

Abstract
FSAP (Factor VII-activating protease) is a novel plasma-derived serine protease that regulates haemostasis as well as vascular cell proliferation. FSAP undergoes autoactivation in the presence of polyanionic macromolecules such as heparin and RNA. Competition experiments suggest that RNA and heparin bind to the same or overlapping interaction sites. A proteolysis approach, where FSAP was hydrolysed into smaller fragments, was used to identify the polyanion-binding site. The EGF (epidermal growth factor)-like domains EGF2 and EGF3 of FSAP are the major interaction domains for RNA. The amino acids Arg170, Arg171, Ser172 and Lys173 within the EGF3 domain were essential for this binding. This is also the region with the highest positive net charge in the protein and is most probably located in an exposed loop. It is also highly conserved across five species. Disruption of disulphide bridges led to the loss of RNA and heparin binding, indicating that the three-dimensional structure of the EGF3 domain is essential for binding to negatively charged heparin or RNA. The identification of polyanion-binding sites will help to define the role of FSAP in the vasculature.
Keywords: epidermal growth factor-like domain, Factor VII-activating protease, haemostasis, heparin, hyaluronic acid binding protein 2, polyanion binding
Abbreviations: CHO cell, Chinese-hamster ovary cell; EGF, epidermal growth factor; FSAP, Factor VII-activating protease; ME, 2-mercaptoethanol; TBS, Tris-buffered saline; tPA, tissue plasminogen activator; uPA, urokinase-type plasminogen activator
INTRODUCTION
FSAP (Factor VII-activating protease) is a serine protease that circulates in its inactive single-chain form in human plasma at a concentration of 12 μg/ml [1,2]. FSAP is synonymous with hyaluronic acid binding protein 2 or plasma hyaluronic acid binding protein [3,4]. It consists of three EGF (epidermal growth factor)-like domains, a kringle domain and a serine protease domain. FSAP has a calculated molecular mass of 60.2 kDa but migrates at approx. 75 kDa in SDS/PAGE analysis most probably due to glycosylation within its heavy chain [1]. Once purified in its single-chain form, the zymogen is autocatalytically cleaved into the active two-chain form including a 46 kDa chain, composed of amino acids Gly23 to Arg313, and a 29 kDa chain, composed of amino acids Ile314 to Phe560 [1,5,6]. This autocatalysis is accelerated by polyanionic compounds such as hyaluronic acid, heparin or heparan sulphate [1,5,6].
The Marburg I polymorphism of FSAP, a single amino acid substitution from glycine to glutamate at position 511, was found to be associated with carotid stenosis [7] and venous thromboembolism [8]. FSAP can activate Factor VII [9] and uPA (urokinase-type plasminogen activator) [1], degrade several matrix proteins such as fibronectin and fibrinogen [10], and activate kininogen and release bradykinin [11]. FSAP reduces cell proliferation of human umbilical vein endothelial cells by binding to and degrading basic fibroblast growth factor [12] and of vascular smooth muscle cells by binding to platelet-derived growth factor BB [13].
Recently, RNA was identified as a novel cofactor for FSAP (auto-) activation [14]. In this regard, a template model was proposed where both zymogen and active FSAP bind simultaneously to an RNA molecule that augments proenzyme conversion. Although both forms of FSAP exhibit comparable affinity in binding to RNA, the respective protein-binding sites for polyanions remained undefined [14]. We have now investigated the interaction of FSAP with RNA and heparin in detail to identify the domains of FSAP involved in these interactions.
EXPERIMENTAL
Materials and reagents
Wild-type CHO cells (Chinese-hamster ovary cells) were cultured in Dulbecco's modified Eagle's medium containing 10% (v/v) fetal calf serum, 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Karlsruhe, Germany) in a humidified atmosphere of 5% CO2 at 37 °C. Total RNA was isolated from confluent CHO cell cultures using the RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Quantity and quality of purified RNA were estimated by using a Gene Quant photometer (Amersham Biosciences, Freiburg, Germany) and electrophoresis analysis on a 1% (w/v) agarose gel followed by ethidium bromide staining [15]. Genomic DNA was isolated from confluent CHO cell cultures using the genomic isolation kit (Genomic-tip) from Qiagen according to the manufacturer's instructions. Yeast tRNA was purchased from Roche. Unfractionated heparin and biotinylated heparin–albumin were purchased from Sigma (Taufkirchen, Germany). Human FSAP from whole plasma was isolated and characterized as described previously [1]. Human plasminogen was purified from whole plasma by affinity chromatography using L-lysine-coupled Sepharose 4B similar as described in [16]. Human uPA was obtained from ZLB-Behring (Marburg, Germany).
