

Abstract
In growth-factor-stimulated signal transduction, cell-surface receptors recruit PI3Ks (phosphoinositide 3-kinases) and Ras-specific GEFs (guanine nucleotide-exchange factors) to the plasma membrane, where they produce 3′-phosphorylated phosphoinositide lipids and Ras-GTP respectively. As a direct example of pathway networking, Ras-GTP also recruits and activates PI3Ks. To refine the mechanism of Ras–PI3K cross-talk and analyse its quantitative implications, we offer a theoretical model describing the assembly of complexes involving receptors, PI3K and Ras-GTP. While the model poses the possibility that a ternary receptor–PI3K–Ras complex forms in two steps, it also encompasses the possibility that receptor–PI3K and Ras–PI3K interactions are competitive. In support of this analysis, experiments with platelet-derived growth factor-stimulated fibroblasts revealed that Ras apparently enhances the affinity of PI3K for receptors; in the context of the model, this suggests that a ternary complex does indeed form, with the second step greatly enhanced through membrane localization and possibly allosteric effects. The apparent contribution of Ras to PI3K activation depends strongly on the quantities and binding affinities of the interacting molecules, which vary across different cell types and stimuli, and thus the model could be used to predict conditions under which PI3K signalling is sensitive to interventions targeting Ras.
Keywords: mathematical modelling, phosphoinositide 3-kinase (PI3K), platelet-derived growth factor (PDGF), Ras, receptor tyrosine kinase, signal transduction
Abbreviations: ERK, extracellular-signal-regulated kinase; GEF, guanine nucleotide-exchange factor; GFP, green fluorescent protein; MKP, mitogen-activated protein kinase phosphatase; NDPK, nucleoside 5′-diphosphate kinase; N-WASP, neural Wiskott–Aldrich syndrome protein; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; TIRF, total internal reflection fluorescence
INTRODUCTION
PI3K (phosphoinositide 3-kinase) and Ras are pivotal players in signal transduction pathways that co-ordinate proliferation, survival and migration in normal and transformed cells. PI3Ks are composed of regulatory and catalytic subunits, and phosphorylate phosphatidylinositol lipids on the D-3 position, producing specific lipid second messengers in cell membranes that recruit and facilitate activation of signalling proteins containing pleckstrin homology and other modular domains [1–4]. Receptor tyrosine kinases and other tyrosine-phosphorylated proteins activate type I PI3Ks by engaging the Src homology 2 domains of the regulatory subunit, relieving inhibition of the catalytic subunit and localizing it in proximity to its plasma membrane substrate, PtdIns(4,5)P2 [5–7]. PDGF (platelet-derived growth factor) receptors, with two closely spaced PI3K-binding sites, bind with particularly high affinity in vitro (Kd∼1 nM or less) [8–11] and strongly activate PI3K activity in cells [12–14]. Receptors also enhance signalling through Ras proteins, which are small membrane-anchored GTPases that cycle between GDP- and GTP-bound states, by promoting Ras GEF (guanine nucleotide-exchange factor) activity and hence an increase in the level of Ras-GTP. It is in this state that Ras binds to the serine/threonine kinase Raf, initiating a well-known kinase cascade that results in the activation of ERK (extracellular-signal-regulated kinase). The direct interaction between Ras and Raf, two known proto-oncogenes, was a major discovery in the delineation of a growth-factor-stimulated cell proliferation pathway [15].
The finding that PI3Ks are also Ras effectors [16] was perhaps the most dramatic evidence that signalling pathways are highly interconnected, rather than linear and independent. Such cross-talk interactions offer multiple avenues for activating and regulating signalling enzymes, which can be thought of as nodes in a large cell regulatory network [17,18]. Like phosphorylated PDGF receptors, Ras-GTP binds directly to PI3K and enhances its enzymatic activity in vitro; in contrast, however, Ras-GTP engages the catalytic subunit, and with somewhat lower affinity (Kd∼100 nM) [16,19]. Oncogenic (G12V) Ras is constitutively GTP-bound and significantly activates PI3K in cells [7,16,19], suggesting that the interaction is sufficient for the activation and membrane localization of PI3K; however, Ras is not universally required for PI3K activation. Dominant-negative (S17N) Ras blocks receptor-mediated modulation of endogenous Ras-GTP, and the resulting effects on growth-factor-stimulated PI3K activation range from partial inhibition [16,20] to no inhibition [7].
These studies indicate that there are two productive routes for PI3K recruitment: (i) binding of the regulatory domain to tyrosine-phosphorylated receptors/adaptor proteins, and (ii) binding of the catalytic domain to Ras-GTP. In a given cell type under various stimulation conditions, however, the extents to which the two interactions contribute to PI3K activation are difficult to parse out. Another unresolved question is whether the two routes are independent or instead lead to the assembly of a stabilized ternary (receptor–PI3K–Ras) complex. In the latter case, the concentrating effect of membrane localization would tend to enhance the second step in the ternary complex assembly [21–23], i.e. the process would exhibit positive co-operativity. To address these quantitative issues, we offer a simple mathematical model of PI3K recruitment and activation. Analysis of the model suggested that the nature of the binding co-operativity could be ascertained in a straightforward way from quantitative measurements, and we proceeded to perform such experiments for the PDGF receptor system. Our results are consistent with a mechanism in which ternary complex assembly occurs with positive co-operativity as described above.
MATERIALS AND METHODS
Equilibrium-binding model of PI3K activation
A quantitative model was formulated based on the interactions of activated receptors (R*) and Ras-GTP (S*) that are free for binding with distinct subunits of cytosolic PI3K (P) (Figure 1A). It is assumed that these interactions occur rapidly on the time scale over which R* and S* change appreciably (minutes). Thus binary (RP and PS) and ternary (RPS) complexes form according to the following pseudo-equilibrium equations:
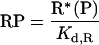 |
(1) |
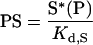 |
(2) |
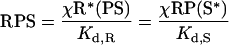 |
(3) |
Species are defined as depicted in Figure 1, Kd,R and Kd,S are dissociation constants for the receptor–PI3K and Ras–PI3K interactions respectively (in units of molecules/cell), and χ is an enhancement factor accounting for co-operativity in the formation of the ternary complex. Microscopic reversibility dictates that the magnitude of the enhancement χ does not depend on the sequence of binding, i.e. one obtains the same overall equilibrium constant for the assembly of RPS from its three components. The total numbers of PI3K (PTot), activated receptor (R*Tot), and Ras-GTP (S*Tot) molecules are specified in the model, yielding the following conservation equations:
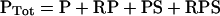 |
(4) |
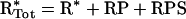 |
(5) |
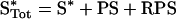 |
(6) |
Finally, the observed PI3K activity, AP, was taken as a weighted sum of the various binding states, with αi defined as the specific activity of species i.
