIV (B). Immunoprecipitates were then subjected to SDS/PAGE and electrotransferred on to nitrocellulose paper followed by Western-blot analyses using anti-COIV (A) or anti-PKCϵ (B) antisera and 125I-labelled Protein A detection (see the Experimental section). Molecular-mass standard migrations (kDa) are indicated on the left side of each autoradiograph. Means±S.E.M. densitometry data were taken from three independent experiments, each from a separate myocyte preparation.
Biphasic effects of hypoxia on CO activity in NCMs
We next determined if the hypoxia-induced PKCϵ–COIV interaction was associated with changes in CO activity. NCMs were exposed to hypoxia for 0–180 min, mitochondria were isolated under isotonic conditions and CO activity was monitored as described in the Experimental section. When mitochondria isolated from NCMs exposed to hypoxia for up to 60 min were monitored in the absence of the detergent D-β-M (n-dodecyl-β-maltoside), less than 5% of the total CO activity was observed (Figure 4, hatched bars). This indicated that the majority of the CO activity resided inside intact mitochondria. In contrast, in assays using D-β-M-solubilized mitochondria, there was a timedependent hypoxia-induced enhancement of CO activity that reached a peak at approx. 10 min and remained elevated at least 2-fold above normoxic control levels for up to 60 min. However, after 90–180 min of hypoxia, the CO activity decreased to background levels, which may reflect dramatic reductions in complex IV activity, as was previously reported after prolonged myocardial ischaemia [40]. The means±S.E.M. of CO activity (expressed as a percentage of normoxic control) after 10, 60 and 180 min of hypoxia were 343±38, 219±16 and 26±11 (n=4, each group P<0.01 versus control) respectively.
Figure 4. Brief hypoxia enhances CO activity in NCMs.

Cells were exposed to hypoxia for the time periods shown followed by isolation of mitochondria (see the Experimental section). Mitochondria were then assayed for CO activity in the absence (hatched bars) or presence (open bars) of 1 mM D-β-M detergent. Results shown are means±S.E.M. for a single experiment conducted in duplicate and are representative of four identical experiments, each taken from a separate myocyte preparation.
In contrast with our findings, however, there have been previous reports suggesting that uncoupling of the mitochondrial ETC contributes to cardiac PC in adult rat hearts. For example, Ganote et al. reported potassium cyanide (KCN) [55] and DNP (2,4-dinitrophenol) [56]-induced cardiac protection against ischaemia/reperfusion injury in ex vivo adult rat hearts. Lesnefsky et al. [57] found that perfusion of adult rat hearts with the complex I inhibitor rotenone for 1 min prior to and during 45 min of ischaemia induced cardiac PC. Additionally, Holmuhamedov et al. [58] reported that the mKATP (mitochondrial ATP-dependent potassium) channel-opening drugs diazoxide and pinacidil had protonophoric effects on adult rat hearts to uncouple mitochondrial membrane potential as a mechanism of PC. The precise reasons for the discrepancy between our results and theirs are incompletely understood, but could involve differences in model systems, the use of neonatal versus adult heart cells and protocol differences. For example, neonatal heart cells rely more heavily on glycolytic metabolism than adult heart cells [59] and they are considerably more resistant to damage related to oxygen deprivation than the adult myocardium. Therefore differences in the mechanisms of PC in neonatal versus adult myocardium may exist. We would also like to point out that these studies [55–58] did not confirm the involvement of the PKCϵ isoenzyme in PC, opening the possibility that they may have been studying an altogether different cardioprotective mechanism(s) than we did. In addition, it is not clear that inhibition of ETC function is the direct cause of protection. In one study, Ganote et al. [56] used 5 mM KCN in Langendorf perfusates to uncouple complex IV prior to reperfusion after 45 min of ischaemia. It is likely that 45 min of ischaemia would by itself inhibit complex IV substantially and therefore the proposed protective mechanism of KCN would be related to its continuous inhibition of complex IV during reperfusion. This method is very different from the one we used, and it is entirely possible that both methods (ours and those of [55–58]) could induce protection, particularly in light of the fact that, in recent studies, cardiac post-conditioning (i.e. pharmacological treatments or brief periods of reperfusion and ischaemia prior to a prolonged reperfusion) in cardiac myocytes has been shown to produce equal protection as the PC methods [60]. These results must be tempered, however, by the exceptionally high level of KCN used in their study. The action of cyanide to inhibit complex IV involves protonation of the cyano anion followed by its binding to the cytochrome a3-Cu2+ binuclear centre of COI. This inhibits O2 binding to that site and is essentially irreversible (predicted t1/2 for CN− release is approx. 138 h) [61], which brings into question how irreversible inhibition of CO could be protective in the long term, as subsequent aerobic respiration would then be inhibited. In addition, 120 μM blood levels of cyanide are fatal in man [62], and KCN potently inhibits other enzymes, including carbonic anhydrase [63], superoxide dismutase [64], phosphorylase [65] and several gluco- and galacto-sidases [66]. There has also been a report suggesting that KCN inhibits stimulus-induced mobility shifts (suggestive of autophosphorylation) of the PKCα, PKCβ2 and PKCϵ isoenzymes in smooth muscle cells [67]. In addition, these authors replaced glucose in ischaemic incubations with 10 mM concentrations of mannitol to maintain the physiological osmotic pressure. They also included mannitol during reperfusions, which does not mimic physiological reperfusion and would necessitate greater dependence on glycogen and fatty acid oxidation. It is also well known that mannitol at this concentration can alter coronary flow, have antioxidant effects and alter nitric oxide levels [68], and mannitol alone has been previously shown to decrease myocardial ischaemia/reperfusion injury [68,69].