FSAP enzyme activity assay
FSAP enzyme activity assays were performed as described previously [14]. In brief, microtitre plate wells were blocked with TBS (Tris-buffered saline; 25 mM Tris/HCl, pH 7.5, and 150 mM NaCl) containing 3% (w/v) BSA for 1 h and washed repeatedly with TBS. The standard assay system consisted of TBS, 20 nM FSAP and 0.2 mM of the chromogenic substrate S-2288 (H-D-isoleucyl-L-prolyl-L-arginine-p-nitroaniline dihydrochloride; Chromogenix, Mölndal, Sweden) and was followed over a period of 30 min at 37 °C at 405 nm in a microplate reader EL 808 (BioTek Instruments, Winooski, VT, U.S.A.).
Biotinylated heparin binding to immobilized FSAP
Microtitre plate wells were coated with 50 μl of a 10 μg/ml FSAP solution in 100 mM sodium carbonate (pH 9.5) at room temperature (22 °C) for 16 h. Wells were washed and non-specific binding sites were blocked with TBS containing 3% BSA for 1 h. Biotinylated heparin–albumin was allowed to bind for 1 h at room temperature in TBS containing 0.1% BSA, after which the plates were washed three times with 100 μl of TBS. Bound biotinylated heparin was detected using peroxidase-conjugated streptavidin (Dako, Glostrup, Denmark) and the immunopure TMB (3,3′,5,5′-tetramethylbenzidine) substrate kit (Pierce Biotechnology, Rockford, IL, U.S.A.) according to the manufacturer's instructions. In competition binding assays, biotinylated heparin (2 μg/ml) was mixed with dilutions of total RNA, genomic DNA, tRNA and heparin.
Plasmin(ogen)- and uPA-mediated proteolysis of human FSAP
Human FSAP (500 μg) was buffer exchanged into PBS using a prepacked PD10 Sephadex G-25 gel filtration column (Amersham Biosciences) and incubated with 10 μg of plasminogen and 10 μg of uPA, to generate in situ plasmin, in PBS at 25 °C for 24 h. Human FSAP fragments generated by proteolysis were identified by SDS/PAGE [17]. These fragments were blotted on to a PVDF membrane according to the manufacturer's instructions (Amersham Biosciences) for N-terminal sequencing.
N-terminal sequencing
The N-terminal sequences of the fragments were determined by automated Edman degradation using an Applied Biosystems 492 pulsed liquid-phase sequencer equipped with an on-line 785A phenylthiohydantoin derivative analyser.
Northwestern-blot analysis
Northwestern-blot analysis was performed essentially as described in [18]. In brief, the membranes were preincubated in buffer A containing 10 mM Tris/HCl (pH 7.5), 50 mM NaCl, 1 mM EDTA and 1× Denhardt's solution (0.02%, w/v, Ficoll, 0.02%, w/v, polyvinylpyrrolidone and 0.02%, w/v, BSA) at 4 °C for 3 h for renaturing blotted FSAP fragments. After blocking with 20 ml of buffer A with 100 μg/ml yeast tRNA (Sigma, Taufkirchen, Germany), the membranes were incubated overnight at 25 °C in the presence of 3 μg of γ-32P-labelled total RNA. Labelling of total RNA was carried out using [γ-32P]ATP (PerkinElmer, Wellesley, MA, U.S.A.) and polynucleotide kinase (Fermentas, Burlington, ON, Canada), according to the manufacturer's instructions. After washing twice with buffer A, the membranes were subjected to autoradiography.
Affinity chromatography on heparin–Sepharose
FSAP and hydrolysed FSAP in 20 mM sodium phosphate (pH 7.4) and 100 mM NaCl were applied on a HiTrap heparin–Sepharose HP column (Amersham Biosciences) equilibrated with the same buffer, and the bound FSAP was eluted with a linear gradient from 0.1 to 1 M NaCl using FPLC (Amersham Biosciences). Eluted proteins were monitored by UV absorbance at 280 nm and were precipitated in each fraction by adding trichloroacetic acid to a final concentration of 10% (w/v) at 0 °C for 60 min. Pellets were washed three times with acetone, solubilized and subjected to SDS/PAGE analysis [17].
Identifying RNA–FSAP complexes in solution
Poliovirus IRES (internal ribosome entry site)-RNA (906 nt) was transcribed in vitro from plasmid pMPolio [14,19] linearized with BsiWI in the presence of [α-32P]UTP using SP6 RNA polymerase. Binding and UV-cross-link reactions as previously outlined [19] were performed in the absence or presence of 3 μg of hydrolysed FSAP with 0.07 pmol of radiolabelled RNA in binding buffer (10 mM Hepes, pH 7.3, containing 34 mM KCl, 70 mM NaCl, 2 mM MgCl2, 10%, v/v, glycerol, 0.05% Nonidet P40, 1 μg/ml tRNA and 1 mM dithiothreitol) in 15 μl reaction volume. Reaction mixtures were incubated for 10 min at 30 °C, irradiated with UV light (254 nm) for 30 min at 0 °C and treated with 0.1 mg/ml RNase for 60 min at 37 °C. After addition of one-third volume of loading buffer [125 mM Tris/HCl, pH 6.8, containing 4%, w/v, SDS, 10%, v/v, ME (2-mercaptoethanol), 20% glycerol, 0.02% Bromophenol Blue and 7 M urea], samples were boiled and subjected to SDS/PAGE on 12% gels followed by autoradiography.