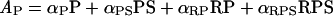 |
(7) |
The model equations were recast in terms of dimensionless parameters (R*Tot/Kd,R, S*Tot/Kd,S, Kd,R/PTot, Kd,S/PTot and χ), and the fraction of PI3K in each state was calculated using Microsoft Excel.
Figure 1. Model based on assembly of receptor, PI3K and Ras-GTP complexes.

(A) Model schematic. Cytosolic PI3K (P) is recruited to the plasma membrane by activated receptors (R*) or Ras-GTP (S*). These interactions produce RP and PS complexes, with equilibrium dissociation constants Kd,R and Kd,S respectively. The ternary RPS complex may then form, and it is postulated that the second binding step is greatly enhanced by induced proximity at the plasma membrane (χ≫1). Alternatively, the two interactions may exhibit no co-operativity (χ=1), negative co-operativity (0<χ<1) or purely competitive binding (χ=0). (B) Hypothetical effects of increasing Ras-GTP levels on the dose–response of PI3K activation. As long as Ras-GTP is sufficient for activating Ras, the basal PI3K activity will tend to be enhanced (1). The maximal PI3K activity can increase or decrease (2), depending on whether the ternary complex RPS exhibits a higher or lower specific activity than the binary RP complex. Finally, the number of activated receptors required to elicit a half-maximal increase in PI3K activation can decrease or increase (3), depending on whether the co-operativity factor χ is greater or less than 1 respectively.
Reagents
All tissue culture reagents were from Invitrogen. Human recombinant and 125I-labelled human PDGF-BB were from Peprotech and Amersham Biosciences respectively. Antibodies against Ras (Y13-259, agarose-conjugated), H-Ras, Akt-1/2 N-terminus, ERK1 C-terminus and MKP-1 (mitogen-activated protein kinase phosphatase 1) were from Santa Cruz Biotechnology. Antibodies against Akt pSer473, ERK pThr202/pTyr204 and PDGF β-receptor pTyr751 were from Cell Signaling Technology. Unless otherwise noted, all other reagents were from Sigma–Aldrich.
Plasmids and retroviral infection
The GFP (green fluorescent protein)–AktPH plasmid and construction of the ΔC1 vector harbouring G12V H-Ras were described previously [24]; other ΔC1-H-Ras constructs were prepared by the same methods. These H-Ras plasmids were used as PCR templates for cloning into the NotI/BamHI sites of the retroviral vector pBM-IRES-Puro, a gift from Dr Steven Wiley and Dr Lee Opresko (Pacific Northwest National Laboratory, Richland, WA, U.S.A.). All constructs were confirmed by DNA sequencing. Packaging cells were transfected by calcium phosphate precipitation for 4 h with 15 μg of pBM-IRES-Puro construct/100 mm plate. After replacing the medium, viral supernatants were collected at 24, 28 and 32 h after transfection, filtered, supplemented with 5 μg/ml polybrene and used for serial infection of NIH 3T3 fibroblasts (A.T.C.C.). These cells were plated at 2×105 cells/60-mm-diameter dish 24 h before infection. This procedure yielded >80% infection efficiency, as judged by control infections with pBM-IRES-EGFP. At 24 h after the last infection, viral supernatants were removed, and target cells were incubated with growth medium containing 2 μg/ml puromycin and selected for 2 days.
Cell culture, lysate preparation and biochemical assays
NIH 3T3 fibroblasts and variant cell lines were cultured at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) foetal bovine serum, 2 mM L-glutamine and the antibiotics penicillin and streptomycin. Ras expression was maintained under puromycin selection and was stable for at least five passages. Cells were serum-starved for 4 h before PDGF stimulation. Detergent lysates were prepared for quantitative immunoblotting, and immunoblots were performed using enhanced chemiluminescence, as described previously [25]. Phospho-Akt and -ERK blots comparing different cell lines were performed in parallel and were exposed at the same time. The Bio-Rad Fluor S-Max system, which gives a linear response with respect to light output, was used. Total protein concentrations were determined using the Micro BCA (bicinchoninic acid) kit (Pierce).
To determine the amounts of Ras-bound GTP, the coupled NDPK (nucleoside 5′-diphosphate kinase; Sigma N2635)–luciferase assay of Boss and colleagues [26,27] was performed essentially as described in [28]. A significant reduction in background was gained by preparing an exhaustively dialysed NDPK stock. This allowed us to pre-incubate 50 μl of GTP/GDP sample with 50 μl of FL-AAM (firefly luciferase/ATP assay mixture; Sigma), supplemented with 100 nM purified ADP, in order to deplete any remaining ATP contamination in the sample or ADP stock. To this, 50 m-units of the NDPK was added to initiate the reaction, and light production was integrated over 10 min in a PerkinElmer 96-well-plate-reading luminometer. Ras-bound GTP+GDP was quantified by first phosphorylating GDP to GTP in the presence of pyruvate kinase and phospho(enol)pyruvate (Sigma P9136 and P7127 respectively) [27]. GDP and GTP standards, pre-treated in this manner, yielded near identical standard curves. These amounts were converted into molecules per cell, with the cell number per plate estimated in parallel by Coulter counting.
PDGF receptor binding experiments and estimation of activated receptor numbers
Cells were plated in 35-mm-diameter dishes, serum-starved for 4 h, then incubated with 125I-PDGF for 5 min at 37 °C. To determine total cell-associated radioactivity, cells were washed rapidly six times with ice-cold PBS then lysed in 1 M NaOH. Surface-bound and internalized 125I-PDGF were distinguished by first incubating the washed cells in ice-cold acid strip buffer (50 mM glycine/HCl, 100 mM NaCl and 2 M urea, pH 3.0) for 10 min, collecting the surface-bound radioactivity, followed by lysis in NaOH. These amounts were corrected for the stripping efficiency (≈93%), determined from control 125I-PDGF-binding experiments at 4 °C [29], and for non-specific binding, assessed in the presence of 100 nM unlabelled PDGF. Radioactivity was quantified by scintillation counting and compared with 125I-PDGF standards. These amounts were converted into molecules per cell, with the cell number per plate estimated in parallel by Coulter counting.