Similarly, in studies using DNP to induce protection, it is also unclear that ETC uncoupling is the mechanism of PC. DNP is a more selective mitochondrial uncoupler than KCN, but it has also been reported to inhibit the sarcolemmal NADPH dehydrogenase [70] and alter the activity of the actomyosin ATPase in skeletal muscle [71]. However, the major complication in the interpretation of these results is that DNP (and metabolic inhibition in general) uncouples ATP production, which will promote the accumulation of AMP in cardiac and other cells. An elevation in the AMP/ATP ratio will activate AMPK (AMP-dependent protein kinase) that, in general, will inhibit enzymatic processes consuming ATP and facilitate activation of those producing ATP [72]. For example, AMPK will phosphorylate and enhance the activities of glycolytic enzymes such as phosphofructokinase 2, which in turn increase glycolytic ATP production. Active AMPK also increases the translocation of glucose transporters to the sarcolemma, which greatly facilitates glucose uptake. Of interest, these actions of DNP have been demonstrated in cardiac myocytes [73]. ETC uncoupling does not appear to be required for this protection, as agonists that activate AMPK without mitochondria uncoupling produce the same effects [72]. Furthermore, these treatments also activate p38 MAPK [73], which (in the absence of irreversible mitochondrial uncoupling) also produces cardioprotection [19–21]. AMPK also inhibits acetyl-CoA carboxylase and HMG-CoA reductase (3-hydroxy-3-methylglutaryl-CoA reductase), which will increase fatty acid oxidation in heart and also lower cholesterol levels. In this context, it is important to note that statins (HMG-CoA reductase inhibitors) have previously been reported to have protective actions on the myocardium independent of mitochondrial ETC uncoupling, indicating that certain statin-like actions may also be evoked as a component of DNP actions. AMPK also increases the gene expression of numerous metabolic enzymes, including bioenergetic mitochondrial proteins [72].
In the studies by Lesnefsky et al. [57], 60 μM concentration of the irreversible ETC complex I inhibitor rotenone administered to ex vivo rat hearts prior to prolonged ischaemia (without washout or reperfusion) prevented ROS production and inhibition of complex IV activity. It is very possible that different signalling mechanisms could be induced by their experimental conditions from those evoked in our NCM model of hypoxic PC, particularly since their study involved continuous incubation with rotenone throughout the index ischaemia period. We propose that PC may occur via inhibition of complex I with rotenone and via an independent mechanism involving enhancement of complex IV. The observed phenotype may also be highly dependent on the model system (neonatal versus adult) and the timing (pretreatment versus continuous throughout index ischaemia) of the PC stimulus.
In the studies by Holmuhamedov et al. [58] using diazoxide and pinacidil, there was no evaluation of extensive drug time courses, and complex IV activity was not directly monitored. It is possible that early times of mKATP channel opener treatment may have revealed the enhancement of CO activity that we observed. There is also controversy over the role of the mKATP channel and the actions of diazoxide and pinacidil in the induction of PC [74]. In addition to diazoxide/pinacidil effects on mKATP channels, there have been reports of this drug class to directly inhibit ETC complex II, promote the opening of the MPTP, induce changes in mitochondrial matrix volumes, cause release of mitochondrial Ca2+, adenylate kinase and cytochrome c and other effects [74]. There has also been a report demonstrating a lack of ETC uncoupling and increased O2 consumption (as could occur with enhanced cytochrome oxidase activity) in mKATP channel activator-induced PC [74]. We therefore believe that the evidence supporting an uncontested role for mitochondrial ETC uncoupling to play a direct role in cardiac PC is controversial and does not rule out the possibility that enhanced CO activity could contribute to cardioprotective mechanisms.
Hypoxia-induced elevation of CO activity is attenuated by a PKCϵ-selective translocation inhibitor
We next determined if the enhancement of CO activity observed following brief hypoxia exposures was mediated by the PKCϵ isoenzyme (Figure 5). NCMs were first incubated in the presence of 1 μM concentrations of control or PKCϵ-selective translocation inhibitor peptides for 30 min. To facilitate cell entry, these peptides were chemically coupled with the HIV-Tat carrier peptide as described previously [37]. Cells were then subjected to a 30 min hypoxia exposure. In the carrier control and δV1-1 peptide groups, we observed 2.8±0.6 and 2.7±0.2 (n=2)-fold enhancements of CO activity following a 30 min exposure to hypoxia. There was no statistically significant difference between hypoxia-induced enhancement of CO activities in the carrier control versus the δV1-1 group (P<0.7). In contrast, the PKCϵ-selective translocation inhibitor (ϵV1-2) markedly attenuated the hypoxia-induced increase in CO activity to just 0.9±0.2-fold (P<0.04) above normoxic control levels (Figure 5). Our results are therefore consistent with the PKCϵ isoenzyme mediating hypoxia-induced enhancement of CO activity under conditions that induce PC in NCMs.