Molecular modelling of the EGF3 domain of FSAP
Sequences similar to the FSAP-EGF3 domain were identified using BLAST-P (Basic Local Alignment Search Tools for Proteins 2.2.12; http://www.ncbi.nlm.nih.gov/BLAST/) using the Brookhaven Protein Data Bank. This analysis revealed that the EGF domain of tPA (tissue plasminogen activator) and the EGF1 domain of Factor IX exhibit high similarity to EGF3 of FSAP. The solution structure of tPA-EGF (PDB code 1tpg) and Factor IX EGF1 (PDB code 1ixa) were used as a template to obtain a model for EGF3 of FSAP using the Swiss Model Alignment Interface (http://swissmodel.expasy.org/). Pictures were generated with 3D-Mol viewer, a component of Vector NTI Advance 9.1.0 (Invitrogen).
RESULTS AND DISCUSSION
FSAP activation through binding to RNA and heparin
In order to analyse the interaction between FSAP and polyanions, we tested several natural polyanionic compounds as cofactors for FSAP activation. For heparin and total RNA, mainly composed of rRNA and mRNA, chain length of approx. 1000–5000 nt, a maximal induction of the FSAP activity of approx. 3.5-fold above basal level was detected at a concentration of 3–30 and 10–100 μg/ml respectively (Figure 1A). In contrast, genomic DNA and tRNA (short-chain RNA, chain length of ∼80 nt) increased FSAP activity only at very high concentrations (above 100 μg/ml) approx. 2-fold over basal levels. RNA and heparin exhibited a bell-shaped concentration-dependent FSAP activation curve that is characteristic for a template-dependent mechanism of enzyme activation (Figure 1A) [14]. In competition experiments, we found that RNA is a strong competitor for heparin binding to immobilized FSAP (Figure 1B), suggesting overlapping or identical interaction sites. Interestingly, total RNA showed approx. 80% inhibition of heparin binding at a concentration of 10 μg/ml, whereas tRNA showed an inhibition of 60% at a concentration of 1 mg/ml (Figure 1B). This is in agreement with our previous observation that RNA molecules longer than approx. 200 nt are more effective cofactors than shorter molecules [14].
Figure 1. Analysis of FSAP activity in the presence of total RNA, genomic DNA, tRNA and heparin.
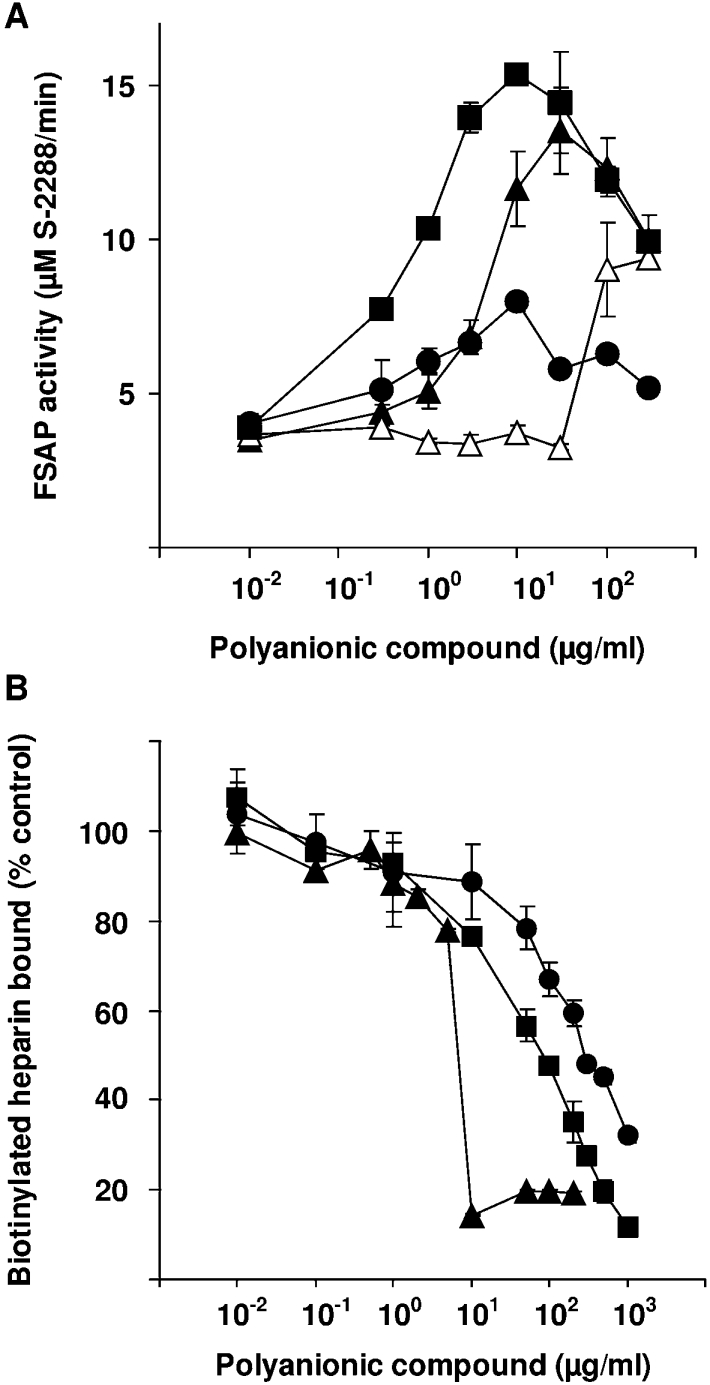
(A) The activity of FSAP (20 nM) was analysed in the presence of different concentrations of total RNA (▲), genomic DNA (△), tRNA (●) and heparin (■). FSAP activity is expressed as substrate S-2288 turnover (μM/min) at maximal reaction velocity. (B) Biotinylated heparin (2 μg/ml) was mixed with dilutions of total RNA (▲), tRNA (●) and heparin (■), and these mixtures were added to FSAP-coated wells for 1 h. The wells were washed and biotinylated heparin binding was measured. Results represent means±S.D.
Identification of FSAP domains important for RNA and heparin binding using hydrolysed FSAP
