

Abstract
Eosinophil peroxidase is a haem enzyme of eosinophils that is implicated in oxidative tissue injury in asthma. It uses hydrogen peroxide to oxidize thiocyanate and bromide to their respective hypohalous acids. Nitrite is also a substrate for eosinophil peroxidase. We have investigated the mechanisms by which the enzyme oxidizes nitrite. Nitrite was very effective at inhibiting hypothiocyanous acid (‘cyanosulphenic acid’) and hypobromous acid production. Spectral studies showed that nitrite reduced the enzyme to its compound II form, which is a redox intermediate containing FeIV in the haem active site. Compound II does not oxidize thiocyanate or bromide. These results demonstrate that nitrite is readily oxidized by compound I, which contains FeV at the active site. However, it reacts more slowly with compound II. The observed rate constant for reduction of compound II by nitrite was determined to be 5.6×103 M−1·s−1. Eosinophils were at least 4-fold more effective at promoting nitration of a heptapeptide than neutrophils. This result is explained by our finding that nitrite reacts 10-fold faster with compound II of eosinophil peroxidase than with the analogous redox intermediate of myeloperoxidase. Nitration by eosinophils was increased 3-fold by superoxide dismutase, which indicates that superoxide interferes with nitration. We propose that at sites of eosinophilic inflammation, low concentrations of nitrite will retard oxidant production by eosinophil peroxidase, whereas at higher concentrations nitrogen dioxide will be a major oxidant formed by these cells. The efficiency of protein nitration will be decreased by the diffusion-controlled reaction of superoxide with nitrogen dioxide.
Keywords: eosinophil peroxidase, hydrogen peroxide, myeloperoxidase, nitrite, superoxide dismutase, thiocyanate
Abbreviations: DTPA, diethylenetriaminepenta-acetic acid; HPA, hydroxyphenylpropanoic acid; TNB, 5-thio-2-nitrobenzoic acid
INTRODUCTION
Interactions of nitric oxide and nitrite with peroxidases have become an active focus of research since it was shown that these haem enzymes oxidize nitrite and promote nitration of tyrosine and tyrosyl residues [1,2]. It is now apparent that peroxidases are responsible for protein nitration in many inflammatory pathologies and the presence of 3-nitrotyrosine does not necessarily implicate peroxynitrite as the reactive nitrogen species [3,4]. Eosinophil peroxidase, an abundant protein of eosinophils, is capable of oxidizing nitrite [5,6]. In doing so, it can catalyse nitration of tyrosine and tyrosyl residues on proteins [6]. It promotes nitration by oxidizing nitrite to nitrogen dioxide, which reacts with tyrosyl radicals to form 3-nitrotyrosine [6,7]. Eosinophil peroxidase has been shown to mediate protein nitration in allergic airway inflammation in mice [8,9] and is responsible for the high content of 3-nitrotyrosine in proteins from the airways of asthmatics [7,10]. Hence, eosinophil peroxidase is likely to contribute to oxidative stress in asthma through the production of reactive nitrogen species.
Eosinophil peroxidase belongs to the same superfamily of enzymes as myeloperoxidase and lactoperoxidase [11]. The main physiological substrates for this enzyme are presumed to be thiocyanate and bromide [12–15]. Furtmüller et al. [15] have revealed that, like other peroxidases, the native or ferric form of the enzyme (EP3+) reacts with hydrogen peroxide to form a compound I species (reaction 1). This is an unstable redox intermediate that, in the absence of reducing substrates, decays to a compound II-like intermediate (compound II*). Presumably, this occurs through reduction of the haem by one of its amino acid residues (reaction 2). Alternatively, compound I reacts rapidly with bromide or thiocyanate to regenerate the native enzyme and produce hypobromous acid and hypothiocyanous acid (HOSCN; ‘cyanosulphenic acid’) respectively (reaction 3) [15]. Thiocyanate is five times more preferred as a substrate for compound I than bromide. Organic substrates (RH) such as ascorbate and tyrosine reduce compound I to compound II via one-electron reactions, and are also capable of converting compound II back to the ferric enzyme (reactions 4 and 5) [16]. Turnover of compound II is rate-determining.