Figure 5. Hypoxia-induced increases in CO activity are attenuated by introduction of a PKCϵ-selective translocation inhibitor.

NMCs were preincubated with HIV-Tat coupled control (left) or PKC isoenzyme-selective translocation inhibitor peptides as described in the Experimental section. Next, cells were given normoxic (N) or hypoxia (Hx) exposures for 20 min and mitochondria were isolated from Optiprep density gradients as described in the Experimental section. CO activity was then monitored in all groups using a spectrophotometric method, which detects the oxidation of ferrocytochrome c to ferricytochrome c (see the Experimental section). Shown are mean±half range data for a single experiment assayed for CO activity in duplicate. Results are typical of two identical experiments, each taken from a different myocyte preparation.
Hypoxic PC of NCMs protects COIV levels
To determine if PC had effects on COIV levels that could be detected following prolonged index, hypoxic cells were subjected to control, PC+9 h index hypoxia or 9 h index hypoxia alone. Next, mitochondria isolated from each treatment group were solubilized and subjected to Western-blot analyses. Equal amounts of mitochondrial protein from each treatment group were used for these analyses. Similar results were obtained using whole cell lysates (results not shown), ruling out the possibility that differences in COIV subcellular localization could explain the index hypoxia-induced decline in COIV levels. Figure 6 (lanes 1 and 2 versus lanes 5 and 6) shows a clear decline in COIV levels when normoxic controls were compared with cells receiving a 9 h index hypoxia (a 61% decrease; n=4). In contrast, cells receiving a 30 min hypoxic PC step prior to the 9 h index hypoxia showed only a 13% (n=4) decrease in COIV levels. The preservation of COIV levels in the PC+9 h hypoxia (compared with the cells receiving 9 h hypoxia alone) was statistically significant (P<0.02). Since we observe augmentations of CO activity (Figure 5) and enhanced COIV–PKCϵ interactions (Figure 3) during much shorter PC periods (and COIV levels are not affected under those conditions (results not shown), the PC-induced enhancement of these parameters cannot be explained by changes in COIV levels during the early phases of PC. However, when PC is administered before prolonged hypoxia, an additional protection may occur by preserving COIV levels and ETC function. Determination of whether the PC-induced protection of COIV levels involves modification of COIV nuclear gene expression (i.e. COIV is coded for in the cell nucleus) or protein synthesis, stability, degradation or import into mitochondria will require further study.
Figure 6. Hypoxic PC preserves COIV levels in NCMs.

NCMs were subjected to control (normoxic), PC (30 min hypoxic/30 min reperfusion period) followed by 9 h of (index) hypoxia (PC+9 h) or 9 h of hypoxia alone. Cells were lysed and mitochondria were isolated from Percoll/Optiprep gradients as described in the Experimental section. COIV levels were then monitored in mitochondria isolated from each treatment group by Western blotting using anti-COIV antisera. Similar results were obtained whether whole cell lysates or purified mitochondria were used, indicating that the observed changes occur inside mitochondria. Shown in the upper part of the Figure is a representative autoradiograph in which samples from two independent experiments, each conducted on a separate myocyte prepapration, are shown. Note that each histogram bar corresponds to two gel lanes (one from each experiment). Lanes 1, 3 and 5 are from the same experiment and lanes 2, 4 and 6 are from a second experiment. The histogram shows means±S.E.M. densitometry values taken from four independent experiments conducted on separate myocyte preparations.