In order to identify the domains of FSAP critical for RNA and heparin binding, we used a proteolysis approach to obtain fragments of FSAP. In agreement with published results [6], we found that FSAP undergoes autoactivation followed by inactivation, through hydrolysis, of its serine protease domain (Figure 2A). Digestion with in situ generated plasmin resulted in a complex hydrolysis pattern (Figure 2A). Fragments of hydrolysed FSAP resolved by SDS/PAGE and blotted on to PVDF membrane (Figure 2B) were analysed by N-terminal sequencing (Supplementary Table 1 at http://www.BiochemJ.org/bj/394/bj3940687add.htm). This led to the determination of preferential proteolytic sites (Supplementary Table 1) and identification of the corresponding fragments (F1–F19) of hydrolysed FSAP.
Figure 2. SDS/PAGE analysis of human FSAP hydrolysed by in situ generated plasmin.

(A) Purified human FSAP (lane A) consists of approx. 98% inactive single-chain (sc) and 2% active two-chain form composed of the heavy chain (hc, amino acids 23–313) and the light chain (lc, amino acids 314–560). Upon incubation in PBS (pH 7.4), human FSAP undergoes complete autoactivation followed by further inactivation by hydrolysis of the light chain, resulting in fragments of approx. 16 and 10 kDa (lane B). Incubation of human FSAP with plasminogen and uPA (leading to in situ generation of plasmin) resulted in a complex hydrolysis pattern (lane C). Without incubation of the samples with ME, several chains of hydrolysed FSAP are connected through disulphide bridges. (B) For N-terminal sequencing, hydrolysed human FSAP was incubated in the presence (lane 1, 50 μg of protein) or absence (lane 2, 5 μg of protein; lane 3, 50 μg of protein) of ME for 5 min at 95 °C respectively and separated by SDS/PAGE. The protein fragments were blotted on to a PVDF membrane and stained with Coomassie Blue. Bands F1–F19 were analysed by N-terminal sequencing. Molecular mass standards are indicated in kDa.
Hydrolysed FSAP was resolved by electrophoresis, and after blotting on to a membrane, the polyanion-binding capability of the individual fragments (F1–F19) was investigated by Northwestern-blot analysis. The bands F10, F11, F12, F13, F14, F15 and F16 bound strongly to radiolabelled total RNA (Figure 3). It should be noted that in the FSAP fragments F10–F19, several chains were still connected by disulphide bridges. Band F17 corresponds to the band F14 but starts at residue Val111 and lacks the amino acids Arg170, Arg171, Ser172 and Lys173 (Supplementary Table 1). Although band F14 binds RNA, band F17 does not do so, probably because of the missing basic amino acid residues (Figure 3). Additionally, bands F18 and F19, corresponding to the first and the second half of the protease domain, were not able to bind RNA. If disulphide bridges of FSAP were reduced, only weak RNA binding could be observed to band F7 composed of the EGF2 and EGF3 domains (Figure 3). It should be noted that bands F1 and F2 also harbouring the EGF2 and EGF3 domains do not bind RNA in this analysis, probably because the renaturation of the binding site is favoured in the smaller fragment F7. Taken together, these results suggest that the RNA binding is mediated mainly by the EGF2 and EGF3 domains and that the amino acids Arg170, Arg171, Ser172 and Lys173 are essential for this binding. Additionally, the importance of the disulphide bridges for binding to RNA shows that an intact three-dimensional structure is essential.
