

Abstract
A GlcNase (exo-β-D-glucosaminidase) was purified from culture supernatant of Amycolatopsis orientalis subsp. orientalis grown in medium with chitosan. The enzyme hydrolysed the terminal GlcN (glucosamine) residues in oligomers of GlcN with transglycosylation observed at late reaction stages. 1H-NMR spectroscopy revealed that the enzyme is a retaining glycoside hydrolase. The GlcNase also behaved as an exochitosanase against high-molecular-mass chitosan with Km and kcat values of 0.16 mg/ml and 2832 min−1. On the basis of partial amino acid sequences, PCR primers were designed and used to amplify a DNA fragment which then allowed the cloning of the GlcNase gene (csxA) associated with an open reading frame of 1032 residues. The GlcNase has been classified as a member of glycoside hydrolase family 2 (GH2). Sequence alignments identified a group of CsxA-related protein sequences forming a distinct GH2 subfamily. Most of them have been annotated in databases as putative β-mannosidases. Among these, the SAV1223 protein from Streptomyces avermitilis has been purified following gene cloning and expression in a heterologous host and shown to be a GlcNase with no detectable β-mannosidase activity. In CsxA and all relatives, a serine-aspartate doublet replaces an asparagine residue and a glutamate residue, which were strictly conserved in previously studied GH2 members with β-galactosidase, β-glucuronidase or β-mannosidase activity and shown to be directly involved in various steps of the catalytic mechanism. Alignments of several other GH2 members allowed the identification of yet another putative subfamily, characterized by a novel, serine-glutamate doublet at these positions.
Keywords: amino sugar, chitosan hydrolysis, csxA gene, exo-glucosaminidase, glycoside hydrolase, Streptomyces
Abbreviations: CBM, carbohydrate-binding module; GH, glycoside hydrolase family; GlcN, D-glucosamine; GlcNAc, N-acetylglucosamine; GlcNase, exo-β-D-glucosaminidase; ORF, open reading frame; p-NP, p-nitrophenyl; SP-Sepharose, sulphopropyl-Sepharose; YME medium, yeast/malt extract medium
INTRODUCTION
Chitin is a linear polysaccharide formed of β-(1-4)-linked GlcNAc (N-acetyl-D-glucosamine) residues. It is widely distributed in Nature as the major structural component of fungal cell walls, exoskeletons in arthropods and the cell walls of some microscopic algae. The main pathway of chitin degradation by micro-organisms begins with combined actions of endochitinases and exochitinases, resulting essentially in N,N′-diacetylchitobiose production. The dimers are then transported inside the cell, processed by phosphorylation and further hydrolysed into monomers by intracellular 6-phospho-N-acetyl-β-D-glucosaminidase, or first hydrolysed by an extracellular exo-N-acetyl-β-D-glucosa-minidase into GlcNAc units, which are then transported inside the cells in a phosphorylation-coupled manner.
Chitosan, a partly or totally N-deacetylated chitin derivative, is much less abundant in Nature. It is found in the cell walls of some phytopathogenic fungi belonging to genera such as Fusarium, Phytophthora and Mucor [1]. Chitosan is mainly composed of D-glucosamine residues (GlcN) with a variable content of GlcNAc residues. It can be produced on an industrial scale by alkaline deacetylation of chitin and is presently the subject of intense studies, owing to its great application potential in biomedical, agricultural and environmental fields. However, studies of chitosan metabolism and enzymology are much less extensive than for chitin. Chitosan is recognized as substrate by several endochitinases, which catalyse the hydrolytic cleavage of rare GlcNAc–GlcNAc links and more frequent GlcN–GlcNAc or GlcNAc–GlcN links. Enzymes classified as chitosanases (EC 3.2.1.132) usually also recognize one of these ‘mixed’ linkages, but hydrolyse mainly the GlcN–GlcN links largely predominant in chitosan. It results that a major proportion of oligosaccharide products obtained after endohydrolysis steps by chitosanases will have GlcN residues at their non-reducing end [2].
A GlcNase (exo-β-D-glucosaminidase) enzyme is required to complete the hydrolysis of these oligomeric chitosan forms into monomers. To date, very few studies have been dedicated to such enzymes. There is no EC number associated with this activity. The first GlcNase was characterized by Nanjo et al. [3]. It was produced extracellularly by the actinomycete Nocardia orientalis (present name Amycolatopsis orientalis subsp. orientalis) in a medium with colloidal chitosan as carbon source. The enzyme hydrolysed specifically β-D-glucosaminide groups from the non-reducing end and had no activity against β-D-N-acetylglucosaminides. The measured initial rates of hydrolysis were very similar for short chitosan oligosaccharides (GlcN)2 or (GlcN)3 and for high-molecular-mass chitosan. Thus this enzyme was also designated as an exochitosanase. Later, enzymes with similar properties were purified from filtrates of filamentous fungi cultures: Trichoderma reesei PC-3-7 [4], Penicillium funiculosum KY616 [5] and Aspergillus oryzae IAM2660 [6]. The enzyme from Penicillium was shown to have transglycosylation activity towards chitosan using alcohols as substrate, which presupposes a mechanism of action with retention of the anomeric form. Such a mechanism was clearly demonstrated by NMR studies for the GlcNase from Trichoderma reesei PC-3-7 [4]. However, almost 15 years after their first description, no structural data are available so far for GlcNases/exochitosanases.
Recently, Tanaka et al. [7,8] reported a novel pathway of chitin utilization from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. This pathway insures a complete degradation of chitin oligomers into GlcN monomers through a combined activity of a diacetylchitobiose deacetylase, acting on the non-reducing-end GlcNAc residue of the chitin oligomers, or on monomeric GlcNAc, and a GlcNase, a homodimeric, intracellular enzyme cleaving the terminal GlcN residue from the nonreducing end. The deduced amino acid sequence of the GlcNase showed homology with that of GH (glycoside hydrolase family) 35 and GH42. Tanaka et al. [7] suggested, however, that the primary sequence of the archaeal enzyme may be not representative of other previously studied GlcNases, considering important differences in substrate specificity (the T. kodakaraensis GlcNase acted very inefficiently as an exochitosanase on high-molecular-mass chitosan), quaternary structure (homodimeric versus monomeric) and localization (intracellular versus extracellular).
In the present paper we report, for the first time, the primary structure of a GlcNase with exochitosanase activity and show that this enzyme and other related proteins belong to a distinct subfamily within GH2.
MATERIALS AND METHODS
Bacterial strains, vectors and general cloning procedures
Amy. orientalis subsp. orientalis IFO12806 and Streptomyces avermitilis MA-4680 were obtained from the American Type Culture Collection, Manassas, VA, U.S.A. (strains A.T.C.C. 19795 and 31267). Both were routinely propagated on YME (yeast/malt extract) medium [yeast extract, (4 g/litre), glucose (4 g/litre), malt extract (10 g/litre)] with shaking (300 rev./min) at 30 °C. A partial gene library of Amy. orientalis chromosomal DNA was constructed in pUC19 plasmid using Escherichia coli XL-10 Gold competent cells (Stratagene, La Jolla, CA, U.S.A.) as transformation host. PCR fragments were cloned by the TA-cloning procedure using the pCR 2.1 vector and INVαF′ competent E. coli cells (Invitrogen, Carlsbad, CA, U.S.A.). Production of proteins from cloned genes was done using Strep. lividans TK24, propagated and transformed with plasmid DNA as described in [9]. For expression in Strep. lividans, the genes were subcloned into the shuttle vector pFD666 [10].