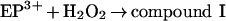 |
(1) |
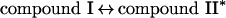 |
(2) |
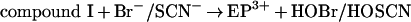 |
(3) |
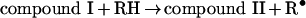 |
(4) |
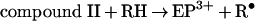 |
(5) |
Hazen and co-workers [6,7,10] have performed detailed studies on the oxidation of nitrite by eosinophil peroxidase. They demonstrated that bromide and thiocyanate are 4- and 12-fold more preferred than nitrite as substrates for the enzyme. These results imply that at physiological concentrations of these anions, eosinophil peroxidase should oxidize nitrite in vivo [6]. Detection of nitrogen dioxide supports a mechanism involving sequential oxidation of nitrite by compound I and compound II (reactions 4 and 5) [7], which is analogous to that for nitrite oxidation by myeloperoxidase [17,18]. In the present study, our aims were to determine the rate constants for reaction of nitrite with the redox intermediates of eosinophil peroxidase, establish how nitrite affects oxidant production by the enzyme and assess the influence superoxide and tyrosine have on the ability of eosinophils to nitrate proteins.
EXPERIMENTAL
Materials
Carboxymethyl-Sepharose, Percoll and Ficoll-Paque™ PLUS solution were obtained from Amersham Biosciences (Uppsala, Sweden). Acetyl-Ser-Gln-Asn-Tyr-Pro-Val-Val-amide, Ala-Pro-Arg-Leu-Arg-Phe-Tyr-Ser, DTPA (diethylenetriaminepenta-acetic acid), L-tyrosine, superoxide dismutase, dextran and PMA were purchased from Sigma (St. Louis, MO, U.S.A.). 3-Nitro-L-tyrosine was from Fluka (Buchs, Switzerland). D,L-Methionine and sodium bromide were from BDH Chemicals (Poole, Dorset, U.K.). Hexadecyltrimethylammonium chloride was from Acros Organics (Morris Plains, NJ, U.S.A.). Magnetic separation (MACS) CD16 immunomagnetic beads and cell separation (CS) depletion columns were from Miltenyl Biotec (Bergisch Gladbach, Germany). COMPLETE™ protease inhibitor cocktail tablets, inhibitory to a large spectrum of serine proteases, were from Roche Molecular Biochemicals. Solutions of sodium nitrite were prepared daily and kept on ice. Hydrogen peroxide solutions were prepared daily by diluting a 30% stock, and the concentration was calculated by measuring its absorbance at 240 nm (ϵ240 43.6 M−1·cm−1) [19].
Eosinophil peroxidase was purified from eosinophils isolated from horse blood by the method of Jorg et al. [20] as outlined previously [14]. With 2 nM eosinophil peroxidase, the concentration of chloride and bromide in reaction systems contributed from the enzyme preparation was calculated at 0.6 mM and 0.3 μM respectively. The preparation contained minimal myeloperoxidase as shown by haem staining of native PAGE [14,21]. The preparation of eosinophil peroxidase was unable to oxidize chloride, which indicated that it was not contaminated with myeloperoxidase (results not shown). For spectral studies, where high concentrations of enzyme were required, the preparation was dialysed into 50 mM sodium phosphate buffer (50 mM sodium dihydrogen phosphate and disodium hydrogen phosphate buffer, pH 7.0) containing 100 mM NaCl, 0.02% cetyltrimethylammonium chloride and 2% (v/v) glycerol.
Methods
Measurement of hydrogen peroxide utilization by eosinophil peroxidase
The utilization of hydrogen peroxide by eosinophil peroxidase in the presence of bromide, thiocyanate or nitrite was measured continuously with a YSI 2510 oxidase probe fitted to a YSI model 25 oxidase meter (Yellow Springs Instruments, Yellow Springs, OH, U.S.A.) [22]. The electrode was calibrated against known concentrations of hydrogen peroxide. Glutathione was included in the reaction mixtures to scavenge the hypohalous acids and prevent them from interfering with the electrode signal [23]. All reactions were started with the addition of eosinophil peroxidase. For experiments containing nitrite, the electrode was equilibrated at each concentration of nitrite.