Figure 3. Interaction of hydrolysed human FSAP with radiolabelled total RNA.

Hydrolysed human FSAP was incubated in the absence (lane A) or in the presence (lane B) of ME for 5 min at 95 °C, separated by SDS/PAGE and blotted on to a PVDF membrane. Proteins were stained with Coomassie Blue, photographed, completely destained with 70% (v/v) ethanol, and equilibrated in binding buffer. The membrane was incubated overnight at 25 °C in the presence of γ-32P-labelled total RNA. After extensive washing, radiolabelled RNA bound to the membrane was detected by autoradiography. Bands interacting with RNA are indicated with an arrow. Band F17 was unable to bind RNA and is indicated with an asterisk. Bands F10, F11, F12, F13, F14, F15 and F16 showed a high binding capacity for RNA and band F7 showed a weak binding capacity for RNA. Intact disulphide bridges were necessary for efficient RNA binding. Molecular mass standards are indicated in kDa.
To investigate further the nature of these binding interactions, we then determined the potential of hydrolysed FSAP to bind to a heparin–Sepharose column. Hydrolysed and native FSAP bound to and could be eluted from the heparin–Sepharose column under similar ionic strength conditions (Figure 4). As described above for RNA, the binding of hydrolysed FSAP to heparin was completely abolished when disulphide bridges were reduced (Figure 4). SDS/PAGE analysis of the elution peak for hydrolysed FSAP revealed bands corresponding to F11 and F12. F10 is most probably converted into F11 and F12 by autocatalytic activity of FSAP in contact with the heparin matrix and the amount of the other fragments of FSAP is, most probably, below the detection limit of the staining method. The binding of FSAP fragments to RNA in the filter binding assays was more robust and amenable to analysis than their binding to the heparin column.
Figure 4. Heparin-binding analysis of hydrolysed human FSAP.

Hydrolysed human FSAP (hydr. FSAP, −ME) eluted from the heparin column at a similar ionic strength as native human FSAP (native FSAP). In contrast, there was no heparin binding of hydrolysed FSAP when pretreated with 100 mM ME for 15 min at room temperature (hydr. FSAP, +ME). Elution was followed by analysis of UV absorbance at 280 nm. Conductivity indicates the increase in NaCl concentration. SDS/PAGE analysis of the flow-through (lane 1), wash fraction (lane 2) and elution peak (lane 3) was performed and the proteins were detected by silver staining. F11 and F12 bands interacting with heparin are indicated with an arrow and molecular mass standards are indicated in kDa.
In order to circumvent any immobilization-related artifacts, we analysed the interaction of hydrolysed FSAP fragments with radiolabelled RNA in solution. Using a UV-cross-link approach, we were able to detect FSAP fragments directly interacting with RNA in the solution phase. FSAP fragments migrating at 46 and 28 kDa, representing fragments F1 and F2 respectively, bound RNA (Figure 5). F1 represents the FSAP fragment composed of EGF1, EGF2 and EGF3 domains and the kringle domain. F2 represents the FSAP fragment composed of EGF1, EGF2 and EGF3 domains without the kringle domain. Radiolabelled EGF2 and EGF3 fragment of FSAP (∼10 kDa) should also be detected but is most probably overshadowed by the co-migrating hydrolysed RNA (results not shown).
Figure 5. Interaction of hydrolysed human FSAP with radiolabelled RNA in solution.
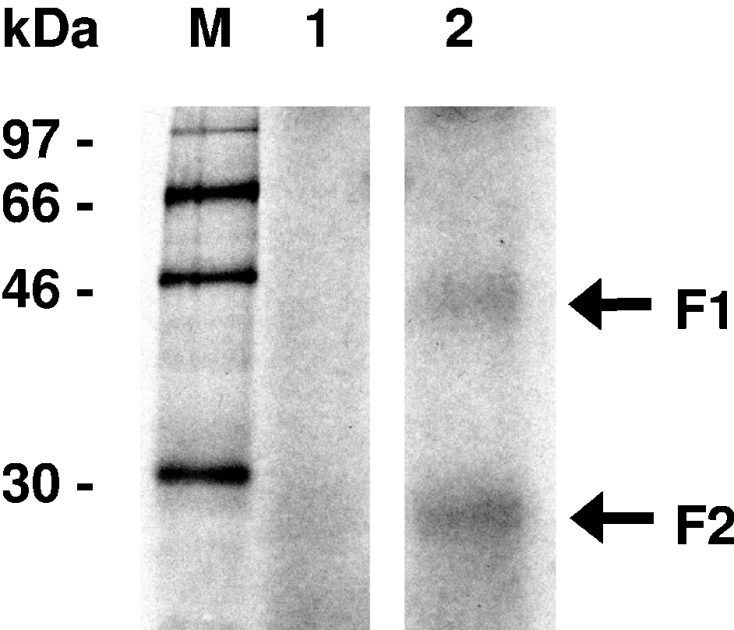
[α-32P]UTP-labelled poliovirus RNA was incubated at 30 °C for 10 min in the absence (lane 1) or presence of hydrolysed human FSAP (lane 2) in solution. Samples were irradiated with UV light (254 nm), treated with 0.1 mg/ml RNase A, denatured and separated on a denaturing SDS/12% polyacrylamide gel. For detection of protein bound to radiolabelled RNA, an X-ray film was exposed for 16 h. Size markers indicate positions of 14C-labelled marker proteins (M). Arrows indicate the bands F1 and F2. Molecular mass standards are indicated in kDa.