Enzyme production and purification
For native GlcNase production, Amy. orientalis was grown in YME medium for 24 h at 30 °C. The mycelium was recovered by centrifugation (3000 g, 10 min, 4 °C) and inoculated (1 ml of mycelial pellet per 100 ml of medium) into a medium consisting of K2HPO4 (4.5 g/l), MgSO4·7H2O (0.2 g/l), FeSO4·7H2O (0.01g/l), CaCl2·2H2O (0.01g/l), (NH4)2SO4 (0.1 g/l), trace-elements solution [9] (1 ml/l), chitosan (6%, N-acetylated, finely ground; 10g/l), malt extract (10 g/l), pH 6.8, with shaking at 30 °C for 60 h. At harvesting, the mycelium was eliminated by centrifugation (3000 g, 20 min, 4 °C). All the subsequent steps were done at 4 °C. The supernatant was clarified by filtration (0.8 μm-mesh-size filter) and the proteins were fractionated with (NH4)2SO4. The 50–90%-satd.-(NH4)2SO4-precipitated fraction was dissolved in 50 mM Tes buffer, pH 7.7 (buffer A) and dialysed overnight against the same buffer. The retained material was applied (70 ml/h) on to an SP (sulphopropyl)-Sepharose Fast Flow column (17.0 cm×2.6 cm) (Amersham Biosciences, Baie d'Urfé, QC, Canada) equilibrated with buffer A. The column was washed with 200 ml of buffer A and protein elution was achieved with a 0–0.7 M NaCl gradient in buffer A. Two glucosaminidase activity peaks were observed: one was eluted in the flow-through (GlcNase I) and the second was eluted at 0.3 M NaCl (GlcNase II).
The GlcNase I fractions were dialysed against 50 mM sodium acetate buffer, pH 5.5 (buffer B) and applied (100 ml/h) to an SP-Sepharose Fast Flow column (17.0 cm×2.6 cm). The column was washed with 100 ml of buffer B and the proteins were eluted with a 0–1 M NaCl gradient. GlcNase was recovered at 0.5 M NaCl. The active fractions were dialysed against 1 mM sodium phosphate buffer, pH 6.8 (buffer C) and applied (50 ml/h) to a hydroxyapatite (Bio-Gel HTP; Bio-Rad, Hercules, CA, U.S.A.) column (6.0 cm×2.6 cm). The column was washed with 70 ml of buffer C and the proteins were eluted with a 1–5 mM gradient of unbuffered MgCl2. GlcNase I activity was recovered as a large peak at the end of the gradient. To concentrate the protein, the active fractions were diluted 10-fold with buffer B, applied to a mini-column (1 ml; HiTrap SP XL; Amersham Biosciences) and GlcNase activity was eluted in a total volume of 8.5 ml of 1 M NaCl in buffer B. This material was applied (13 ml/h) to a Macro-Prep SE 100/40 gel-filtration column (90 cm×1.6 cm) (Bio-Rad) and eluted with buffer B.
The GlcNase II fractions were dialysed against buffer C and applied (50 ml/h) to a Bio-Gel HTP hydroxyapatite column (6.0 cm×2.6 cm). The column was washed with 70 ml of buffer C and the proteins were eluted with a 1–5 mM gradient of unbuffered MgCl2. GlcNase activity was recovered at 2.5–4.5 mM MgCl2 and dialysed against buffer B.
For GlcNase production in a heterologous host, the spores of Strep. lividans TK24 (pFD666-csxA) were inoculated (106/ml) in 50 ml of YME medium for 72 h at 30 °C with shaking. Pelleted cells were transferred to 0.8 litre of M14 minimal medium [11] supplemented with 0.2% glucosamine hydrochloride and 0.8% chitosan (finely ground) as carbon sources. After 6 days of culture, the supernatant was adjusted to pH 4.2 with 100% acetic acid, filtered through 0.8-μm- and 0.2-μm-mesh-size filters and loaded directly on an SP-Sepharose column (8.0 cm×2.6 cm) equilibrated with 50 mM sodium acetate buffer, pH 4.2 (buffer D). Elution was performed with a 0–1M NaCl gradient in buffer D. Most of the activity was eluted from 360 to 440 mM NaCl. The fractions with activity were pooled (32 ml) and dialysed overnight against buffer C. The retained material was loaded on a Bio-Gel HTP column (8.0 cm×1.6 cm) equilibrated with buffer C. Elution was done with an unbuffered 0–1M MgCl2 gradient. GlcNase activity was recovered at ≈0.1 M MgCl2 and dialysed against buffer B.
For short-term storage, enzymes were kept in buffer B at 4 °C. For long-term storage, 50% (v/v) glycerol was added and enzymes were kept at −20 °C.
Biochemical assays
Enzyme activity was determined with (GlcN)2 as substrate. A standard GlcNase assay was performed combining 100 μl of 2 mM (GlcN)2 in 50 mM sodium acetate buffer, pH 5.3, with 100 μl of appropriately diluted enzyme sample. The reaction was allowed to proceed for 10 min at 37 °C and was stopped by the addition of 25 μl of 0.2 M sodium tetraborate. The GlcN liberated from the dimer was N-acetylated by addition of 25 μl of acetylation reagent (1.5% acetic anhydride in acetone, freshly prepared) followed by 10 min incubation at room temperature. The resulting N-acetylglucosamine was determined by the method of Reissig et al. [12] modified as follows. A 50 μl portion of 0.8 M potassium tetraborate was added and tubes were incubated for 3 min in boiling water. Then, 1.5 ml of chromogenic reagent [0.5% 4-(N,N-dimethylamino)benzaldehyde and 1.3% HCl in 100% acetic acid] was added and the mixture incubated for 20 min at 37 °C. Absorbance was measured at 585 nm.
Considering that one cleaved molecule of (GlcN)2 resulted in the release of two GlcN monomers, one unit of enzyme activity was defined as the amount that liberated 2 μmol of glucosamine from (GlcN)2/min under the above conditions. This is equivalent to the release of 1 μmol of glucosamine/min (at least under initial conditions) from other substrates such as longer oligomers, chitosan or the chromogenic substrate p-NP (p-nitrophenyl) β-D-glucosaminide.
All other oligomeric substrates were used at 1 mM. Chitosan was used at 1 mg/ml. Commercial chitosan was purified before use in enzyme assays by the following steps: dissolution in 0.2 M acetic acid, precipitation with 0.2 M KOH, filtration, washing with 0.5 M KCl, dissolution in 0.2 M acetic acid, precipitation with 10 vol. of 100% ethanol, recovery by centrifugation and freeze-drying. With chitosan substrate, activity was estimated by two methods: liberation of GlcN (as above) or production of reducing sugar (as in standard endo-chitosanase assay) [13]. In the latter case, the reducing sugars were determined by the method of Lever [14], as modified by Schep et al. [15].
Chromogenic p-NP glycosides were used at a final concentration of 1 mM. When these substrates were used, the reaction was stopped by addition of 1 vol. of 0.5 M glycine, pH 11, and the activity was calculated from the amount of p-nitrophenol liberated, measured by A405.
To examine the course of oligosaccharide hydrolysis, the enzymatic reaction was monitored by TLC. A 3 μl portion of the enzyme solution (0.48 μM) was added to the (GlcN)4 or (GlcN)5 solution (12.3 mM) in 50 mM sodium acetate buffer, pH 5.0, and incubated at 37 °C. After an appropriate reaction time, an aliquot withdrawn from the reaction mixture was mixed with the same volume of 0.1 M NaOH, and then spotted on the TLC aluminium plate (Silica gel 60; Merck). The chromatographic separation was conducted with a 28% ammonia/propan-1-ol (1:2, v/v) solvent system. The sugar spots were visualized by spraying with ninhydrin reagent, followed by heating at 90 °C.