In our previous kinetic model of PDGF receptor dynamics in NIH 3T3 cells [25], R, C1 and C2 are free receptors, 1:1 ligand–receptor complexes and 1:2 dimerized complexes on the cell surface respectively. The total cell-associated ligand is thus given by LTot=C1+C2+Li, where Li is the internalized ligand. By conservation, Li=(R0−R−C1−2C2)/2. The model and measurements were aligned by minimizing the sum of absolute deviations, with R0 as the adjustable parameter, giving the number of activated receptors for any condition (R*Tot=2C2).
Estimation of 3′-phosphoinositide turnover rate constant
The rate of 3′-phosphoinositide turnover was estimated from the decay of TIRF (total internal reflection fluorescence) intensity in living NIH 3T3 cells expressing GFP–AktPH, a 3′-phosphoinositide-binding fusion construct, upon successive treatments with PDGF and wortmannin as described in detail previously [25,30].
Estimation of the receptor activation level yielding half-maximal PI3K recruitment
The quantity R*1/2 is defined by eqn (12) below. If one assumes that the free Ras-GTP level S* is roughly constant for each Ras variant cell line (but different across cell lines), then each has its own value of Kdeff. Eqns (7), (9), (10) and (11) can be manipulated to obtain the following:
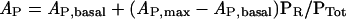 |
(8) |
The receptor-bound PI3K, PR, is determined from the quantities R*Tot, PTot and Kdeff (eqn 11, rearranged using the quadratic formula). As a function of PDGF concentration, R*Tot was taken from the analysis presented in Figure 2. Hence, R*1/2 is robustly estimated as the value of R*Tot that yields PR/PTot=1/2, or PI3K activation at the midpoint between its basal and maximum values (eqn 8), and its estimated value is insensitive to the assumed value of PTot (provided that PTot<2R*1/2; eqn 12). A value of PTot=600/cell was arbitrarily assumed for all cell lines, and the Akt phosphorylation dose–response curves were subjected to a standard threeparameter (AP,basal, AP,max and R*1/2) fit using KaleidaGraph (Synergy Software).
Figure 2. PDGF receptor activation and Ras-GTP generation in NIH 3T3 fibroblasts.

(A) Theoretical calculations and measurements of cell-associated PDGF (△, ▽) and receptor activation (●) (means±S.E.M.; data from [25]), as a function of PDGF concentration (t=5 min), were aligned as described in the Materials and methods section. Total cell-associated (△) and internalized (broken curve; ▽) PDGF were determined in four separate 125I-PDGF-binding experiments. (B) Ras-associated GTP was quantified at the peak (2.5 min, ●) and plateau (15 min, ○) following addition of PDGF to the concentrations indicated (means±S.E.M.; n=3). Absolute amounts were estimated from GTP standards.
RESULTS
The degree to which receptor and Ras inputs co-operate to recruit PI3K can be assessed using a simple equilibrium-binding model
Based on the binding of different PI3K subunits by activated receptors and Ras-GTP, it was reasoned that a ternary receptor–PI3K–Ras complex could plausibly form in two steps (Figure 1A). Moreover, the plasma membrane localization of both PI3K-binding partners suggested that the second step in the process is greatly enhanced because of the concentrating effect of membrane recruitment, which is expected to enhance the rate of binding by a factor, χ, of ∼100–1000 [21–23]. An allosteric co-operativity, suggested from in vitro experiments [19,31], would synergize with this membrane localization effect. On this basis, a mathematical model was formulated (see the Materials and methods section). Binding is assumed to occur rapidly relative to the rates at which receptor activation and Ras-GTP levels change [25,32], such that quasi-equilibrium relationships can be posed (eqns 1–3). In this respect, the model is similar to the description of co-operativity in the activation of N-WASP (neural Wiskott–Aldrich syndrome protein) in vitro [33], with two important distinctions: our model conserves the total numbers of PI3K, activated receptor and Ras-GTP molecules (eqn 4–6), and it allows the various PI3K species to possess different specific activities (eqn 7). Although the model was formulated based on the hypothesis that receptors and Ras recruit PI3K in a co-operative manner (χ≫1), it should be noted that the model does not exclude the possibilities that the two interactions are independent (χ=1) or competitive (χ=0). Indeed, the model was used to rule out these alternative possibilities.
The model predicts that the presence of Ras can have an impact on three distinct aspects of the dose–response curve for receptor-mediated PI3K activation (Figure 1B). First, as long as Ras binding is sufficient for PI3K activation (αPS>αP), the basal PI3K activity is an increasing function of the unoccupied Ras-GTP level, S*:
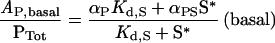 |
(9) |
On the other hand, with a large number of activated receptors, all PI3K p85–p110 heterodimers are recruited from the cytosol into receptor-bound forms (RP and RPS), and the observed PI3K activity achieves a plateau.
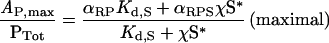 |
(10) |
In previous work, we noted that PI3K/Akt activation is saturated at concentrations of PDGF that give submaximal PDGF receptor phosphorylation in NIH 3T3 fibroblasts [25]. Our interpretation, consistent with biochemical experiments performed by others [8], was that high concentrations of PDGF do indeed drive all of the available PI3K into receptor complexes, although it should be noted that another study reported a transient low-affinity PDGF receptor–PI3K interaction in fibroblasts [34]. From eqn (10), it is seen that the presence of Ras can increase, decrease or have no effect on the maximal PI3K activity, depending on whether the specific activity α of the ternary RPS complex is greater than, less than or equal to that of the RP complex respectively.
Finally, and most importantly, increasing levels of Ras-GTP tend to shift the dose–response curve towards lower PDGF concentrations when the ternary RPS complex forms in a co-operative manner (χ>1). Conversely, if the interactions are competitive, Ras-GTP will tend to shift the dose–response curve towards higher PDGF concentrations. To illustrate this conclusion mathematically, manipulation of the model equations reveals that formation of the receptor complexes, RP and RPS, is governed by an effective dissociation constant, which encapsulates the influence of Ras-GTP.
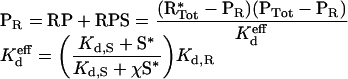 |
(11) |
Half-maximal PI3K recruitment by receptors (PR=PTot/2) is achieved when the receptor activation level satisfies the following relationship:
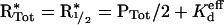 |
(12) |
It is readily seen that Kdeff and thus R*1/2 are decreasing functions of Ras-GTP when χ>1, a conclusion upon which we based our experimental design.