Modelling of the FSAP EGF3 domain
The available solution structure of the closely related EGF domain of human tPA [20] (PDB code 1tpg) was used to model EGF3 of FSAP using SWISS-MODEL [21–23] (Figure 6, and Supplementary Figure 1A at http://www.BiochemJ.org/bj/394/bj3940687add.htm). This model indicates that the positively charged amino acid stretch from residue Arg167 to residue Lys173 (RHKRRSK) is located in an exposed loop. In the case of tPA, this loop binds to cellular uptake receptors [20], whereas in FSAP, it is likely to be involved in interactions with polyanions. It should be noted that, although the overall similarity between the two structures is approx. 50%, in the loop itself the similarity is rather low, precluding any further conclusions regarding the accessibility of the individual amino acids. A similar result was obtained when EGF3 of FSAP was modelled using the available solution structure of the EGF1 domain of human coagulation Factor IX (PDB code 1ixa) (Supplementary Figure 1B at http://www.BiochemJ.org/bj/394/bj3940687add.htm). Hence, amino acids Arg170, Arg171, Ser172 and Lys173 within the EGF3 domain are the likely polyanion-binding segments. The proximity of Lys183 and Lys185 to this loop suggests that they may also be involved in interactions with negatively charged glycosaminoglycans and RNA (Figure 6 and Supplementary Figure 1). Alignment of FSAP from five different species indicates that this region is highly conserved and polyanion-binding properties can be expected in other species as well (Figure 7B).
Figure 6. Model of the three-dimensional structure of the EGF3 domain of human FSAP.

The available solution structure of the closely related EGF domain of human tPA (PDB code 1tpg) was used to model EGF3 of FSAP using SWISS-MODEL. Residues Arg167 to Lys173, Lys183 and Lys185 are indicated.
Figure 7. Schematic structure of human FSAP and the polyanion-binding domains.

(A) The structure of FSAP including identified proteolysis sites (indicated by vertical arrows) and selected forms of FSAP capable of RNA binding are shown schematically. Experimentally identified N-terminal ends are indicated by horizontal arrows and the predicted C-terminal ends are indicated by open circles. The amino acid stretch from residue Arg167 to residue Lys173 (RHKRRSK) has a high average positive charge and is also the site within the EGF3 (E3) domain that is essential for RNA binding, since deletion of the amino acids Arg170, Arg171, Ser172 and Lys173 in hydrolysed FSAP (F17) resulted in complete loss of RNA binding. 1Bands corresponding to the molecular mass of F11 and F12 were also able to bind to the heparin column. 2RNA binding was detected in solution but not by Northwestern-blot analysis. 3Weak RNA binding was detected by Northwestern-blot analysis. (B) Alignment of FSAP from human (Swiss-Prot accession number Q14520), chimpanzee (GenBank® accession number XP_508042), bovine (Swiss-Prot accession number Q5E9Z2), mouse (Swiss-Prot accession number Q8K0D2), rat (Swiss-Prot accession number Q6L711) and chicken (GenBank® accession number XP_421761) indicates that this region is highly conserved in evolution.
Conclusions
We identified the EGF2 and EGF3 domains of FSAP as the major interaction domain for RNA (Figure 7). Amino acids Arg170, Arg171, Ser172 and Lys173 are essential for RNA binding. Furthermore, this interaction is dependent on the three-dimensional structural organization of the protein since reducing disulphide bridges led to loss of RNA as well as heparin binding. Competition between RNA and heparin for binding to FSAP suggests that identical or overlapping binding sites are involved. One RNA-binding motif identified in ribosomal proteins or coat proteins of RNA viruses consists of a short region of positively charged amino acids rich in arginine [24]. Heparin-binding sites are commonly observed on the external surface of proteins and correspond to shallow pockets of arginine and lysine [25]. Both these observations are in accordance with the binding site identified in the present study.