Protein concentration was estimated by the method of Bradford [16], using the Bio-Rad Protein Assay kit with BSA as standard. For N-terminal sequencing, the protein was subjected to SDS/10%-(w/v)-PAGE in 25 mM Tris/250 mM glycine/0.1% SDS buffer. After migration, the protein was transferred to a PVDF membrane (Millipore, Billerica, MA, U.S.A.) using 25 mM Tris/192 mM glycine/20% methanol as transfer buffer. The N-terminal protein sequence was determined by Edman degradation on a Procise sequencer (Applied Biosystems, Foster City, CA, U.S.A.). After trypsin digestion, internal sequences were determined by an MS/MS (tandem MS) method on an LCQ Deca XP ion-trap installation (Thermo Electron, Waltham, MA, U.S.A.).
1H-NMR spectroscopy
The oligosaccharide substrate was freeze-dried three times from 2H2O, and then dissolved in 0.5 ml of 2H2O. The pH was adjusted to 5.0. The substrate solution was placed in a 5-mm-diameter NMR tube, and 60 μl of the enzyme solution (0.3 μM) were added to start the enzymatic reaction. The NMR tube was immediately set into a 5 mm probe on a JEOL EX-270 instrument, which was thermostatically controlled at 25 °C. After an appropriate reaction time, the accumulation of a 1H-NMR spectrum was started. One accumulation required 3 min. The substrate concentration was 8.3 mM.
Gene cloning
Standard molecular-biology methods [17] were used for DNA cloning, restriction analysis and DNA–DNA hybridization. Genomic DNA from actinomycetes was prepared using Proteinase K [9]. To amplify a GlcNase-specific DNA fragment from the genome of Amy. orientalis, two PCR primers were designed on the basis of partial amino acid sequences of GlcNase I (5′-TGGAAGGCCACATCGAACC-3′ and 5′- GAGCGGTGGTACTGCTTGG-3′). PCR amplification was done using 150 ng of genomic DNA, 2.5 mM dNTPs, 10 pmol of each primer, 10% DMSO, 0.5 unit of Taq polymerase in 1×Taq buffer (Pharmacia) under the following conditions: initial denaturation for 5 min at 94 °C, followed by 30 cycles of denaturation for 1 min at 94 °C, annealing for 1 min (touchdown of five cycles from 60 to 52 °C, then 25 cycles at 50 °C), extension for 1 min at 72 °C, and final extension for 10 min at 72 °C. The amplified fragment (≈0.5 kb) was then used as hybridization probe in Southern blots with genomic fragments of Amy. orientalis or Strep. avermitilis and in colony hybridization.
To clone the full-length GlcNase-encoding gene from Amy. orientalis, chromosomal DNA was digested with PvuII, blotted from agarose gel on nylon membrane and probed with the GlcNase-specific PCR fragment. A positive signal was obtained with 6–7 kb fragments. Partial gene libraries including these fragments were generated after PvuII digestion, agarose-gel electrophoresis, extraction of the appropriately sized DNA fragments and ligation into a plasmid vector previously digested with SmaI. Positive clones were identified by colony hybridization on Hybond-N+ membrane (Amersham Biosciences), following the manufacturer's recommendations, with the GlcNase-specific PCR-fragment probe. DNA sequencing has shown, however, that the isolated PvuII fragment contained only the 5′ region of the GlcNase gene. The remaining part was cloned following the same procedure and a 4 kb PstI fragment was isolated. The complete GlcNase gene was reconstituted by ligating a NotI–EcoRI segment from the PvuII fragment to an EcoRI-PstI segment from the PstI fragment. The ligation product was introduced into a NotI+PstI-digested pFD666 vector, giving the pFD666-CsxA plasmid.
For expression of the SAV1223 protein, a 6.2 kb PstI fragment including the corresponding gene was isolated from chromosomal DNA of Strep. avermitilis using the same cloning steps as that described above. The cloned fragment was then excised from the plasmid vector with HindIII and XbaI, subcloned in pFD666 vector and transformed into Strep. lividans TK24.
Bioinformatic procedures
The relatedness of primary structures of proteins was estimated by sequence alignment with the T-Coffee program [18]. The same program generated phylogenetic trees which were then visualized with TreeView [11]. For the identification of putative catalytic residues and other highly conserved residues in the GH2 family, we searched the Superfamily database (version 1.67) [19] for the best structural template for the TIM-barrel modules extracted from a set of 16 representative GH2 protein sequences. This template was then used for a structure-based alignment using the 3DCoffee program [20]. To validate the obtained alignment by an independent method, secondary-structure motifs in the analysed protein modules were predicted with PSIPRED [21] and overlapped with the structure-based alignment.
Reagents
GlcN oligomers (GlcN)2–6 were obtained from MJS BioLynx Inc. (Brockville, ON, Canada). Chitosan (6% N-acetylated) was from ISM Biopolymer (Granby, Canada). Restriction enzymes were from New England Biolabs (Beverly, MA, U.S.A.). The chromogenic substrate pNP 2-amino-2-deoxy-β-D-glucopyranoside was from Rose Scientific (Edmonton, AB, Canada). The mono-N-acetylated chitotetraose [(GlcN)3-GlcNAc] used as substrate in 1H-NMR spectroscopy studies was obtained by the method of Mitsutomi et al. [22]. All the other reagents and enzyme substrates were commercially available products of analytical grade. Culture-media components were from Difco (Mississauga, Canada).
RESULTS
Screening for extracellular GlcNase activity
A total of 40 chitosanolytic actinomycete strains from the collection of our laboratory, isolated from various soil samples, were screened for the production of extracellular GlcNase by culture in chitosan liquid medium and detection of GlcNase in the culture supernatant. Surprisingly, this activity turned out to be uncommon, as no strain was found to express it at a significant level. We then decided to characterize the GlcNase produced by Amy. orientalis IFO12806 (=A.T.C.C. 19795), a strain that has been the subject of a previous study by Nanjo et al. [3].
Purification and biochemical characterization of native GlcNase
From the work of Nanjo et al. [3], it was expected that Amy. orientalis would produce a GlcNase that could be purified in four chromatographic steps and characterized by an alkaline pI of 8.8. Trying to simplify the purification method, we used a cation-exchange resin equilibrated at pH 7.7 as the first chromatographic step. Unexpectedly, the total GlcNase activity was separated in two distinct fractions on this column. Most of the activity (GlcNase I) was retrieved in the flow-through, whereas a second, minor, peak (GlcNase II) was eluted at 0.3 M NaCl, showing the expected chromatographic behaviour. Three subsequent chromatographic steps were necessary to purify GlcNase I, whereas only one more step was sufficient for GlcNase II.
Both native enzymes showed a maximum activity at pH 5.3 when assayed in a series of citrate/phosphate/borate buffers. Among a series of buffers assayed at this optimal pH, maximal activity was obtained in 50 mM sodium acetate buffer (results not shown), which was subsequently adopted as the standard assay buffer. Substrate specificities of both native GlcNases were almost identical. Both enzymes hydrolysed with similar efficiency oligomeric substrates of GlcN in the tested range (dimer to hexamer) as well as high-molecular-mass chitosan. Among the p-NP derivatives tested, only the p-NP β-D-glucosaminide was hydrolysed to a significant extent, but about 600 times less efficiently than (GlcN)2. No activity was detectable against p-NP derivatives of β-D-glucopyranoside, β-Dxylopyranoside, β-D-mannoside, β-D-glucuronide, β-D-galactoside, β-D-N-acetylgalactosaminide, β-D-N-acetylglucosaminide and α-D-N-acetylglucosaminide. The optimum temperature determined for a 10 min reaction was about 60 °C for both enzymes. From gel-filtration chromatography it was concluded that the enzyme is a monomeric protein. Partial amino acid sequencing showed that both proteins have the same N-terminal sequence, namely AAGNATPIPGYVNIQ. All these data suggested that GlcNase I and GlcNase II are two fractions of the same protein which differ essentially in their chromatographic behaviour.