PI3K recruitment is limited by the number of activated receptor, Ras-GTP, or PI3K molecules
The formation of signalling complexes is limited by the availability of binding partners. For example, the maximum number of RPS complexes that can form is the lowest value among PTot, R*Tot and S*Tot. The actual numbers of the various complexes depend on these quantities and other model parameters. We therefore sought to quantify, as a function of PDGF concentration, the average numbers of activated PDGF receptor and Ras-GTP molecules in our mouse fibroblasts.
A validated model of PDGF receptor activation in these cells [25] gives the value of R*Tot in normalized units for any condition, and 125I-PDGF binding experiments were subsequently conducted to convert this quantity into an approximate number per cell. The amounts of surface-bound, internalized and total cell-associated ligand (0.5 or 1 nM PDGF, 5 min) were predicted from the kinetic model, and the relevant values were aligned with the binding data to yield an estimate of approx. 200000 PDGF receptors/cell before stimulation (Figure 2A), consistent with previous estimates [35]. The value of R*Tot was taken as twice the calculated number of receptor dimers.
The numbers of Ras-GTP molecules in our cells were also quantified, using an assay that allows the determination of absolute amounts (Figure 2B). The peak value was reached within 2–3 min, followed by a decrease to a plateau at 10–15 min (Figure 2B, and results not shown). Significantly, the measurements indicated that our cells contain approx. 10000 Ras-GTP molecules even before stimulation. The sum of Ras-GTP and Ras-GDP in our cells was found to be approx. 400000 molecules/cell (results not shown). The basal GTP/(GTP+GDP) ratio (≈2.5%) is similar to that measured by others in murine fibroblasts [36,37].
In the range of PDGF concentrations that give the greatest gains in PI3K activation (approx. 0.1 nM [25]), it is thus predicted that there are >10-fold more Ras-GTP molecules than activated PDGF receptors in our cells. This analysis suggests that, until all of the PI3K is recruited, it is the availability of activated receptors that limits complex formation. Ras-GTP is apparently in excess in our cells.
Modulation of Ras-GTP levels by retroviral infection
To ascertain the degree of co-operation between PDGF receptors and Ras in the activation of PI3K, we modulated the levels of Ras-GTP in our cells through the expression of dominant-negative (S17N) or constitutively active (G12V) H-Ras, introduced by retroviral infection and puromycin selection; control cells were infected with virus lacking an insert (Figure 3). Ras expression was confirmed by immunoblotting with anti-H-Ras antibodies (Figure 3A), and Ras-GTP measurements followed the expected trend (S17N<control≪G12V; Figure 3B). Immunoblotting with phospho-specific PDGF β-receptor antibodies (pTyr751; one of the PI3K-binding sites) was used to confirm that the Ras variants did not significantly alter PDGF receptor phosphorylation (Figure 3C).
Figure 3. Characterization of NIH 3T3 variants with various levels of Ras-GTP.

Dominant-negative (S17N) or constitutively active (G12V) H-Ras was introduced in NIH 3T3 cells by retroviral infection and selection, with empty vector serving as a control. (A) H-Ras expression. Cell lysates (25 μg of total protein) were subjected to SDS/PAGE and blotted for H-Ras. (B) Ras-GTP levels. Cell variants were unstimulated (grey bars) or stimulated with PDGF (black bars). Ras-GTP levels were measured as in Figure 2(B) and were normalized by the level in unstimulated control cells (approx. 18000 molecules/cell, determined from GTP standards). Values are means±S.E.M.; n=2. (C) PDGF receptor phosphorylation. Cells were stimulated with the indicated concentrations of PDGF (nM) for 5 min, and lysates were probed with anti-phospho-PDGFβ-receptor (pTyr751) antibodies. (D) ERK phosphorylation. Lysates were prepared from cells stimulated with the indicated concentrations of PDGF for 5 min, subjected to SDS/PAGE and probed with anti-phospho-ERK (pThr202/pTyr204) antibodies. The blot shown is representative of three independent experiments. (E) MKP-1 expression. Lysates were prepared from cells stimulated with the indicated concentrations of PDGF (nM) for 5 min, subjected to SDS/PAGE and probed with anti-MKP-1 antibodies.
To confirm that changes in Ras-GTP level were significant for modulation of downstream signalling, we assessed the dose–responses of ERK phosphorylation as the output of the Raf–MEK (mitogen-activated protein kinase/ERK kinase)–ERK cascade (Figure 3D). As expected, PDGF-stimulated ERK phosphorylation was significantly inhibited by S17N Ras, while ERK phosphorylation in G12V Ras cells was elevated in the absence of PDGF and was insensitive to PDGF stimulation. Total ERK amounts, as a fraction of the total protein concentration, were roughly the same in all samples (results not shown). Interestingly, the ERK phosphorylation level in G12V Ras cells was lower than in maximally stimulated control cells, which might be attributed to a well-known feedback loop in which activated ERK induces expression of MKP dual-specificity phosphatases [38,39]. Indeed, we confirmed that G12V Ras expression is accompanied by robust expression of MKP-1 in our cells (Figure 3E).
Co-operative activation of PI3K/Akt in PDGF-stimulated fibroblasts
Akt phosphorylation was used as a well-documented biochemical readout of PI3K signalling [40]. At 5 min of PDGF stimulation, Akt activity reaches its peak in our cells, and our previous model predicts that Akt activation is directly proportional to PI3K activity under these conditions [25]. The PDGF dose–responses of Akt phosphorylation (p-Akt) in control, S17N Ras and G12V Ras cells were quantitatively determined by immunoblotting samples from three independent experiments; the p-Akt band intensity of each sample was normalized by that of total Akt (t-Akt), assessed in parallel (Figures 4A and 4B). Compared with the dose–response in control cells, S17N Ras shifted the dose–response to higher PDGF concentrations, such that the degree of inhibition varied with PDGF dose. At the lowest dose (0.05 nM), the normalized p-Akt level was inhibited by approx. 40%, and inhibition by S17N Ras was also statistically significant at 0.1 nM PDGF. G12V Ras cells exhibited enhanced PI3K/Akt activation in the absence of PDGF (approx. 6-fold compared with control), although the level was significantly lower than that elicited by maximal PDGF stimulation [41]. When stimulated maximally with PDGF, Akt phosphorylation levels were similar in control and S17N Ras cells, but approx. 1.5-fold higher in G12V Ras cells (Figure 4B).
Figure 4. Co-operative activation of PI3K/Akt by PDGF receptors and Ras-GTP.