FSAP is known to activate Factor VII as well as pro-uPA and FSAP itself is activated by polyanionic polymers such as heparin and RNA. In this aspect, FSAP is quite similar to Factor XII which is also activated by different types of negatively charged polymers [26]. Factor XII also has a dual role in haemostasis since it activates both the contact phase of blood coagulation as well as fibrinolysis [27–29]. The domains of Factor XII responsible for binding polyanions are within the N-terminal heavy chain, including a positively charged amino acid stretch of residues 40–47 (HKCTHKGR) that is essential for polyanion binding [30,31]. The information that FSAP has similar domains for binding to polyanions suggests a role similar to Factor XII. With the identification of the RNA and heparin binding domain, it is possible to develop peptides or antibodies that might interfere with the effects of FSAP in haemostasis and cell proliferation and thereby help to elucidate the functions of FSAP.
Online data
Acknowledgments
The excellent and skilful technical assistance of Hans-Günter Welker, Thomas Schmidt-Wöll and Horst Thiele (Justus-Liebig-Universität Giessen) is greatly appreciated. We also thank Dr Malgorzata Wygrecka (Justus-Liebig-Universität Giessen) and Katja Altincicek (Giessen, Germany) for helpful comments on this paper and Gernot Kieseritzky (Free University of Berlin, Berlin, Germany) for help with the three-dimensional models. This project was supported in part by a grant (Ka1468/2-2) to S. M. K. from the Deutsche Forschungsgemeinschaft (Bonn, Germany).
References
- 1.Kannemeier C., Feussner A., Stöhr H. A., Weisse J., Preissner K. T., Römisch J. Factor VII and single-chain plasminogen activator-activating protease: activation and autoactivation of the proenzyme. Eur. J. Biochem. 2001;268:3789–3796. doi: 10.1046/j.1432-1327.2001.02285.x. [DOI] [PubMed] [Google Scholar]
- 2.Römisch J. Factor VII activating protease (FSAP): a novel protease in hemostasis. Biol. Chem. 2002;383:1119–1124. doi: 10.1515/BC.2002.121. [DOI] [PubMed] [Google Scholar]
- 3.Choi-Miura N. H., Tobe T., Sumiya J. I., Nakano Y., Sano Y., Mazda T., Tomita M. Purification and characterization of a novel hyaluronan-binding protein (PHBP) from human plasma: it has three EGF, a kringle and a serine protease domain, similar to hepatocyte growth factor activator. J. Biochem. (Tokyo) 1996;119:1157–1165. doi: 10.1093/oxfordjournals.jbchem.a021362. [DOI] [PubMed] [Google Scholar]
- 4.Sumiya J. I., Asakawa S., Tobe T., Hashimoto K., Saguchi K. I., Choi-Miura N. H., Shimizu Y., Minoshima S., Shimizu N., Tomita M. Isolation and characterization of the plasma hyaluronan-binding protein (PHBP) gene (HABP2) J. Biochem. (Tokyo) 1997;122:983–990. doi: 10.1093/oxfordjournals.jbchem.a021861. [DOI] [PubMed] [Google Scholar]
- 5.Etscheid M., Hunfeld A., Konig H., Seitz R., Dodt J. Activation of proPHBSP, the zymogen of a plasma hyaluronan binding serine protease, by an intermolecular autocatalytic mechanism. Biol. Chem. 2000;381:1223–1231. doi: 10.1515/BC.2000.150. [DOI] [PubMed] [Google Scholar]
- 6.Choi-Miura N. H., Tobe T., Sumiya J. I., Nakano Y., Sano Y., Mazda T., Tomita M. Proteolytic activation and inactivation of the serine protease activity of plasma hyaluronan binding protein. Biol. Pharm. Bull. 2001;24:448–452. doi: 10.1248/bpb.24.448. [DOI] [PubMed] [Google Scholar]
- 7.Willeit J., Kiechl S., Weimer T., Mair A., Santer P., Wiedermann C. J., Roemisch J. Marburg I polymorphism of factor VII-activating protease: a prominent risk predictor of carotid stenosis. Circulation. 2003;107:667–670. doi: 10.1161/01.cir.0000055189.18831.b1. [DOI] [PubMed] [Google Scholar]
- 8.Hoppe B., Tolou F., Radtke H., Kiesewetter H., Dorner T., Salama A. Marburg I polymorphism of factor VII-activating protease is associated with idiopathic venous thromboembolism. Blood. 2005;105:1549–1551. doi: 10.1182/blood-2004-08-3328. [DOI] [PubMed] [Google Scholar]
- 9.Römisch J., Feussner A., Vermohlen S., Stöhr H. A. A protease isolated from human plasma activating factor VII independent of tissue factor. Blood Coagulation Fibrinolysis. 1999;10:471–479. [PubMed] [Google Scholar]
- 10.Choi-Miura N. H., Yoda M., Saito K., Takahashi K., Tomita M. Identification of the substrates for plasma hyaluronan binding protein. Biol. Pharm. Bull. 2001;24:140–143. doi: 10.1248/bpb.24.140. [DOI] [PubMed] [Google Scholar]