Significance of mitochondrial PKCϵ–COIV interactions in PC
In the present study, we have identified a PC-induced PKCϵ–COIV interaction in NCM mitochondria (Figure 3) that is associated with a 2–4-fold enhancement of CO activity (Figure 4). This is the first report of hypoxia-induced modulation of CO activity by the PKCϵ isoenzyme. We do not believe that CO activity is enhanced throughout the entire prolonged (9 h) index ischaemia. In fact, Figure 4 illustrates a dramatic reduction in CO activity following 90–180 min of hypoxia. We therefore hypothesize that early enhancement of CO activity during short (≤60 min) hypoxia exposure sets in motion a protective response that can be observed following 9 h of hypoxia in NCMs. It should be noted, however, that our study employed NCMs, which are highly resistant to hypoxic damage when compared with adult cardiac cells. In addition, we did not examine the effects of reperfusion after index hypoxia, which is where a major component of ischaemia/reperfusion injury occurs in the adult myocardium. Future studies will be required to determine if the responses observed in the present study occur in the adult myocardium and to determine their relationship to protective mechanisms operational in adult mammalian ischaemia/reperfusion injury. We therefore propose a novel potential mechanism of PC involving PKCϵ regulation of the ETC via interactions with COIV. PKCϵ enhancement of CO activities should improve the efficiency of electron flow from cytochrome c to molecular oxygen and enhance proton efflux out of the mitochondrial matrix [75]. These combined activities aid in maintenance of the mitochondrial inner membrane electrochemical gradient and aerobic respiration. In support of our hypothesis, improved preservation of ATP levels following adenosine and other PC exposures has been previously reported [76] and could be related (at least in part) to changes in complex IV activity. Also of interest, CO inhibition shifts other ETC complexes to a more reduced state, which favours the production of superoxide from ETC complexes I and III [39,40]. Therefore enhanced CO activities should reduce ROS production via improved electron flow through complexes I and III and, consequently, prevent oxidative damage to heart cells. CO activity also declines during cardiac ischaemia or metabolic inhibition by cyanide or elevated levels of carbon monoxide or nitric oxide [39,40]. PKCϵ activation and modulation of CO in these states may, therefore, attenuate CO inhibition and ROS production.
It is well known that the PKCϵ isoenzyme induces cardioprotection during cardiac PC [2,19,20,24,28,29,31,33–37,49]. Many mitochondrial signalling events have been proposed to contribute to this protection including the formation of signalling modules between PKCϵ and MAPKs [19], inhibition of the MPTP [33] and elevation of mitochondrial ROS [77,78]. Our work does not challenge any of these potential mechanisms of PC. Instead, we hypothesize that PKCϵ modulation of CO could work in synergy with many of them. For example, PKCϵ inhibition of the MPTP [33] preserves mitochondrial cytochrome c levels necessary for complex IV activity. Enhanced CO activity and decreased ROS production may also prevent or delay MPTP opening by indirectly affecting mitochondrial membrane potentials. It is also likely that enhanced energetics and decreased ROS production would influence many other mitochondrial PKCϵ signalling events as well. We therefore propose potential roles for the PKCϵ isoenzyme in NCM mitochondrial energetics and ROS production, which may be of significance in myocardial ischaemia/reperfusion damage, metabolic inhibition by cyanide, carbon monoxide or nitric oxide and in protection induced by cardiac PC.
Online Data
Acknowledgments
We thank the Emory University Microchemical Facility for synthesis of Tat-coupled PKC inhibitory peptides. This research was supported by grants from the American Heart Association (no. 0355244B) and the National Heart Lung and Blood Institute (no. HL076805).
References
- 1.Murry C. E., Jennings R. B., Reimer K. A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 2.Gray M. O., Karliner J. S., Mochly-Rosen D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg M., Zhang H. L., Steinberg S. F. Hypoxia alters the subcellular distribution of protein kinase C isoforms in neonatal rat ventricular myocytes. J. Clin. Invest. 1997;99:55–61. doi: 10.1172/JCI119133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Light P. E., Sabir A. A., Allen B. G., Walsh M. P., French R. J. Protein kinase C-induced changes in the stiochiometry of ATP binding activates cardiac ATP-sensitive K+ channels: a possible mechanistic link to ischemic preconditioning. Circ. Res. 1996;79:399–406. doi: 10.1161/01.res.79.3.399. [DOI] [PubMed] [Google Scholar]