To explain the differences between the two proteins, we started from the observation that a relatively important background of GlcN was always measured in reference samples of GlcNase II (incubated in the absence of added substrate) when assayed for activity under various conditions. Such a background was not observed with GlcNase I. We postulated that GlcNase II could be tightly linked to chitosan present in culture media during the enzyme production by Amy. orientalis. To test this possibility, we verified the chromatographic behaviour of GlcNase II applied on the cation-exchange column after a 150 min preincubation at 37 °C, to allow for the eventual hydrolysis of the bound chitosan into monomers. We found that the preincubated GlcNase II was no longer adsorbed to the sulphopropyl-derivatized cation exchanger at pH 7.7. Accordingly, GlcNase I preincubated with chitosan (1 mg/ml) at 4 °C for 120 min acquired the capacity to bind to this resin at pH 7.7. We conclude that the GlcNase is able to tightly bind the chitosan substrate and that this binding is stable for many days at 4 °C and is conserved through the various purification steps. The binding phenomenon results in a shift of the isoelectric point of the protein–substrate complex, changing its chromatographic properties.
Further biochemical characterization was done with GlcNase I only. The determination of kinetic parameters with the glucosamine dimer failed because of the insufficient sensitivity of the colorimetric assay for GlcNAc. Within the detection limit, the lowest (GlcN)2 substrate concentration that could be tested was 125 μM. At this concentration, the enzyme's initial velocity was still equal to 35% of velocity measured at saturating substrate concentration (2 mM). Instead, and for the first time in the literature, the Km and kcat constants for exochitosanase activity could be determined with chitosan as substrate using the more sensitive reducing sugar assay [14,15], giving 0.16 mg/ml and 2832 min−1 respectively.
When the enzymatic hydrolysis of (GlcN)5 was monitored by TLC (Figure 1A), the enzyme at first produced GlcN and (GlcN)4, which was then hydrolysed to GlcN and (GlcN)3. The trisaccharide was further hydrolysed to GlcN and (GlcN)2. The end product was GlcN. The result clearly indicates that the enzyme is GlcNase. Figure 1(B) shows the TLC profile showing the enzymatic hydrolysis of (GlcN)4. The course of the product formation was similar to that obtained with the substrate (GlcN)5. In this case, however, the spots for the transglycosylation product (GlcN)5 clearly appeared, indicating that the enzyme catalyses transglycosylation as well as hydrolysis.
Figure 1. TLC profiles showing the enzymatic hydrolysis of (GlcN)5 (A) and (GlcN)4 (B).

A 3 μl portion of the GlcNase solution (0.48 μM) was added to 100 μl of the substrate solution (12.3 mM) in 50 mM sodium acetate buffer, pH 5.0. The reaction mixture was incubated at 37 °C. Numerals in the chromatogram indicate the degree of polymerization of individual oligosaccharide products.
In conclusion, the purification and biochemical characterization of the GlcNase from the strain A.T.C.C. 19795 indicated that it produces extracellularly only one enzyme with such an activity in a medium with chitosan as the main carbon source and that this enzyme is very similar to, if not identical with, the enzyme characterized in 1990 [3], despite the fact that the bacterial strain studied originated from a different source.
Mechanism of enzymatic hydrolysis of (GlcN)3-GlcNAc as determined by 1H-NMR spectroscopy
To examine the catalytic mechanism of the enzyme, we determined the anomeric form of the reaction products from the enzymatic hydrolysis of (GlcN)3-GlcNAc. The left-hand panel of Figure 2 shows the time-dependent profiles of the anomeric proton region of 1H-NMR spectra of the enzymatic reaction mixture. The doublet signal derived from the β-form of the GlcN monomer H1 appeared immediately after beginning the enzymatic reaction. However, the signal from the α-form did not appear until 30 min has elapsed. The relative signal areas of the α- and β-forms were calculated and plotted against the reaction time (the right-hand panel of Figure 2). The result suggests that the enzyme produces the β-form, which is then converted into the α-form by mutarotation. The enzyme was found to be a retaining glycoside hydrolase.
Figure 2. Time course of 1H signals during (GlcN)3-GlcNAc hydrolysis.

The relative peak areas were calculated from the NMR spectra (on the left) and plotted against reaction times (on the right). ◆, the α-form of GlcN; ■, the β-form of GlcN.
Cloning of the GlcNase gene and enzyme production in a heterologous host
Besides the N-terminal sequence, further sequencing of a fragment resulting from GlcNase digestion with trypsin gave the internal sequence AQ[IL]SQYENVR. PCR primers derived from these sequences allowed us to amplify a fragment of about 1.6 kb from the genomic DNA of Amy. orientalis, from which a subsequence of about 0.5 kb was used as a hybridization probe to clone the full-length gene. The GlcNase gene was named csxA to emphasize the chitosanase exo-type activity of the corresponding protein (the symbol csn was previously proposed for chitosanases of endo-type) [23,24]. Activity was observed in the supernatant of a Strep. lividans transformant culture harbouring the plasmid pFD666-csxA, whereas no activity was found when the cells were transformed with the vector plasmid alone. The enzyme was purified from culture supernatant with a final recovery of 48.3% and a 3.7-fold purification by a simple two-step procedure summarized in Table 1, omitting the ammoniumprecipitation step. The GlcNase was purified to apparent homogeneity, as judged from Coomassie Blue-stained SDS/polyacrylamide gels (Figure 3). The GlcNases purified from the native and the heterologous hosts migrated similarly on SDS/PAGE and seemed to have a very similar, if not identical, molecular mass estimated at 103.5 kDa. The identity of the enzyme from the heterologous source was confirmed by its ability to generate monomeric GlcN from oligomeric substrates as well as from chitosan.
Table 1. Purification of GlcNase from culture of recombinant Strep. lividans pFD666-csxA.
| Step | Total activity (units) | Total protein (mg) | Specific activity (units/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Crude extract | 573 | 52.8 | 10.9 | 100 | − |
| SP-Sepharose | 548 | 23.9 | 22.9 | 95.6 | 2.1 |
| Hydroxyapatite | 277 | 6.9 | 40.1 | 48.3 | 3.7 |
Figure 3. SDS/PAGE analysis of purified GlcNases.
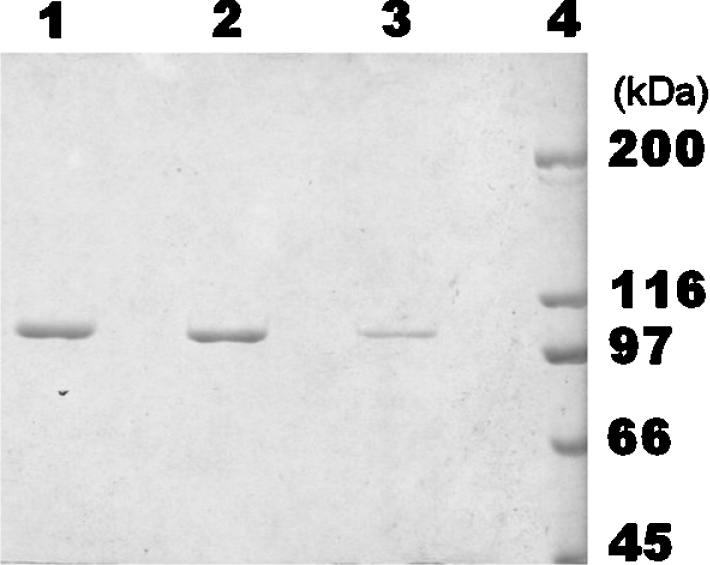
The SDS/6%-(w/v)-PAGE gel was stained with Coomassie Brillant Blue. Lane 1, purified GlcNase I (1.5 μg); lane 2, purified GlcNase II (1.0 μg); lane 3, purified recombinant GlcNase (0.5 μg); lane 4, ‘High Range Protein Molecular Weights’ (Bio-Rad).