Spectral analysis of eosinophil peroxidase
The visible absorption spectrum of eosinophil peroxidase was recorded during the oxidation of bromide with or without nitrite, or during the oxidation of nitrite alone, by using a Beckman 7500 diode array spectrophotometer. Each spectrum was recorded over 2 s and is an average of 20 spectra.
Eosinophil and neutrophil isolation
Venous blood (50–100 ml) was obtained from healthy volunteers and collected into 20 μl of heparin (written informed consent was obtained from the volunteers). Neutrophils were isolated by the method of Böyum [24] using Ficoll-Paque™ PLUS separation. Eosinophils were isolated from a leucocyte pellet obtained either by Ficoll-Paque™ PLUS separation or by discontinuous Percoll gradient separation of whole blood by the method of Cramer et al. [25]. Eosinophils were separated from neutrophils in the leucocyte pellets by CD16-negative selection by the method of Hansel et al. [26]. Eosinophil purity >95% was determined by Giemsa staining of cytospin preparations. Cell viability was >99% as determined by Trypan Blue exclusion.
Measurement of superoxide production by eosinophils and neutrophils
Neutrophils (0.5×106 ml−1) or eosinophils (0.5×106 ml−1) in PBS containing 1 mM CaCl2, 0.5 mM MgCl2, 100 μg/ml glucose, 20 μg/ml catalase and 100 μM cytochrome c were stimulated with 100 ng/ml PMA at 37 °C. The rate of O2− generation was determined by continuously monitoring the change in absorbance at 550 nm (Δϵ550 21100 M−1·cm−1) [27,28].
Measurement of hypobromous acid production by eosinophils
Human eosinophils were incubated in PBS under the conditions described above except that 100 μM bromide and 10 mM taurine were added to the buffer. After 30 min of stimulation with PMA, accumulated taurine bromamine was assayed by using TNB (5-thio-2-nitrobenzoic acid) [22].
Nitration of tyrosyl residues in peptides
Nitration of acetyl-Ser-Gln-Asn-Tyr-Pro-Val-Val-amide by purified eosinophil peroxidase was carried out at 21 °C in 50 mM sodium phosphate buffer (pH 7.4) containing 20 μM DTPA, 100 μM peptide and nitrite as indicated. Reactions were started by the addition of 100 μM hydrogen peroxide, stopped after 1 h with catalase (20 μg/ml) and then analysed by HPLC (see below). Neutrophils and eosinophils (1×106 ml−1) were incubated at 37 °C with 100 μM acetyl-Ser-Gln-Asn-Tyr-Pro-Val-Val-amide or Ala-Pro-Arg-Leu-Arg-Phe-Tyr-Ser in PBS containing 5 mM glucose, 1 mM CaCl2, 0.5 mM MgCl2 and nitrite (0–1 mM). Cells were stimulated with PMA (100 ng/ml) and the reactions were stopped after 1 h by adding 20 μg/ml catalase. Cells were pelleted by centrifugation at 10000 g for 3 min. COMPLETE™ protease inhibitors (0.8 mg/ml) were then added. The supernatant fraction was stored at −20 °C until analysed by HPLC.
HPLC
Products of nitration reactions were analysed by HPLC as described previously [17]. Tyrosine, 3-nitrotyrosine, peptides and nitrated peptides were monitored by UV detection (Philips PU 4120 diode array detector and Waters 996 Photodiode array detector) at 274 nm and identification of products was made by the comparative time of elution with standards and by spectral matching. The peak for the nitrated peptides has previously been shown to contain 3-nitrotyrosyl residues by showing that they had the appropriate absorption spectrum for 3-nitrotyrosine and analysing the hydrolysed material using GLC with MS [17]. Standards for the nitrated peptides were not available, so quantification of products was made by comparison of the area under the curve for each peak at 274 nm relative to a standard curve for 3-nitrotyrosine.