Dose–responses of Akt phosphorylation (pSer473) at 5 min of PDGF stimulation were measured by quantitative immunoblotting for the cell variants described in Figure 3. (A) Representative immunoblot. (B) Quantification of phosphorylated Akt levels (p-Akt), normalized by total Akt content (t-Akt). Values are means±S.E.M.; n=3: vector control (●); S17N H-Ras (▽); G12V H-Ras (△). Asterisks denote significantly higher or lower values compared with the control cells for the same PDGF dose, based on Student's t test (*, P<0.05; **, P<0.01). The curves are best fits of the co-operative assembly model, assuming that Ras-GTP is in excess (S*≈S*Tot). (C) Best-fit values (±S.E.M.) of R*1/2, the number of activated receptors yielding half-maximal recruitment of PI3K into receptor-bound complexes (eqn 12).
The Akt phosphorylation data were fitted to the equilibrium-binding model by non-linear regression to extract the apparent value of R*1/2 (eqn 12, assuming that S* and thus Kdeff for each cell line is approximately constant), the number of activated receptors required to recruit half of the p85–p110 PI3K heterodimers into receptor complexes (Figure 4C). This value was found to be approx. 1.7 times higher for S17N Ras cells than for control cells. Because Ras-GTP levels are reduced in S17N Ras cells, we conclude that ternary complex assembly occurs by a co-operative mechanism (χ>1). A less predictable finding was that the best-fit R*1/2 values for G12V Ras cells were only slightly smaller (by approx. 20%), and within the standard errors, compared with control cells (Figure 4C). This observation is not inconsistent with the model, however, as outlined in the next section.
It was reasoned that the increase in maximally stimulated Akt phosphorylation observed in G12V Ras cells compared with control and S17N Ras cells might be attributed to regulation of 3′-phosphoinositide turnover, as opposed to enhanced PI3K activation. This possibility was assessed in living NIH 3T3 cells transiently co-transfected with a fluorescent, 3′-phosphoinositide lipid-binding probe (GFP–AktPH) and either G12V Ras or empty vector, imaged using TIRF microscopy (Figure 5). These cells were first stimulated with 10 nM PDGF for 10 min, followed by a high dose of wortmannin to rapidly block 3′-phosphoinositide production, and the 3′-phosphoinositide turnover rate constant was inferred from the ensuing exponential decay in TIRF intensity [25,30] (Figure 5A). 3′-Phosphoinositide turnover was found to be significantly slower in the G12V Ras cell population (Figure 5B), and, in fact, the ratio of the mean apparent rate constants (0.59 min−1 for control and 0.36 min−1 for G12V Ras) can account for the observed 1.5-fold change in maximally stimulated Akt phosphorylation. Together with our previous observation that PDGF stimulation does not significantly alter 3′-phosphoinositide turnover in NIH 3T3 cells [30], this suggests that PI3K activation in response to maximal PDGF stimulation is not dramatically affected by modulation of Ras-GTP.
Figure 5. Reduced 3′-phosphoinositide turnover rate in G12V Ras-expressing cells.

Individual NIH 3T3 cells were co-transfected with GFP–AktPH and either G12V Ras or control vector and stimulated with 10 nM PDGF for 10 min, followed by inhibition of PI3K with wortmannin, and the ensuing decay in 3′-phosphoinositide level was monitored in real time using TIRF microscopy [25,30]. A total of seven control and 23 G12V Ras cells, from 4 days of experiments, were analysed. (A) Normalized TIRF time courses, following PI3K inhibition, for the control cells. (B) Whisker plot of estimated first-order 3′-phosphoinositide turnover rate constants for the two cell populations, depicting the median, upper and lower quartiles, and range of values for each population. Turnover was significantly slower in G12V Ras cells (P<0.001, Student's t test).
Sensitivity of PI3K activation to receptor and Ras inputs
The tendency to form binary and ternary complexes of receptors, PI3K and Ras and their relative lipid kinase activities are important considerations that can be assessed through an analysis of the equilibrium-binding model. Figure 6 shows calculated PI3K activation as a function of the normalized receptor activation and Ras-GTP levels, with different values of the other constant parameters. Here we assume a value of the co-operativity parameter, χ, of 100 or 1000, consistent with the expected magnitude of binding enhancement through the concentrating effect of membrane recruitment. Increasing levels of Ras-GTP will thus tend to shift the dose–response leftward, towards lower receptor activation (eqns 10 and 11).
Figure 6. Model calculations and analysis.

The hypothetical PI3K activity was calculated in dimensionless form (AP/αRPSPTot) from eqns (1)–(7), as a function of dimensionless receptor (R*Tot/Kd,R) and Ras (S*Tot/Kd,S) binding sites. The values of the latter are 0, 0.01, 0.1, 1 and 10. Relative binding constants are as follows. (A) and (B): Kd,R/PTot=1, Kd,S/PTot=100, χ=100; (C) and (D): Kd,R/PTot=10, Kd,S/PTot=100, χ=100; (E) and (F): Kd,R/PTot=10, Kd,S/PTot=100, χ=1000. The binary complexes, RP and PS, were assumed to be either less active than (αRP=αPS=0.5αRPS; A, C and E) or as active as (αRP=αPS=αRPS; B, D and F) the ternary RPS complex. The ♦ symbols signify the position of the R*1/2 value for each curve (eqn 12), and the broken lines show the range of R*1/2 values across the spectrum of Ras-GTP levels.
Eqn (12) also shows that the magnitude of the shift is limited by the stoichiometry of binding (R*1/2=PTot/2). In our cells, we found that control and G12V Ras cells exhibit similar R*1/2 values, consistent with this limit. The apparent value is approx. 600 activated receptors/cell (Figure 4C), or approx. 1 nM on a whole-cell basis. Across the spectrum of Ras-GTP values, the range of possible R*1/2 values is determined by the dissociation constant for the receptor interaction relative to the PI3K concentration (Kd,R/PTot); or comparing cells containing no Ras (R*1/2=PTot/2+Kd,R) with those at the stoichiometric binding limit, the fold change is (1+2Kd,R/PTot). For example, when Kd,R=PTot (Figures 6A and 6B), the maximum span of R*1/2 values is a factor of 3. When Kd,R=10PTot (Figures 6C–6F), the range is 21. In our experiments, we found a shift in R*1/2 of approx. 2-fold between cells expressing S17N Ras and G12V Ras, suggesting that Kd,R is at least roughly the same magnitude as PTot or larger in our cells. As predicted from eqn (12), the value of χ determines how much Ras-GTP is required to produce a significant shift and to achieve near-stoichiometric binding (compare Figures 6C and 6D with Figures 6E and 6F). If the Ras-GTP level in control cells is sufficient to give stoichiometric PI3K recruitment, yet the basal PI3K signalling is low (S*Tot/Kd,S<1), we conclude that the value of χ must be large (χ≫10).