Sequence analysis of the GlcNase-encoding gene csxA
Sequence analysis of the csxA gene (Figure 4) reveals an ORF (open reading frame) of 1032 amino acid residues with a calculated molecular mass of 110557 Da. A probable ribosome-binding site is located eight nucleotides upstream from the proposed translation start site, GTG. Further upstream, a palindromic sequence is found, similar to motifs found upstream from endochitosanase genes from Streptomyces sp. N174 [24] and Kitasatospora sp. N106 (formerly Nocardioides sp. N106) [25]. This palindrome is also present upstream from the ORF SAV1223 from Strep. avermitilis (see the Discussion). The csxA gene is preceded by an ORF belonging to COG3012 [26], grouping ‘uncharacterized proteins conserved in bacteria’. This ORF fragment shares similarity with SAV6631 from Strep. avermitilis and SCO1677 from Strep. coelicolor A3(2).
Figure 4. CsxA: DNA and protein sequence.

Nucleotides and amino acids are numbered on the right-hand side. The stop codon is indicated by an asterisk. The amino acid sequence determined by protein sequencing and used for PCR primer design are underlined. The signal sequence is depicted in boldface. DNA palindromic sequences corresponding to a putative operator and terminator are indicated by arrows.
CsxA is produced extracellularly ([3]; the present study). Accordingly, a well-defined signal peptide is detected with the SignalP 3.0 program [27]. However, the program predicted a signal peptidase cleavage site between the residues 32 and 33, whereas the observed N-terminus is at residue 47 (Figure 4).
A search with the BLAST program (version 2.2.10) [28] revealed that the deduced amino acid sequence of CsxA exhibits similarity to a few hundreds of protein sequences found in databases (not shown). Similarly, CsxA appears to be a member of GH2. The segment similar to other GH2 members ends at residues 897–899 and is followed by a putative CBM (carbohydrate-binding module) belonging to family 6, a feature unique among GH2 members. The closest CsxA relatives are ORFs extracted from genomic databases. The majority of them have not yet been characterized at the biochemical level. Most of them are annotated as putative β-mannosidases, except the BAD99604 protein from Hypocrea jecorina (formerly Trichoderma reesei), recently submitted to the database, which is annotated as a GlcNase. The amino acid sequence showing the greatest similarity to CsxA is SAV1223 from the genome of Strep. avermitilis [29]. This ORF is shorter than that of CsxA as it does not have a CBM. SAV1223 exhibits 62.5% identity with, and 86.2% similarity to, CsxA in an 893-residue overlap. The other close relatives originate from sequenced fungal genomes and are diverse in their modular structure (Figure 5). They all share three modules found in all GH2 members: the glyco hydro 2N or sugar-binding domain, the glyco hydro 2 or immunoglobulin-like β-sandwich domain and the glyco hydro 2C or TIM-barrel domain [PFAM database (Protein Families Database of Aligments and Hidden Markov Models (http://www.sanger.ac.uk/Software/Pfam/)] [30]. Interestingly, despite their high mutual similarity, only some of these modules were recognized after a search completed in the PFAM database. Some of these ORFs include other modules less commonly found in glycoside hydrolases (Figure 5).
Figure 5. Modular structure of CsxA and related ORFs.

ORFs are designated by their gene names (CsxA; SAV1223) or their database entry. Wide boxes, modules identified by databases searches and alignments; narrow boxes, regions of similarity not corresponding to known modules; lines, low similarity segments; shaded boxes, segments recognized by search in PFam database; S, signal peptides identified experimentally (CsxA) or recognized by the SignalP algorithm; 2N, glyco hydro 2N or sugar-binding domain; 2, glyco hydro 2 or immunoglobulin-like β-sandwich domain; 2C, glyco hydro 2C or TIM-barrel domain; CBM6, carbohydrate-binding module family 6; P and P′, peptidase_M20 or peptidase family M20/M25/M40; D, M20_dimer or peptidase dimerization domain; *, serine-rich linker.
To determine the position of CsxA-related ORFs inside GH2, we chose to compare individual domains rather than full-length protein sequences. We extracted the sequences of TIM-barrel domains from 25 GH2 members. These domains include the catalytic amino acids and several other residues conserved throughout this family [31,32]. Besides CsxA-related ORFs, we only analysed GH2 members whose β-galactosidase, β-glucuronidase or β-mannosidase activities were confirmed by biochemical studies. A phylogenetic tree was derived by sequence analysis with the T-Coffee multiple sequence alignment package [18]. As shown in Figure 6, CsxA and the related ORFs form a distinct subfamily inside GH2. Interestingly, similar trees with four main branches are obtained from the alignments of other domains from the same set of proteins (sugar-binding domains or immunoglobulin-like β-sandwich domains) (results not shown). It indicates that each of the three domains is adapted to the interaction with the specific substrate characteristic of each GH2 subfamily.
Figure 6. Phylogenetic analysis of the (α/β)8-barrel sequences of GH2 family members.

The name of the originating organism and the database entry are indicated. Cs, CsxA and related ORFs; CsS, Strep. avermitilis (NP 822398); CsAo, Amy. orientalis (the present work; csxA, AAX62629); CsH, H. jecorina (BAD99604); CsN: Neurospora crassa (XP_331434.1); CsM, Magnaporthe grisea (MG05864.4); CsG, Gibberella zeae (FG02314.1); CsAn, Asp. nidulans (AN2824.2); Ga, β-galactosidases from: GaK, Kluyveromyces lactis (P00723; BGAL_KLULA); GaL, Lactococcus lactis (Q48727; BGAL_LACLA); GaEcl, Enterobacter cloacae (Q47077, BGAL_ENTCL); GaEco, E. coli (P00722, BGAL_ECOLI); GaA, Arthrobacter sp. (Q59140, GAL_ARTSB); GaC, Clostridium acetobutylicum (P24131, BGAL_CLOAB); Gl, β-glucuronidases from: GlC, Canis familiaris (dog) (O18835, BGLR_CANFA); GlF, Felis silvestris catus (cat) (O97524, BGLR_FELCA); GlM, Mus musculus (mouse) (P12265, BGLR_MOUSE); GlH, Homo sapiens (human) (P08236, BGLR_HUMAN); GlE, E. coli (P05804, BGLR_ECOLI); GlS: Staphylococcus sp. (patent PCT/US98/19217); GlL, Lactobacillus gasseri (AAK07836, Q9AHJ8_9LACO); Ma, β-mannosidases from: MaA, A. niger (Q9UUZ3, MANBA_ASPNG); MaB, Bos taurus (ox) (Q29444, MANBA_BOVIN); MaH, Homo sapiens (O00462, MANBA_HUMAN); MaM, Mus musculus (Q8K2I4_MOUSE); MaT, Thermobifida fusca (Q8KLI9_THEFU); MaC, Cellulomonas fimi (AAD42775, Q9XCV4_CELFI).
In GH2 as well in families 1, 5, 10 and 17 (members of the clan GH-A) [31–33], the catalytic functions are attributed to a nucleophile, which is a glutamate residue located close to the C-terminus of the seventh β-strand, and an acid/base residue, which is a glutamate residue (always preceded by an asparagine residue) located close to the C-terminus of the fourth β-strand. The putative catalytic residues in CsxA and related proteins were identified through structure-based alignment. A search in the Superfamily database (version 1.67) [19] identified domain 3 from the three-dimensional structure of human β-glucuronidase (Protein Data Bank entry 1BHG; residues 329–632) [34] as the best template for structure-guided alignment with the TIM-barrel-domain sequences from CsxA and related ORFs. The alignment itself was then performed with the 3DCoffee program [20] and allowed to define the putative catalytic residues in comparison with known catalytic residues formally identified in other members of GH2 [35–39] (Figure 7). This revealed an unexpected trait of the TIM-barrel domains in CsxA and related ORFs: whereas the putative glutamate nucleophiles seemed to be strictly conserved in all the analysed GH2 members, a doublet Ser-Asp instead of Asn-Glu was observed at the location of the putative acid/base residue. This suggests that an aspartate residue instead of a glutamate residue could play the role of the catalytic acid/base residue in CsxA. This major difference reinforces the distinct character of the exochitosanase-related proteins inside GH2. The alignment allowed the identification to several other residues strictly conserved in GH2, some of which have not been described previously (Table 2).