- 5.Nayeem M. A., Elliot G. T., Shah M. R., Hastillo-Hass S. L., Kukreja R. C. Monophosphoryl lipid A protects adult rat cardiac with induction of heat shock proteins: a cellular model of pharmacologic preconditioning. J. Mol. Cell. Cardiol. 1997;29:2305–2310. doi: 10.1006/jmcc.1997.0452. [DOI] [PubMed] [Google Scholar]
- 6.Rowland R. T., Meng X., Cleveland J. C., Meldrum D. R., Harken A. H., Brown J. M. Cardioadaptation induced by cyclic ischemic preconditioning is mediated by translational regulation of de novo protein synthesis. J. Surg. Res. 1997;71:155–160. doi: 10.1006/jsre.1997.5142. [DOI] [PubMed] [Google Scholar]
- 7.Liu G. S., Thornton J., VanWinkle D. M., Stanley A. W., Olsson R. A., Downey J. M. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- 8.Shizukuda Y., Iwamoto T., Mallet R. T., Downey H. F. Hypoxic preconditioning attenuates stunning by repeated coronary artery occlusions in dog heart. Cardiovasc. Res. 1993;27:559–564. doi: 10.1093/cvr/27.4.559. [DOI] [PubMed] [Google Scholar]
- 9.Randall M. D., Gardener S. M., Bennett T. Enhanced cardiac preconditioning in the isolated heart of transgenic ((mReN 2) 27) hypertensive rat. Cardiovasc. Res. 1997;33:400–409. doi: 10.1016/s0008-6363(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 10.Stowe D. F., Fujita S., Bosnjak Z. J. Improved contractility and coronary flow in isolated hearts after 1-day hypothermic preservation with isoflurane is not dependent on K(ATP) channel activation. Anesthesiology. 1998;88:233–244. doi: 10.1097/00000542-199801000-00032. [DOI] [PubMed] [Google Scholar]
- 11.Tuschida A., Liu G. S., Mullane K., Downey J. M. Acadesine lowers temporal threshold for the myocardial infarct size limiting effect of preconditioning. Cardiovasc. Res. 1993;27:116–120. doi: 10.1093/cvr/27.1.116. [DOI] [PubMed] [Google Scholar]
- 12.Meldrum D. R. Mechanisms of cardiac preconditioning ten years after the discovery of ischemic preconditioning. J. Surg. Res. 1997;73:1–13. doi: 10.1006/jsre.1997.5187. [DOI] [PubMed] [Google Scholar]
- 13.Lu E. X., Chen S. X., Yuan M. D., Hu T. H., Zhon H. C., Luo W. J., Li G. H., Xu L. M. Preconditioning improves myocardial preservation in patients undergoing open heart operations. Ann. Thorac. Surg. 1997;64:1320–1324. doi: 10.1016/S0003-4975(97)00838-2. [DOI] [PubMed] [Google Scholar]
- 14.Meldrum D. R., Cleveland J. C., Jr, Sheridan B. C., Rowland R. T., Banerjee A., Harken A. H. Cardiac preconditioning with calcium: clinically accessible myocardial protection. J. Thorac. Cardiovasc. Surg. 1996;112:778–786. doi: 10.1016/S0022-5223(96)70065-X. [DOI] [PubMed] [Google Scholar]
- 15.Liang B. T. Protein kinase C-mediated preconditioning of cardiac myocytes: role of adenosine receptor and K(ATP) channel. Am. J. Physiol. 1997;273:H847–H853. doi: 10.1152/ajpheart.1997.273.2.H847. [DOI] [PubMed] [Google Scholar]
- 16.Hoag J. B., Qian Y. Z., Nayeem M. A., D'Angelo M., Kukreja R. C. ATP-sensitive potassium channel mediates delayed ischemic protection by heat stress in rabbit heart. Am. J. Physiol. 1997;273:H2458–H2464. doi: 10.1152/ajpheart.1997.273.5.H2458. [DOI] [PubMed] [Google Scholar]
- 17.Cleveland J. C., Jr, Meldrum D. R., Rowland R. T., Sheridan B. C., Banerjee A., Harken A. H. The obligate role of protein kinase C in mediating clinically accessible cardiac preconditioning. Surgery. 1996;120:345–352. doi: 10.1016/s0039-6060(96)80308-4. [DOI] [PubMed] [Google Scholar]
- 18.Rehring T. F., Friese R. S., Cleveland J. C., Meng X., Robertson F. G., Harken A. H., Banerjee A. Alpha-adrenergic preservation of myocardial pH during ischemia is protein kinase C isoform-dependent. J. Surg. Res. 1996;63:324–327. doi: 10.1006/jsre.1996.0269. [DOI] [PubMed] [Google Scholar]
- 19.Baines C. P., Zhang J., Wang G. W., Zheng Y. T., Xiu J. X., Cardwell E. M., Bolli R., Ping P. Mitochondrial PKCepsilon and MAPK form modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ. Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 20.Li R. C., Ping P., Zhang J., Wead W. B., Cao X., Gao J., Zheng Y., Huang S., Han J., Bolli R. PKCepsilon modulates NF-kappaB and AP-1 via mitogen-activated protein kinases in adult rabbit cardiomyocytes. Am. J. Physiol. 2000;279:H1679–H1689. doi: 10.1152/ajpheart.2000.279.4.H1679. [DOI] [PubMed] [Google Scholar]
- 21.Yue Y., Qin Q., Cohen M. V., Downey J. M., Critz S. D. The relative order of mK(ATP) channels, Free radicals, and p38 MAPK in preconditioning's protective pathway in rat heart. Cardiovasc. Res. 2002;55:681–689. doi: 10.1016/s0008-6363(02)00452-2. [DOI] [PubMed] [Google Scholar]