As long as binding to Ras is sufficient for PI3K activation (αPS>αP), the basal PI3K level will be an increasing function of the Ras-GTP level in cells (eqn 9). In our G12V Ras cells, the basal Akt phosphorylation was found to be approx. 10% of maximal (Figure 4B). This indicates that the specific activity of the PS complex is at least 10% of the maximum (αPS/αRPS≥0.1) in our cells, although our data cannot distinguish between partial recruitment and partial activation in the basal state (compare Figures 6E and 6F, for example). The maximal PI3K activity is also a function of Ras-GTP when the specific activity of the RP complex is different from that of the ternary RPS complex (αRP¬αRPS); however, the sensitivity of the plateau value to changes in the Ras-GTP level depends on the value of χ (eqn 10; compare Figures 6C and 6E). Thus our conclusion that the maximal PI3K activation levels were similar among the cell variants does not preclude the possibility that receptors and Ras together yield the highest specific PI3K activity, as suggested from experiments with purified proteins [19,31].
DISCUSSION
Propagation of intracellular signals generally requires the recruitment of signalling proteins to the plasma membrane, where they are activated and/or gain access to their substrates. For enzymes such as PI3K, this localization is critical for the control of cell migration, phagocytosis and other spatially co-ordinated processes [42,43]. Transmembrane receptors, receptor-bound adaptors, lipids and lipid-tethered G-proteins/GTPases are among the signalling molecules that serve as binding sites at the membrane, recognized by proteins that have certain modular interaction domains/motifs [44,45]. When a cytosolic protein may simultaneously interact with two such membrane-associated molecules, assembly of the ternary complex is thermodynamically favoured because of the reduced volume of the membrane-proximal space. Allosteric co-operativity, whereby one domain is exposed or better oriented upon binding of the other, is not necessary for this effect, but would enhance it, as noted in the regulation of protein kinase C [46], Raf-1 [47,48] and N-WASP [33]. In these and other such studies, the presence of a multimolecular complex is inferred from measurements of protein recruitment or activation, as the direct isolation of such complexes is hindered by their low abundance and transient lifetime, and/or the need for an intact membrane. In the light of these difficulties, quantitative analysis of complex formation might hinge on a useful mathematical description of the system that captures the possible mechanisms involved.
An equilibrium-binding model was formulated and used to analyse the activation of PI3K in PDGF-stimulated fibroblasts, and, in the context of this model, we find that PDGF receptors and Ras-GTP recruit PI3K to the plasma membrane in a co-operative fashion, through the formation of a receptor–PI3K–Ras complex. We also assert that the magnitude of the co-operativity is such that PI3K binding is roughly stoichiometric, limited by the availability of activated PDGF receptors and PI3K at low and high PDGF concentrations, respectively. Thus Ras-GTP apparently plays an important yet permissive role, as expression of G12V Ras does not drastically alter the concentration of PDGF required for half-maximal PI3K/Akt activation in our cells. Interestingly, however, we found that G12V Ras expression yields a modest reduction in the rate of 3′-phosphoinositide turnover, presumably by affecting the abundance or activity of lipid phosphatases.
In general, our analysis of the model indicates that the inhibition or amplification of Ras signalling will have the most influence when the affinity of PI3K for the phosphorylated receptor/adaptor protein is relatively weak. The magnitude of the effect depends not only on the receptor system being studied, but also on the concentration of the stimulus and expression levels of receptors and PI3Ks in the particular cell lines used.
In this analysis, it is important to distinguish complex formation from enzyme activation. Experiments with purified proteins suggest that the receptor–PI3K–Ras complex possesses the highest PI3K activity, and, if so, Ras-GTP and PDGF will always enhance PI3K signalling. We find again, however, that the magnitude of this effect depends on the parameters of the specific system. If the Ras-GTP levels could be carefully titrated, the saturation of PI3K activity in the absence of PDGF would reveal the relative activity of the PI3K–Ras complex in the cell; a complete analysis would also consider the various isoforms of Ras and PI3Ks. In cells stimulated maximally with PDGF, the model prediction is that the saturation of PI3K activity would occur at much lower Ras-GTP levels. Hence, the observation that the maximal PI3K activity was insensitive to the Ras-GTP level in our cells does not necessarily mean that the receptor–PI3K interaction is sufficient for full activation.
It has long been a curiosity that receptor-mediated increases in Ras-GTP tend to be modest in magnitude yet functionally important. A potential explanation is that Ras-GTP in compartments other than the plasma membrane may blur the comparison of whole-cell Ras-GTP amounts [49,50]. A more significant possibility is that, through coupling of an activated receptor to GEF activity, Ras-GTP is much more concentrated in the vicinity of the receptor compared with the average. Indeed, it is not possible to obtain from eqns (10) and (11) relative R*1/2 values of 1.7X, 1X and 0.8X given corresponding Ras-GTP concentrations of 0.5X, 1X and 15X, as observed on a whole-cell basis in PDGF-stimulated S17N Ras, control and G12V Ras cells respectively (Figures 4C and 3B). Although Ras can presumably form PS complexes anywhere in the membrane, the relevant Ras-GTP concentration driving RPS formation is that seen by the activated receptor. In the context of the equilibrium-binding model, which ignores spatial gradients, such differences between local and average Ras-GTP densities are effectively lumped in the value of χ. Thus, although this simplification does not affect our qualitative conclusions, a more complete model would account for the local dynamics explicitly.
At least two mechanisms could contribute to a local Ras-GTP accumulation effect, and we have performed an initial exploration of these effects through modelling (see the Supplement at http://www.BiochemJ.org/bj/393/bj3930235add.htm). First, if the receptor-mediated GEF reaction were diffusion-limited, one would observe a gradient of high-to-low Ras-GTP emanating from the receptor, with close to 100% Ras-GTP next to the receptor [51,52]. Secondly, hindrance of Ras diffusion by periodic barriers or corrals, as described by Kusumi and colleagues [53,54], would simultaneously enhance the rate of intermolecular collisions in the receptor corral and allow more time for the GTPase reaction in other corrals; similar systems have been considered theoretically [55–57]. Thus Ras-GTP levels could be heterogeneous even when the GEF reaction is not intrinsically diffusion-limited. In the light of recent observations that suggest that Ras diffusion is indeed constrained [58–60], we consider it plausible that receptor–PI3K–Ras complexes form at ‘point-blank range’.