Figure 7. Assignment of putative catalytic residues in CsxA and related ORFs through structure-guided alignment of TIM-barrel domains from a representative set of GH2 sequences.


Abbreviations are as in Figure 6. Alignment was obtained by the 3DCoffee structure-guided-alignment method [20] using domain 3 from human β-glucuronidase as template. Secondary structures are shaded in dark grey (α-helices) or light grey (β-sheets). Secondary structures formally identified in crystallized proteins are shown for human β-glucuronidase (PDB ID: 1bhg:A) and E. coli β-galactosidase LacZ (PDB ID: 1DPO:A). These two proteins are indicated by bold labels GlH and GaEco respectively. Secondary structures in other proteins were predicted by the PSIPRED algorithm [21]. The putative catalytic acid/base and nucleophile residues are indicated with asterisks. Catalytic residues formally identified by mechanistic studies [35–39] are underlined. Other strictly conserved residues are indicated with triangles. Numbering refers to the primary structure of CsxA (as in Figure 4).
Table 2. Strictly conserved residues in the GH2 family.
These residues are strictly conserved according to an extensive alignment (not shown) including all the proteins from Figure 6 and Supplementary Figure 1 (http://www.BiochemJ.org/bj/394/bj3940675add.htm).
| Conserved residues | |||||
|---|---|---|---|---|---|
| Enzyme… Species… | β-Glucuronidase (LacZ) H. sapiens | β-Galactosidase (Man2A) E. coli | β-Mannosidase (CsxA) C. fimi | β-Glucosaminidase Amy. orientalis | Remarks |
| Gly347 | Gly353 | Gly326 | Gly360 | First identification | |
| Asn379 | Asn385 | Asn358 | Asn389 | First identification | |
| Arg382 | Arg388 | Arg361 | Arg392 | Catalytic nucleophile activator [51] | |
| Asp397 | Asp403 | Asp376 | Asp407 | First identification | |
| Gly400 | Gly406 | Gly379 | Gly410 | First identification | |
| Glu540 | Glu537 | Glu519 | Glu541 | Catalytic nucleophile [35–39] | |
| Gly583 | Gly564 | Gly608 | Gly638 | First identification | |
| Trp587 | Trp568 | Trp612 | Trp642 | Substrate binding [52] | |
SAV1223 from Strep. avermitilis is a GlcNase
The ORF SAV1223, which is the closest relative of CsxA, was defined as a ‘putative β-mannosidase’ in the genome annotation of Strep. avermitilis MA-4680 [29] on the basis of sequence similarity. To extend the biochemical data to at least one other member of the proposed new GH2 subfamily, we cloned the SAV1223 gene in a closely related heterologous host, Strep. lividans. The SAV1223 protein was purified to apparent homogeneity by the same two-step protocol as that used for CsxA (Table 1). In the first chromatographic step we observed the same phenomenon of chitosan binding as was observed for CsxA, indicating that this interaction is not dependent on the presence of the carbohydrate-binding module. The SAV1223 protein showed GlcNase activity, generating monomeric GlcN from (GlcN)2. It also behaved as an exochitosanase, with an apparent Km of 0.04 mg/ml and a kcat of 330 min−1. The optimal pH was of 5.3, as for CsxA. No β-mannosidase activity was detected. We tested the same set of p-NP derivatives as for CsxA, but the only one hydrolysed by SAV1223 was the p-NP β-D-glucosaminide, with a specific activity 500 times lower than for (GlcN)2. The poor recognition of the p-NP β-D-glucosaminide is a common trait of both enzymes and was also observed by Nanjo et al. [3]. As the subdivision of GH2 members into subfamilies is clearly related to their respective enzymatic activities (Figure 6), our data suggest that the new CsxA-related subfamily represents a cluster of enzymes with GlcNase/exochitosanase activity.
DISCUSSION
When first created [40], GH2 grouped β-galactosidases and β-glucuronidases, including such extensively studied proteins as LacZ and GusA from E. coli, widely used as reporter proteins in molecular biology. Cloning and sequencing of the first β-mannosidase [41] extended the family to include these enzymes, first the lysosomal proteins from mammalian species, then similar enzymes of bacterial and fungal origin. With the present work, we further extend the enzymatic-activity spectrum of GH2 to include GlcNases/exochitosanases. This illustrates once again the extreme adaptability of the TIM-barrel enzymes, one of the evolutionary most successful protein folds [42–44].
At the primary-structure level, GH2 is now divided into four subclasses, each dedicated to the hydrolysis of a particular glycosidic link. These subfamilies encompass taxonomic barriers, as the same subfamily can group enzymes from bacteria as well as from mammals. GlcNases/exochitosanases are, however, less widely distributed than the other three activities: after BLAST analysis of 230 prokaryotic genomes and 68 eukaryotic genomes for which sequencing data were completed at the time the present paper was submitted, the taxonomic spectrum of CsxA-related ORFs was found to be limited to two Gram-positive actinobacteria and five filamentous fungi. Interestingly, this corroborates the available biochemical data on these enzymes which have so far only been purified and characterized from members of these two taxonomic groups. Furthermore, this taxonomical distribution is very similar to that of endochitosanases. Despite their molecular diversity (endochitosanases have been assigned to GH families 5, 8, 46, 75 and 80), the endochitosanases were essentially purified and characterized from Gram-positive bacteria (both G+C-rich and A+T-rich branch) and filamentous fungi.
The fact that none of the chitosanolytic actinomycetes from our collection produced detectable extracellular GlcNase, whereas they all produce endochitosanases, indicates that direct uptake of chitosan oligosaccharides (mostly dimers and trimers) is the preferred way of transport of products of chitosan hydrolysis inside the cell, at least among actinomycetes. In other words, chitosan does not need to be hydrolysed into monomers to be used efficiently as a carbon and nitrogen source. GlcNase is thus not essential for chitosan metabolism for most actinomycetes and, perhaps, other chitosanolytic bacteria. For instance, no GlcNase activity has been found so far in Gram-positive bacilli, and no CsxA homologues have been detected in sequenced bacillar genomes, whereas many of them have endochitosanase genes (CAZy database) [45]. We believe, however, that the two GlcNases studied in the present work are involved in chitosan metabolism: a direct functional link between exochitosanases and endochitosanases is suggested by the fact that several genes originating from actinomycetes share a palindromic sequence localized a few tens of base-pairs upstream from the translation start site. The motif present in csxA is highly similar to the ones identified previously in the endochitosanase genes from Streptomyces sp. N174 and Kitasatospora sp. N106 [24,25]. In the latter species, this sequence was shown to be the site of an interaction with an aminosugar-dependent regulatory protein (M.-P. Dubeau and R. Brzezinski, unpublished work). In Strep. avermitilis, this DNA motif defines a putative regulon of four genes, possibly dedicated to chitosan degradation: the exochitosanase (SAV1223), an endochitosanase belonging to GH46 (SAV2015), another putative endochitosanase, member of GH75 (SAV1850) and a gene encoding a putative regulatory protein (SAV5384) belonging to the ROK family [46] and highly similar to NagC, a repressor involved in the regulation of amino-sugar metabolism in E. coli [47].
The enzymes belonging to GH2 are retaining enzymes. We observe the same mechanism for this GlcNase. It seems that the proposed difference in the catalytic acid/base residue (an aspartate in the CsxA subfamily and a glutamate in the other three subfamilies) did not result in a change of the general mechanism of hydrolysis, which remains typical of the whole GH2.