- 11.Etscheid M., Beer N., Fink E., Seitz R., Dodt J. The hyaluronan-binding serine protease from human plasma cleaves HMW and LMW kininogen and releases bradykinin. Biol. Chem. 2002;383:1633–1643. doi: 10.1515/BC.2002.184. [DOI] [PubMed] [Google Scholar]
- 12.Etscheid M., Beer N., Kress J., Seitz R., Dodt J. Inhibition of bFGF/EGF-dependent endothelial cell proliferation by the hyaluronan-binding protease from human plasma. Eur. J. Cell Biol. 2003;82:597–604. doi: 10.1078/0171-9335-00349. [DOI] [PubMed] [Google Scholar]
- 13.Kannemeier C., Al-Fakhri N., Preissner K. T., Kanse S. M. Factor VII activating protease (FSAP) inhibits growth factor-mediated cell proliferation and migration of vascular smooth muscle cells. FASEB J. 2004;18:728–730. doi: 10.1096/fj.03-0898fje. [DOI] [PubMed] [Google Scholar]
- 14.Nakazawa F., Kannemeier C., Shibamiya A., Song Y., Tzima E., Schubert U., Koyama T., Niepmann M., Trusheim H., Engelmann B., et al. Extracellular RNA is a natural cofactor for the (auto-)activation of Factor VII-activating protease (FSAP) Biochem. J. 2005;385:831–838. doi: 10.1042/BJ20041021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. New York: John Wiley and Sons; 1987. Current Protocols in Molecular Biology. [Google Scholar]
- 16.Brockway W. J., Castellino F. J. Measurement of the binding of antifibrinolytic amino acids to various plasminogens. Arch. Biochem. Biophys. 1972;151:194–199. doi: 10.1016/0003-9861(72)90488-2. [DOI] [PubMed] [Google Scholar]
- 17.Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Chen X., Sadlock J., Schon E. A. RNA-binding patterns in total human tissue proteins: analysis by northwestern blotting. Biochem. Biophys. Res. Commun. 1993;191:18–25. doi: 10.1006/bbrc.1993.1178. [DOI] [PubMed] [Google Scholar]
- 19.Ochs K., Zeller A., Saleh L., Bassili G., Song Y., Sonntag A., Niepmann M. Impaired binding of standard initiation factors mediates poliovirus translation attenuation. J. Virol. 2003;77:115–122. doi: 10.1128/JVI.77.1.115-122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith B. O., Downing K. A., Driscoll P. C., Dudgeon T. J., Campbell I. D. The solution structure and backbone dynamics of the fibronectin type I and the epidermal growth factor-like pair of modules of tissue-type plasminogen activator. Structure. 1995;3:823–833. doi: 10.1016/s0969-2126(01)00217-9. [DOI] [PubMed] [Google Scholar]
- 21.Schwede T., Kopp J., Guex N., Peitsch M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 23.Peitsch M. C. Protein modeling by e-mail. Bio/Technology. 1995;13:658–660. [Google Scholar]
- 24.Tan R., Frankel A. D. Structural variety of arginine-rich RNA-binding peptides. Proc. Natl. Acad. Sci. U.S.A. 1995;92:5282–5286. doi: 10.1073/pnas.92.12.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capila I., Linhardt R. J. Heparin–protein interactions. Angew. Chem. Int. Ed. Engl. 2002;41:390–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 26.Hojima Y., Cochrane C. G., Wiggins R. C., Austen K. F., Stevens R. L. In vitro activation of the contact (Hageman factor) system of plasma by heparin and chondroitin sulfate E. Blood. 1984;63:1453–1459. [PubMed] [Google Scholar]
- 27.Pixley R. A., Colman R. W. Factor XII: Hageman factor. Methods Enzymol. 1993;222:51–65. doi: 10.1016/0076-6879(93)22007-3. [DOI] [PubMed] [Google Scholar]
- 28.Colman R. W., Schmaier A. H. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997;90:3819–3843. [PubMed] [Google Scholar]
- 29.Braat E. A. M., Dooijewaard G., Rijken D. C. Fibrinolytic properties of activated FXII. Eur. J. Biochem. 1999;263:904–911. doi: 10.1046/j.1432-1327.1999.00593.x. [DOI] [PubMed] [Google Scholar]
- 30.Citarella F., Ravon D. M., Pascucci B., Felici A., Fantoni A., Hack C. E. Structure/function analysis of human factor XII using recombinant deletion mutants: evidence for an additional region involved in the binding to negatively charged surfaces. Eur. J. Biochem. 1996;238:240–249. doi: 10.1111/j.1432-1033.1996.0240q.x. [DOI] [PubMed] [Google Scholar]
- 31.Citarella F., te Velthuis H., Helmer-Citterich M., Hack C. E. Identification of a putative binding site for negatively charged surfaces in the fibronectin type II domain of human factor XII: an immunochemical and homology modeling approach. Thromb. Haemostasis. 2000;84:1057–1065. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.