Online Data
Acknowledgments
We acknowledge Chun-Chao Wang for providing technical assistance. This research was supported through grants to J.M.H. from NIH (National Institutes of Health; R01-GM067739) and NSF (National Science Foundation; BES-0111434).
References
- 1.Fruman D. A., Meyers R. E., Cantley L. C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 2.Rameh L. E., Cantley L. C. The role of phosphoinositide 3-kinase lipid products in cell function. J. Biol. Chem. 1999;274:8347–8350. doi: 10.1074/jbc.274.13.8347. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B., Waterfield M. D. Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 4.Vanhaesebroeck B., Leevers S. J., Ahmadi K., Timms J., Katso R., Driscoll P. C., Woscholski R., Parker P. J., Waterfield M. D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 5.McGlade C. J., Ellis C., Reedijk M., Anderson D., Mbamalu G., Reith A. D., Panayotou G., End P., Bernstein A., Kazlauskas A., et al. SH2 domains of the p85α subunit of phosphatidylinositol 3-kinase regulate binding to growth factor receptors. Mol. Cell. Biol. 1992;12:991–997. doi: 10.1128/mcb.12.3.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shoelson S. E., Sivaraja M., Williams K. P., Hu P., Schlessinger J., Weiss M. A. Specific phosphopeptide binding regulates a conformational change in the PI 3-kinase SH2 domain associated with enzyme activation. EMBO J. 1993;12:795–802. doi: 10.1002/j.1460-2075.1993.tb05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klippel A., Reinhard C., Kavanaugh W. M., Apell G., Escobedo M., Williams L. T. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 1996;16:4117–4127. doi: 10.1128/mcb.16.8.4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kazlauskas A., Cooper J. A. Phosphorylation of the PDGF receptor β-subunit creates a tight binding site for phosphatidylinositol-3 kinase. EMBO J. 1990;9:3279–3286. doi: 10.1002/j.1460-2075.1990.tb07527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashishian A., Kazlauskas A., Cooper J. A. Phosphorylation sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. EMBO J. 1992;11:1373–1382. doi: 10.1002/j.1460-2075.1992.tb05182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klippel A., Escobedo J. A., Fantl W. J., Williams L. T. The C-terminal SH2 domain of p85 accounts for the high affinity of the binding of phosphatidylinositol 3-kinase to phosphorylated platelet-derived growth factor β receptor. Mol. Cell. Biol. 1992;12:1451–1459. doi: 10.1128/mcb.12.4.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ottinger E. A., Botfield M. C., Shoelson S. E. Tandem SH2 domains confer high specificity in tyrosine kinase signaling. J. Biol. Chem. 1998;273:729–735. doi: 10.1074/jbc.273.2.729. [DOI] [PubMed] [Google Scholar]
- 12.Auger K. R., Serunian L. A., Soltoff S. P., Libby P., Cantley L. C. PDGF-dependent tyrosine phosphorylation stimulates production of novel phosphoinositides in intact cells. Cell. 1989;57:167–175. doi: 10.1016/0092-8674(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 13.Hawkins P. T., Jackson T. R., Stephens L. R. Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase. Nature (London) 1992;358:157–159. doi: 10.1038/358157a0. [DOI] [PubMed] [Google Scholar]
- 14.Jackson T. R., Stephens L. R., Hawkins P. T. Receptor specificity of growth factor-stimulated synthesis of 3-phosphorylated inositol lipids in Swiss 3T3 cells. J. Biol. Chem. 1992;267:16627–16636. [PubMed] [Google Scholar]
- 15.Avruch J., Zhang X., Kyriakis J. M. Raf meets Ras: completing the framework of a signal transduction pathway. Trends Biochem. Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez-Viciana P., Warne P. H., Dhand R., Vanhaesebroeck B., Gout I., Fry M. J., Waterfield M. D., Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature (London) 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 17.Bray D. Intracellular signaling as a parallel distributed process. J. Theor. Biol. 1990;143:215–231. doi: 10.1016/s0022-5193(05)80268-1. [DOI] [PubMed] [Google Scholar]
- 18.Weng G., Bhalla U. S., Iyengar R. Complexity in biological signaling systems. Science. 1999;284:92–96. doi: 10.1126/science.284.5411.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Viciana P., Warne P. H., Vanhaesebroeck B., Waterfield M. D., Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 20.Klinghoffer R. A., Duckworth B., Valius M., Cantley L., Kazlauskas A. Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence Ras activity. Mol. Cell. Biol. 1996;16:5905–5914. doi: 10.1128/mcb.16.10.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haugh J. M., Lauffenburger D. A. Physical modulation of intracellular signaling processes by locational regulation. Biophys. J. 1997;72:2014–2031. doi: 10.1016/S0006-3495(97)78846-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kholodenko B. N., Hoek J. B., Westerhoff H. V. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 23.McLaughlin S., Wang J. Y., Gambhir A., Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 24.Haugh J. M., Codazzi F., Teruel M., Meyer T. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J. Cell Biol. 2000;151:1269–1279. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park C. S., Schneider I. C., Haugh J. M. Kinetic analysis of platelet-derived growth factor receptor/phosphoinositide 3-kinase/Akt signaling in fibroblasts. J. Biol. Chem. 2003;278:37064–37072. doi: 10.1074/jbc.M304968200. [DOI] [PubMed] [Google Scholar]
- 26.Scheele J. S., Rhee J. M., Boss G. R. Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: comparison of parental and Ras-overproducing NIH 3T3 fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 1995;92:1097–1100. doi: 10.1073/pnas.92.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma P. M., Egawa K., Huang Y., Martin J. L., Huvar I., Boss G. R., Olefsky J. M. Inhibition of phosphatidylinositol 3-kinase activity by adenovirus-mediated gene transfer and its effect on insulin action. J. Biol. Chem. 1998;273:18528–18537. doi: 10.1074/jbc.273.29.18528. [DOI] [PubMed] [Google Scholar]
- 28.Haugh J. M., Huang A. C., Wiley H. S., Wells A., Lauffenburger D. A. Internalized epidermal growth factor receptors participate in the activation of p21ras in fibroblasts. J. Biol. Chem. 1999;274:34350–34360. doi: 10.1074/jbc.274.48.34350. [DOI] [PubMed] [Google Scholar]
- 29.Wiley H. S., Cunningham D. D. The endocytic rate constant: a cellular parameter for quantitating receptor-mediated endocytosis. J. Biol. Chem. 1982;257:4222–4229. [PubMed] [Google Scholar]