- 22.Qin Q., Downey J. M., Cohen M. V. Acetylcholine but not adenosine triggers preconditioning through PI3-kinase and a tyrosine kinase. Am. J. Physiol. 2002;283:H2322–H2330. doi: 10.1152/ajpheart.00476.2002. [DOI] [PubMed] [Google Scholar]
- 23.Ping P., Zhang J., Zheng Y. T., Li R. C., Dawn B., Tang X. L., Takano H., Balafanova Z., Bolli R. Demonstration of selective protein kinase C-dependent activation of src and lyk tyrosine kinases during ischemia preconditioning in conscious rabbits. Circ. Res. 1999;85:542–550. doi: 10.1161/01.res.85.6.542. [DOI] [PubMed] [Google Scholar]
- 24.Song C., Vondriska T. M., Wang G. W., Klein J. B., Cao X., Zhang J., Kang Y. J., D'souza S., Ping P. Molecular conformation dictates signalling module formation: example of PKCepsilon and Src tyrosine kinase. Am. J. Physiol. 2002;282:H1166–H1171. doi: 10.1152/ajpheart.00830.2001. [DOI] [PubMed] [Google Scholar]
- 25.Oldenburg O., Quin Q., Sharma A. R., Cohen M. V., Downey J. M., Benoit J. N. Acetylcholine leads to free radical production dependent on K(ATP) channels, G(I) proteins, phosphatidylinositol 3-kinase and tyrosine kinase. Cardiovasc. Res. 2002;55:544–552. doi: 10.1016/s0008-6363(02)00332-2. [DOI] [PubMed] [Google Scholar]
- 26.Liu X., Engelman R. M., Moraru I. I., Rousou J. A., Flack J. E., Deaton D. W., Mualik N., Das D. K. Heat shock a new approach for myocardial preservation in cardiac surgery. Circulation. 1992;86(Suppl. 5):LL358–LL363. [PubMed] [Google Scholar]
- 27.Vegh A., Papp J. G., Szekere S. L., Parratt J. R. Prevention by an inhibitor of the L-arginine nitric oxide pathway of the antiarrythmic effects of bradykinin in anesthetized dogs. Br. J. Pharmacol. 1993;110:18–29. doi: 10.1111/j.1476-5381.1993.tb13764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balafonova Z., Bolli R., Zhang J., Zheng Y., Pass J. M., Bhatnagar A., Tang X. L., Wang O., Cardwell E., Ping P. Nitric oxide (NO) induces nitration of protein kinase C epsilon, facilitating PKCepsilon translocation via enhanced PKCepsilon-RACK2 interactions. A novel mechanism of NO-triggered activation of PKCepsilon. J. Biol. Chem. 2002;277:15021–15027. doi: 10.1074/jbc.M112451200. [DOI] [PubMed] [Google Scholar]
- 29.Ping P., Takano H., Zhang J., Tang X. L., Qiu Y., Li R. C., Banerjee S., Dawn B., Balafonova Z., Bolli R. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: a signalling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ. Res. 1999;84:587–604. doi: 10.1161/01.res.84.5.587. [DOI] [PubMed] [Google Scholar]
- 30.Sun J. Z., Tang X. L., Park S. W., Qui Y., Turrens J. F., Bolli R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J. Clin. Invest. 1996;97:562–576. doi: 10.1172/JCI118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ping P., Zhang J., Qiu Y., Tang X. L., Manchikalapudi S., Cao X., Bolli R. Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ. Res. 1997;81:404–414. doi: 10.1161/01.res.81.3.404. [DOI] [PubMed] [Google Scholar]
- 32.Qiu Y., Ping P., Tang X. L., Manchikalapudi S., Rizvi A., Zhang J., Takano H., Wu W. J., Teschner S., Bolli R. Direct evidence that protein kinase C plays an essential role in the development of late preconditioning against myocardial stunning in conscious rabbits and that epsilon is the isoform involved. J. Clin. Invest. 1998;101:2182–2198. doi: 10.1172/JCI1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baines C. P., Song C., Zheng Y., Wang G., Zhang J., Wang O., Guo Y., Bolli R., Cardwell E. M., Ping P. Protein kinase C ϵ interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pass J. M., Zheng Y., Wead W. B., Zhang J., Li R. C., Bolli R., Ping P. PKC epsilon activation induces dichotomous cardiac phenotypes and modulates PKC epsilon-RACK interactions and RACK expression. Am. J. Physiol. 2001;280:H946–H955. doi: 10.1152/ajpheart.2001.280.3.H946. [DOI] [PubMed] [Google Scholar]
- 35.Saurin A. T., Pennington D. J., Raat N. J. H., Latchman D. S., Owen M. J., Marber M. S. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc. Res. 2002;55:672–680. doi: 10.1016/s0008-6363(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 36.Gray M. O., Zhou H.-Z., Schafhalter-Zoppoth I., Zhu P., Mochly-Rosen D., Messing R. O. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C ϵ. J. Biol. Chem. 2004;279:3596–3604. doi: 10.1074/jbc.M311459200. [DOI] [PubMed] [Google Scholar]
- 37.Chen L., Hahn H., Wu G., Chen C., Liron T., Schechtman D., Cavallaro G., Banci L., Guo Y., Bolli R., et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta and epsilon PKC. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11114–11119. doi: 10.1073/pnas.191369098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capaldi R. A., Marusich M. F., Taanman J. Mammalian cytochrome c oxidase: characterization of enzyme and immunological detection of subunits in tissue extracts and whole cells. Methods Enzymol. 1995;260:117–132. doi: 10.1016/0076-6879(95)60134-1. [DOI] [PubMed] [Google Scholar]