While the presence of an aspartate as putative catalytic acid/base residue is a novelty inside GH2, the change of the residue located immediately upstream was not totally unexpected. Zechel et al. [39] discussed the importance of this residue, an asparagine residue fully conserved in GH2 β-galactosidases, β-glucuronidases and β-mannosidases, which interacts by hydrogen-bonding with a sugar 2-substituent in intermediate complexes formed during catalysis [48]. The 2-substituent in β-D-glucosaminide substrates is an amino group, which possibly explains the replacement of the asparagine residue by a serine residue, a better partner for the establishment of a hydrogen bond with this kind of 2-substituent. In the β-glucosidase CelB from Pyrococcus furiosus belonging to GH1, another TIM-barrel-based family, mutation of this highly conserved asparagine residue into an aspartate or a serine residue resulted in major changes in substrate specificity and catalytic efficiency [49,50].
A close examination of protein sequences assigned to GH2 in recently sequenced genomes reveals that yet another combination of residues can be found at the discussed positions: in proteins Atu2575, BMA0039 and BPSL0611 from the CAZy (carbohydrate-active enzyme) database (originating, respectively from Agrobacterium tumefaciens C58, Burkholderia mallei ATCC 23344 and B. pseudomallei K96243) the putative acid/base residue is a glutamate residue, but it is preceded by a serine residue. This characteristic is shared by several other proteins from the NCBI (National Center for Biotechnology Information) database. Interestingly, the sequences of these proteins cluster as a new branch in the phylogenetic tree of GH2 (see Supplementary Figures 1 and 2 at http://www.BiochemJ.org/bj/394/bj3940675add.htm), which probably indicates that they differ in their catalytic properties from other GH2 members. Most of them, however, are annotated as probable β-mannosidases. In conclusion, we suggest a careful examination of the putative acid/base residue and its upstream neighbour before assigning a possible catalytic function for GH2 members and other TIM-barrel-based glycoside hydrolases.
Online data
Acknowledgments
This work was supported by a strategic research grant from the Natural Sciences and Engineering Research Council of Canada in partnership with ISM Biopolymer (Granby, QC, Canada). We thank Dr Bernard Henrissat [Architecture et Fonction des Macromolécules Biologiques (AFMB), UMR 6098–Centre National de la Recherche Scientifique, Marseille, France] for help with GH-family assignment of CsxA, and Dr Isabelle Boucher (ISM BioPolymers Inc., Granby, QC, Canada) for discussions.
References
- 1.Bartnicki-Garcia S., Nickerson W. J. Isolation, composition, and structure of cell walls of filamentous and yeast-like forms of Mucor rouxii. Biochim. Biophys. Acta. 1962;58:102–119. doi: 10.1016/0006-3002(62)90822-3. [DOI] [PubMed] [Google Scholar]
- 2.Fukamizo T. Chitinolytic enzymes: catalysis, substrate binding, and their application. Curr. Protein Pept. Sci. 2000;1:105–124. doi: 10.2174/1389203003381450. [DOI] [PubMed] [Google Scholar]
- 3.Nanjo F., Katsumi R., Sakai K. Purification and characterization of an exo-β-D-glucosaminidase, a novel type of enzyme, from Nocardia orientalis. J. Biol. Chem. 1990;265:10088–10094. [PubMed] [Google Scholar]
- 4.Nogawa M., Takahashi H., Kashiwagi A., Oshima K., Okada H., Morikawa Y. Purification and characterization of exo-β-D-glucosaminidase from a cellulolytic fungus, Trichoderma reesei PC-3-7. Appl. Microbiol. Biotechnol. 1998;64:890–895. doi: 10.1128/aem.64.3.890-895.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumura S., Yao E., Toshima K. One-step preparation of alkyl β-D-glucosaminide by the transglycosylation of chitosan and alcohol using purified exo-β-D-glucosaminidase. Biotechnol. Lett. 1999;21:451–456. [Google Scholar]
- 6.Zhang X. Y., Dai A. L., Zhang X. K., Kuroiwa K., Kodaira R., Shimosaka M., Okazaki M. Purification and characterization of chitosanase and exo-β-D-glucosaminidase from a Koji mold, Aspergillus oryzae IAM2660. Biosci. Biotechnol. Biochem. 2000;64:1896–1902. doi: 10.1271/bbb.64.1896. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka T., Fukui T., Atomi H., Imanaka T. Characterization of an exo-β-D-glucosaminidase involved in a novel chitinolytic pathway from the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 2003;185:5175–5181. doi: 10.1128/JB.185.17.5175-5181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka T., Fukui T., Fujiwara S., Atomi H., Imanaka T. Concerted action of diacetylchitobiose deacetylase and exo-β-D-glucosaminidase in a novel chitinolytic pathway in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Biol. Chem. 2004;279:30021–30027. doi: 10.1074/jbc.M314187200. [DOI] [PubMed] [Google Scholar]
- 9.Kieser T., Bibb M. J., Buttner M. J., Chater K. F., Hopwood D. A. Norwich: The John Innes Foundation; 2000. Practical Streptomyces genetics. [Google Scholar]
- 10.Denis F., Brzezinski R. A versatile shuttle cosmid vector for use in Escherichia coli and actinomycetes. Gene. 1992;111:115–118. doi: 10.1016/0378-1119(92)90611-r. [DOI] [PubMed] [Google Scholar]
- 11.Page N., Kluepfel D., Shareck F., Morosoli R. Effect of signal peptide alterations and replacement on export of xylanase A in Streptomyces lividans. Appl. Environ. Microbiol. 1996;62:109–114. doi: 10.1128/aem.62.1.109-114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reissig J. L., Storminger J. L., Leloir L. F. A modified colorimetric method for the estimation of N-acetylamino sugars. J. Biol. Chem. 1955;217:959–966. [PubMed] [Google Scholar]
- 13.Boucher I., Dupuy A., Vidal P., Neugebauer W. A., Brzezinski R. Purification and characterization of a chitosanase from Streptomyces N174. Appl. Microbiol. Biotechnol. 1992;38:188–193. [Google Scholar]
- 14.Lever M. A new reaction for colorimetric determination of carbohydrates. Anal. Biochem. 1972;47:273–279. doi: 10.1016/0003-2697(72)90301-6. [DOI] [PubMed] [Google Scholar]
- 15.Schep G. P., Shepherd M. G., Sullivan P. A. Purification and properties of a β-1,6-glucanase from Penicillium brefeldianum. Biochem. J. 1984;223:707–714. doi: 10.1042/bj2230707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J., Russell D. W. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. Molecular Cloning – A Laboratory Manual. [Google Scholar]
- 18.Notredame C., Higgins D. G., Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 19.Madera M., Vogel C., Kummerfeld S. K., Chothia C., Gough J. The SUPERFAMILY database in 2004: additions and improvements. Nucleic Acids Res. 2004;32:D235–D239. doi: 10.1093/nar/gkh117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Sullivan O., Suhre K., Abergel C., Higgins D. G., Notredame C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J. Mol. Biol. 2004;340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 21.Jones D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 22.Mitsutomi M., Takahara S., Kobayashi M., Ueda M., Arai M., Miyatake K. Enzymatic preparation of monoacetylated chitooligosaccharides. Chitin Chitosan Res. 2002;8:228–229. [Google Scholar]