- 30.Schneider I. C., Haugh J. M. Spatial analysis of 3′ phosphoinositide signaling in living fibroblasts. II. Parameter estimates for individual cells from experiments. Biophys. J. 2004;86:599–608. doi: 10.1016/S0006-3495(04)74138-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiménez C., Hernández C., Pimental B., Carrera A. C. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J. Biol. Chem. 2002;277:41556–41562. doi: 10.1074/jbc.M205893200. [DOI] [PubMed] [Google Scholar]
- 32.Haugh J. M., Lauffenburger D. A. Analysis of receptor internalization as a mechanism for modulating signal transduction. J. Theor. Biol. 1998;195:187–218. doi: 10.1006/jtbi.1998.0791. [DOI] [PubMed] [Google Scholar]
- 33.Prehoda K. E., Scott J. A., Mullins R. D., Lim W. A. Integration of multiple signals through cooperative regulation of the N-WASP–Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- 34.Domin J., Dhand R., Waterfield M. D. Binding to the platelet-derived growth factor receptor transiently activates the p85α–p110α phosphoinositide 3-kinase complex in vivo. J. Biol. Chem. 1996;271:21614–21621. doi: 10.1074/jbc.271.35.21614. [DOI] [PubMed] [Google Scholar]
- 35.Grotendorst G. R., Igarashi A., Larson R., Soma Y., Charette M. Differential binding, biological and biochemical actions of recombinant PDGF AA, AB, and BB molecules on connective tissue cells. J. Cell. Physiol. 1991;149:235–243. doi: 10.1002/jcp.1041490209. [DOI] [PubMed] [Google Scholar]
- 36.Gibbs J. B., Marshall M. S., Scolnick E. M., Dixon R. A. F., Vogel U. S. Modulation of guanine nucleotides bound to Ras in NIH3T3 cells by oncogenes, growth factors, and the GTPase activating protein (GAP) J. Biol. Chem. 1990;265:20437–20442. [PubMed] [Google Scholar]
- 37.Gibbs J. B. Determination of guanine nucleotides bound to Ras in mammalian cells. Methods Enzymol. 1995;255:118–125. doi: 10.1016/s0076-6879(95)55014-3. [DOI] [PubMed] [Google Scholar]
- 38.Brondello J., Brunet A., Pouysségur J., McKenzie F. R. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J. Biol. Chem. 1997;272:1368–1376. doi: 10.1074/jbc.272.2.1368. [DOI] [PubMed] [Google Scholar]
- 39.Plows D., Briassouli P., Owen C., Zoumpourlis V., Garrett M. D., Pintzas A. Ecdysone-inducible expression of oncogenic Ha-Ras in NIH 3T3 cells leads to transient nuclear localization of activated extracellular signal-regulated kinase regulated by mitogen-activated protein kinase phosphatase-1. Biochem. J. 2002;362:305–315. doi: 10.1042/0264-6021:3620305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Viciana P., Warne P. H., Khwaja A., Marte B. M., Pappin D., Das P., Waterfield M. D., Ridley A., Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 42.Weiner O. D. Regulation of cell polarity during eukaryotic chemotaxis: the chemotactic compass. Curr. Opin. Cell Biol. 2002;14:196–202. doi: 10.1016/s0955-0674(02)00310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons T. J., Horwitz A. R. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 44.Teruel M. N., Meyer T. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell. 2000;103:181–184. doi: 10.1016/s0092-8674(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 45.Pawson T., Gish G. D., Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001;11:504–511. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 46.Oancea E., Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- 47.Mineo C., Anderson R. G. W., White M. A. Physical association with Ras enhances activation of membrane-bound Raf (RafCAAX) J. Biol. Chem. 1997;272:10345–10348. doi: 10.1074/jbc.272.16.10345. [DOI] [PubMed] [Google Scholar]
- 48.Rizzo M. A., Shome K., Watkins S. C., Romero G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem. 2000;275:23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 49.Hancock J. F. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 50.Bivona T. G., Philips M. R. Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 2003;15:136–142. doi: 10.1016/s0955-0674(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 51.Shea L. D., Omann G. M., Linderman J. J. Calculation of diffusion-limited kinetics for the reactions in collision coupling and receptor cross-linking. Biophys. J. 1997;73:2949–2959. doi: 10.1016/S0006-3495(97)78323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haugh J. M. A unified model for signal transduction reactions in cellular membranes. Biophys. J. 2002;82:591–604. doi: 10.1016/S0006-3495(02)75424-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujiwara T., Ritchie K., Murakoshi H., Jacobson K., Kusumi A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 2002;157:1071–1081. doi: 10.1083/jcb.200202050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murase K., Fujiwara T., Umemura Y., Suzuki K., Iino R., Yamashita H., Saito M., Murakoshi H., Ritchie K., Kusumi A. Ultrafine membrane compartments for molecular diffusion as revealed by single molecule techniques. Biophys. J. 2004;86:4075–4093. doi: 10.1529/biophysj.103.035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saxton M. J. Lateral diffusion in an archipelago: effects of impermeable patches on diffusion in a cell membrane. Biophys. J. 1982;39:165–173. doi: 10.1016/S0006-3495(82)84504-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Melo E. C. C., Lourtie I. M. G., Sankaram M. B., Thompson T. E., Vaz W. L. C. Effects of domain connection and disconnection on the yields of in-plane bimolecular reactions in membranes. Biophys. J. 1992;63:1506–1512. doi: 10.1016/S0006-3495(92)81735-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shea L. D., Linderman J. J. Compartmentalization of receptors and enzymes affects activation for a collision coupling mechanism. J. Theor. Biol. 1998;191:249–258. [Google Scholar]
- 58.Niv H., Gutman O., Kloog Y., Henis Y. I. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J. Cell Biol. 2002;157:865–872. doi: 10.1083/jcb.200202009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lommerse P. H. M., Blab G. A., Cognet L., Harms G. S., Snaar-Jagalska B. E., Spaink H. P., Schmidt T. Single-molecule imaging of the H-Ras membrane-anchor reveals domains in the cytoplasmic leaflet of the cell membrane. Biophys. J. 2004;86:609–616. doi: 10.1016/S0006-3495(04)74139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murakoshi H., Iino R., Kobayashi T., Fujiwara T., Ohshima C., Yoshimura A., Kusumi A. Single-molecule imaging analysis of Ras activation in living cells. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7317–7322. doi: 10.1073/pnas.0401354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.