- 39.Moncada S., Erusalimsky J. D. Does nitric oxide modulate energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 40.Marin-Garcia J., Goldenthal M. J. The mitochondrial organelle and the heart. Rev. Esp. Cardiol. 2002;55:1293–1310. doi: 10.1016/s0300-8932(02)76802-4. [DOI] [PubMed] [Google Scholar]
- 41.Lee I., Salomon A. R., Ficarro S., Mathes I., Lottspeich F., Grossman L. I., Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J. Biol. Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- 42.Bender E., Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- 43.Miyazaki T., Neff L., Tanaka S., Home W. C., Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J. Cell Biol. 2003;160:709–718. doi: 10.1083/jcb.200209098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogbi M., Chew C. S., Pohl J., Stuchlik O., Ogbi S., Johnson J. A. Cytochrome c oxidase subunit IV as a marker of protein kinase Cϵ function in neonatal cardiac myocytes: implications for cytochrome c oxidase activity. Biochem. J. 2004;382:923–932. doi: 10.1042/BJ20040468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson J. A., Gray M. O., Chen C., Karliner J., Mochly-Rosen D. An improved method for the introduction of peptides into neonatal cardiac myocytes. Circ. Res. 1996;79:1086–1099. doi: 10.1161/01.res.79.6.1086. [DOI] [PubMed] [Google Scholar]
- 46.Johnson J. A., Mochly-Rosen D. Inhibition of the spontaneous rate of contraction of neonatal cardiac myocytes by protein kinase C isozymes. A putative role for the ϵ isozyme. Circ. Res. 1995;76:654–663. doi: 10.1161/01.res.76.4.654. [DOI] [PubMed] [Google Scholar]
- 47.Iyengar R., Birnbaumer L. Hormone receptor modulates the regulatory component of adenylyl cyclase by reducing its requirement for Mg2+ and enhancing its extent of activation by guanine nucleotides. Proc. Natl. Acad. Sci. U.S.A. 1982;79:5179–5183. doi: 10.1073/pnas.79.17.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu G. S., Cohen M. V., Mochly-Rosen D., Downey J. M. Protein kinase C epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell. Cardiol. 1999;31:1937–1948. doi: 10.1006/jmcc.1999.1026. [DOI] [PubMed] [Google Scholar]
- 49.Ron D., Luo J., Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of b protein kinase C in vivo. J. Biol. Chem. 1995;270:24180–24187. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 50.Johnson J. A., Gray M. O., Chen C.-H., Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J. Biol. Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- 51.Lawrence K. M., Townsend P. A., Davidson S. M., Carroll C. J., Eaton S., Hubank M., Knight R. A., Stephanou A., Latchman D. S. The cardioprotective effect of urocortin during ischaemia/reperfusion involves the prevention of mitochondrial damage. Biochem. Biophys. Res. Commun. 2004;321:479–486. doi: 10.1016/j.bbrc.2004.06.170. [DOI] [PubMed] [Google Scholar]
- 52.Aizawa K., Turner L. A., Weihrauch D., Bosnjak Z. J., Kwok W. M. Protein kinase C-epsilon primes the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel to modulation by isoflurane. Anesthesiology. 2004;101:381–389. doi: 10.1097/00000542-200408000-00019. [DOI] [PubMed] [Google Scholar]
- 53.Ohnuma Y., Miura T., Miki T., Tanno M., Kuno A., Tsuchida A., Shimamoto K. Opening of mitochondrial KATP channel occurs downstream of PKC-ϵ activation in the mechanism of preconditioning. Am. J. Physiol. 2002;283:H440–H447. doi: 10.1152/ajpheart.00434.2001. [DOI] [PubMed] [Google Scholar]
- 54.Kilts J. D., Grocott H. P., Kwatra M. M. Gαq-coupled receptors in human atrium function through protein kinase C ϵ and δ. J. Mol. Cell. Cardiol. 2005;38:267–276. doi: 10.1016/j.yjmcc.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 55.Ganote C. E., Worstell J., Kaltenbach J. P. Oxygen-induced enzyme release after irreversible myocardial injury. Am. J. Pathol. 1976;84:327–350. [PMC free article] [PubMed] [Google Scholar]
- 56.Ganote C. E., McGarr J., Liu S. Y., Kaltenbach J. P. Oxygen-induced enzyme release. Assessment of mitochondrial function in anoxic myocardial injury and effects of the mitochondrial uncoupling agent 2,4-dinitrophenol (DNP) J. Mol. Cell. Cardiol. 1980;12:387–408. doi: 10.1016/0022-2828(80)90049-8. [DOI] [PubMed] [Google Scholar]