- 23.Fukamizo T., Brzezinski R. Chitosanase from Streptomyces sp. strain N174: a comparative review of its structure and function. Biochem. Cell Biol. 1997;75:687–696. doi: 10.1139/o97-079. [DOI] [PubMed] [Google Scholar]
- 24.Masson J. Y., Denis F., Brzezinski R. Primary sequence of the chitosanase from Streptomyces sp. strain N174 and comparison with other endoglycosidases. Gene. 1994;140:103–107. doi: 10.1016/0378-1119(94)90738-2. [DOI] [PubMed] [Google Scholar]
- 25.Masson J. Y., Boucher I., Neugebauer W. A., Ramotar D., Brzezinski R. A new chitosanase gene from a Nocardioides sp. is a third member of glycosyl hydrolase family 46. Microbiology. 1995;141:2629–2635. doi: 10.1099/13500872-141-10-2629. [DOI] [PubMed] [Google Scholar]
- 26.Tatusov R. L., Natale D. A., Garkavtsev I. V., Tatusova T. A., Shankavaram U. T., Rao B. S., Kiryutin B., Galperin M. Y., Fedorova N. D., Koonin E. V. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bendtsen J. D., Nielsen H., von Heijne G., Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 28.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Ikeda H., Ishikawa J., Hanamoto A., Shinose M., Kikuchi H., Shiba T., Sakaki Y., Hattori M., Omura S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 30.Bateman A., Coin L., Durbin R., Finn R. D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E. L., Studholme D. J., Yeats C., Eddy S. R. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henrissat B., Callebaut I., Fabrega S., Lehn P., Mornon J. P., Davies G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7090–7094. doi: 10.1073/pnas.92.15.7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins J., Lo L. L., Harris G., Pickersgill R. β-Glucosidase, β-galactosidase, family A cellulases, family F xylanases and two barley glycanases form a superfamily of enzymes with 8-fold β/α architecture and with two conserved glutamates near the carboxy-terminal ends of β-strands four and seven. FEBS Lett. 1995;362:281–285. doi: 10.1016/0014-5793(95)00252-5. [DOI] [PubMed] [Google Scholar]
- 33.Durand P., Lehn P., Callebaut I., Fabrega S., Henrissat B., Mornon J. P. Active-site motifs of lysosomal acid hydrolases: invariant features of clan GH-A glycosyl hydrolases deduced from hydrophobic cluster analysis. Glycobiology. 1997;7:277–284. doi: 10.1093/glycob/7.2.277. [DOI] [PubMed] [Google Scholar]
- 34.Jain S., Drendel W. B., Chen Z. W., Mathews F. S., Sly W. S., Grubb J. H. Structure of human β-glucuronidase reveals candidate lysosomal targeting and active-site motifs. Nat. Struct. Biol. 1996;3:375–381. doi: 10.1038/nsb0496-375. [DOI] [PubMed] [Google Scholar]
- 35.Gebler J. C., Aebersold R., Withers S. G. Glu-537, not Glu-461, is the nucleophile in the active site of (lac Z) β-galactosidase from Escherichia coli. J. Biol. Chem. 1992;267:11126–11130. [PubMed] [Google Scholar]
- 36.Islam M. R., Tomatsu S., Shah G. N., Grubb J. H., Jain S., Sly W. S. Active site residues of human β-glucuronidase. Evidence for Glu540 as the nucleophile and Glu451 as the acid–base residue. J. Biol. Chem. 1999;274:23451–23455. doi: 10.1074/jbc.274.33.23451. [DOI] [PubMed] [Google Scholar]
- 37.Stoll D., He S., Withers S. G., Warren R. A. Identification of Glu-519 as the catalytic nucleophile in β-mannosidase 2A from Cellulomonas fimi. Biochem. J. 2000;351:833–838. [PMC free article] [PubMed] [Google Scholar]
- 38.Wong A. W., He S., Grubb J. H., Sly W. S., Withers S. G. Identification of Glu-540 as the catalytic nucleophile of human β-glucuronidase using electrospray mass spectrometry. J. Biol. Chem. 1998;273:34057–34062. doi: 10.1074/jbc.273.51.34057. [DOI] [PubMed] [Google Scholar]
- 39.Zechel D. L., Reid S. P., Stoll D., Nashiru O., Warren R. A., Withers S. G. Mechanism, mutagenesis, and chemical rescue of a β-mannosidase from Cellulomonas fimi. Biochemistry. 2003;42:7195–7204. doi: 10.1021/bi034329j. [DOI] [PubMed] [Google Scholar]
- 40.Henrissat B. Sequence homology between a β-galactosidase and some β-glucosidases. Protein Sequence Data Anal. 1991;4:61–62. [PubMed] [Google Scholar]
- 41.Chen H., Leipprandt J. R., Traviss C. E., Sopher B. L., Jones M. Z., Cavanagh K. T., Friderici K. H. Molecular cloning and characterization of bovine β-mannosidase. J. Biol. Chem. 1995;270:3841–3848. doi: 10.1074/jbc.270.8.3841. [DOI] [PubMed] [Google Scholar]
- 42.Gerlt J. A., Raushel F. M. Evolution of function in (β/α)8-barrel enzymes. Curr. Opin. Chem. Biol. 2003;7:252–264. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- 43.Rigden D. J., Jedrzejas M. J., de Mello L. V. Identification and analysis of catalytic TIM barrel domains in seven further glycoside hydrolase families. FEBS Lett. 2003;544:103–111. doi: 10.1016/s0014-5793(03)00481-2. [DOI] [PubMed] [Google Scholar]
- 44.Pujadas G., Palau J. TIM barrel fold: structural, functional and evolutionary characteristics in natural and designed molecules. Biologia. 1999;54:231–253. [Google Scholar]
- 45.Coutinho P. M., Henrissat B. Carbohydrate-active enzymes: an integrated database approach. In: Gilbert H. J., Davies B., Henrissat B., Svensson B., editors. Recent Advances in Carbohydrate Bioengineering. Cambridge: The Royal Society of Chemistry; 1999. pp. 3–12. [Google Scholar]
- 46.Titgemeyer F., Reizer J., Reizer A., Saier M. H., Jr. Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology. 1994;140:2349–2354. doi: 10.1099/13500872-140-9-2349. [DOI] [PubMed] [Google Scholar]
- 47.Plumbridge J., Pellegrini O. Expression of the chitobiose operon of Escherichia coli is regulated by three transcription factors: NagC, ChbR and CAP. Mol. Microbiol. 2004;52:437–449. doi: 10.1111/j.1365-2958.2004.03986.x. [DOI] [PubMed] [Google Scholar]
- 48.Juers D. H., Heightman T. D., Vasella A., McCarter J. D., Mackenzie L., Withers S. G., Matthews B. W. A structural view of the action of Escherichia coli (lacZ) β-galactosidase. Biochemistry. 2001;40:14781–14794. doi: 10.1021/bi011727i. [DOI] [PubMed] [Google Scholar]
- 49.Kaper T., van Heusden H. H., van L. B., Vasella A., van der O. J., de Vos W. M. Substrate specificity engineering of β-mannosidase and β-glucosidase from Pyrococcus by exchange of unique active site residues. Biochemistry. 2002;41:4147–4155. doi: 10.1021/bi011935a. [DOI] [PubMed] [Google Scholar]
- 50.Lebbink J. H., Kaper T., Kengen S. W., van der O. J., de Vos W. M. β-Glucosidase CelB from Pyrococcus furiosus: production by Escherichia coli, purification, and in vitro evolution. Methods Enzymol. 2001;330:364–379. doi: 10.1016/s0076-6879(01)30389-0. [DOI] [PubMed] [Google Scholar]
- 51.Belaich A., Fierobe H. P., Baty D., Busetta B., Bagnara-Tardif C., Gaudin C., Belaich J. P. The catalytic domain of endoglucanase A from Clostridium cellulolyticum: effects of arginine 79 and histidine 122 mutations on catalysis. J. Bacteriol. 1992;174:4677–4682. doi: 10.1128/jb.174.14.4677-4682.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Juers D. H., Huber R. E., Matthews B. W. Structural comparisons of TIM barrel proteins suggest functional and evolutionary relationships between β-galactosidase and other glycohydrolases. Protein Sci. 1999;8:122–136. doi: 10.1110/ps.8.1.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.