- 57.Lesnefsky E. J., Chen Q., Moghaddas S., Hassan M. O., Tandler B., Hoppel C. L. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 2005;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 58.Holmuhamedov E. L., Jahangir A., Oberlin A., Komarov A., Colombini M., Terzic A. Potassium channel openers are uncoupling protonophores: implications in cardioprotection. FEBS Lett. 2004;568:167–170. doi: 10.1016/j.febslet.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 59.Jarmakani J. M., Nagatomo T., Nakazawa M., Langer G. A. Effect of hypoxia on myocardial high-energy phosphates in the neonatal mammalian heart. Am. J. Physiol. 1978;235:H475–H481. doi: 10.1152/ajpheart.1978.235.5.H475. [DOI] [PubMed] [Google Scholar]
- 60.Hausenloy D. J., Tsang A., Yellon D. M. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc. Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Panda M., Robinson N. C. Kinetics and mechanism for the binding of HCN to cytochrome c oxidase. Biochemistry. 1995;34:10009–10018. doi: 10.1021/bi00031a024. [DOI] [PubMed] [Google Scholar]
- 62.Lovejoy F. H., Jr, Linden C. H. Acute poison and drug overdosage. In: Isselbacher K. J., Braunwald E., Wilson J. D., Martin J., Fauci A. S., Kasper D. L., editors. Harrison's Principles of Internal Medicine. 13th edn. New York: McGraw-Hill; 1994. p. 2451. [Google Scholar]
- 63.Innocenti A., Antel J., Wurl M., Vullo D., Firnges M. A., Scozzafava A., Supuran C. T. Carbonic anhydrase inhibitors. Inhibition of isozymes I, II, IV, V and IX with complex fluorides, chlorides and cyanides. Bioorg. Med. Chem. Lett. 2005;15:1909–1913. doi: 10.1016/j.bmcl.2005.01.082. [DOI] [PubMed] [Google Scholar]
- 64.Inarrea P. Purification and determination of activity of mitochondrial cyanide-sensitive superoxide dismutase in rat tissue extract. Methods Enzymol. 2002;349:106–114. doi: 10.1016/s0076-6879(02)49326-3. [DOI] [PubMed] [Google Scholar]
- 65.Vandebroeck A., Uyttenhove K., Bollen M., Stalmans W. The hepatic glycogenolysis induced by reversible ischaemia or KCN is exclusively catalyzed by phosphorylase a. Biochem. J. 1988;256:685–688. doi: 10.1042/bj2560685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chinchetru M. A., Cabezas J. A., Calvo P. Characterization and kinetics of β-D-gluco/fuco/galactosidase from sheep liver. Comp. Biochem. Physiol. B. 1983;75:719–728. doi: 10.1016/0305-0491(83)90124-4. [DOI] [PubMed] [Google Scholar]
- 67.Kobashi M., Kanayama K., Ishida Y. Effects of metabolic inhibition on phosphorylation levels of PKC isoforms in guinea pig taenia caeci. J. Smooth Muscle Res. 2004;40:85–96. doi: 10.1540/jsmr.40.85. [DOI] [PubMed] [Google Scholar]
- 68.Poullis M. Mannitol and cardiac surgery. Thorac. Cardiovasc. Surg. 1999;47:58–62. doi: 10.1055/s-2007-1013112. [DOI] [PubMed] [Google Scholar]
- 69.Ferreira R., Burgos M., Liesuy S., Molteni L., Milei J., Fecha B. G., Boveris A. Reduction of reperfusion injury with mannitol cardioplegia. Ann. Thorac. Surg. 1989;48:77–84. doi: 10.1016/0003-4975(89)90182-3. [DOI] [PubMed] [Google Scholar]
- 70.Ventura C., Guarnieri C., Bastagli L., Caldarera C. M. Opioids stimulate sarcolemmal NAD(P)H vanadate-sensative dehydrogenase activity. Basic Res. Cardiol. 1988;83:376–383. doi: 10.1007/BF02005823. [DOI] [PubMed] [Google Scholar]
- 71.Salerno V. P., Ribeiro A. S., Dinucci A. N., Mignaco J. A., Sorenson M. M. Specificity and kinetic effects of nitrophenol analogues that activate myosin subfragment 1. Biochem. J. 1997;324:877–884. doi: 10.1042/bj3240877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ingwall J. S., Weiss R. G. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ. Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 73.Pelletier A., Joly E., Prentki M., Coderre L. Adenosine 5′-monophosphate-activated protein kinase and p38 mitogen-activated protein kinase participate in the stimulation of glucose uptake by dinitrophenol in adult cardiomyocytes. Endocrinology. 2005;146:2285–2294. doi: 10.1210/en.2004-1565. [DOI] [PubMed] [Google Scholar]
- 74.Minners J., McLeod J., Sack M. N. Mitochondrial plasticity in classical ischemic preconditioning – moving beyond the mitochondrial KATP channel. Cardiovasc. Res. 2003;59:1–6. doi: 10.1016/s0008-6363(03)00337-7. [DOI] [PubMed] [Google Scholar]
- 75.Gennis R. B. How does cytochrome oxidase pump protons? Proc. Natl. Acad. Sci. U.S.A. 1998;95:12747–12749. doi: 10.1073/pnas.95.22.12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gross H. E., Murphy E., Black R. G., Auchampach J., Steenbergen C. Overexpression of A3 adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H1562–H1568. doi: 10.1152/ajpheart.00335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yellon D. M., Downey J. M. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol. Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 78.Dawn B., Bolli R. Role of nitric oxide in myocardial preconditioning. Ann. N.Y. Acad. Sci. 2002;962:18–41. doi: 10.1111/j.1749-6632.2002.tb04053.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.