RESULTS
Oxidation of nitrite by eosinophil peroxidase and hydrogen peroxide
To determine how readily nitrite is oxidized by eosinophil peroxidase, we monitored the loss of hydrogen peroxide during the course of the reaction using a hydrogen peroxide electrode. This method has the advantage of measuring enzyme activity directly without the presence of chromophores that may react with the peroxidase and attenuate its activity [22,29]. When eosinophil peroxidase was added to hydrogen peroxide and nitrite, there was a steady monophasic decline in the concentration of hydrogen peroxide over time (results not shown). Initial rates of enzyme activity were determined from these traces of hydrogen peroxide consumption. The turnover of the enzyme (i.e. the rate of hydrogen peroxide loss divided by the enzyme concentration) increased with the concentration of nitrite (Figure 1). High concentrations of nitrite were required to promote the loss of hydrogen peroxide. It was not possible to reach a maximum rate of hydrogen peroxide loss because high concentrations of nitrite interfered with the electrode signal. Consequently, we were unable to calculate kinetic parameters for this activity, as done previously for thiocyanate and bromide [14]. However, the slope of the regression line in Figure 1 represents the observed rate constant (kobs) for the rate-determining step in enzyme turnover. The value of for this rate constant was (5.6±0.2)×103 M−1·s−1.
Figure 1. Oxidation of nitrite by eosinophil peroxidase and hydrogen peroxide.
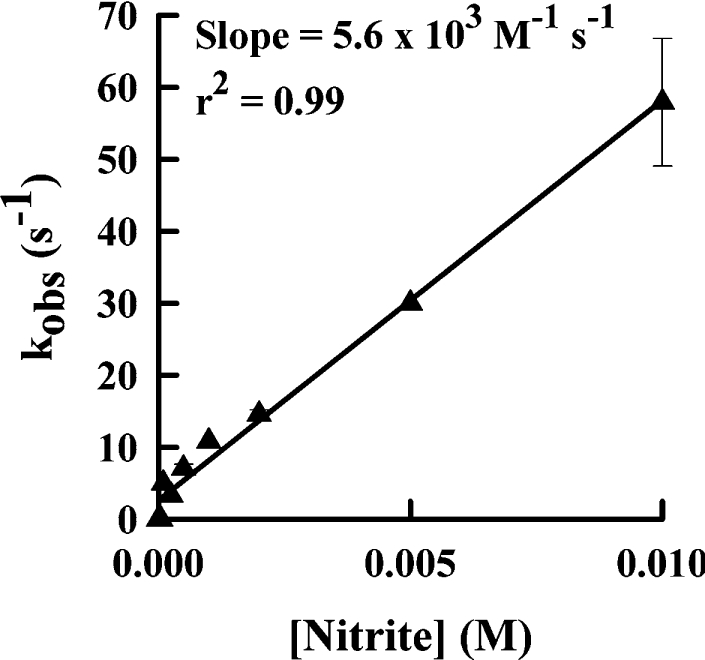
Reactions, carried out at 21 °C, were started by adding 2 nM eosinophil peroxidase to 30 μM hydrogen peroxide in 100 mM sodium phosphate buffer (pH 7.4) containing 20 μM DTPA and various concentrations of nitrite. Steady-state rates were calculated as loss of hydrogen peroxide over 1 min and divided by the enzyme concentration to give the catalytic-centre activity or the observed rate constant (kobs). Results shown are means and ranges for duplicate experiments.
To compare the relative ease at which substrates are oxidized by eosinophil peroxidase, we determined enzyme catalytic-centre activities for nitrite, bromide or thiocyanate, each at a concentration of 100 μM. This concentration is within the physiological range for all the substrates [30]. The catalytic-centre activity was calculated from the steady-state rate of hydrogen peroxide loss. The catalytic-centre activities in the presence of nitrite, bromide and thiocyanate were 5, 45 and 70 s−1 respectively.
Effects of nitrite on oxidation of thiocyanate and bromide
To understand the mechanism by which eosinophil peroxidase oxidizes nitrite, we investigated the effect of nitrite on oxidation of bromide and thiocyanate by the enzyme. Addition of eosinophil peroxidase to hydrogen peroxide and thiocyanate promoted a rapid consumption of the hydrogen peroxide (Figure 2A). Nitrite at 10 μM was a good inhibitor of the loss of hydrogen peroxide. To determine how effective it is at inhibiting eosinophil peroxidase, we measured initial rates of hydrogen peroxide loss at varying concentrations of nitrite. In the presence of 100 μM thiocyanate, the rate of hydrogen peroxide loss over the first 10 s of the reaction was inhibited by 50% (IC50) at a nitrite concentration of 1 μM (Figure 2B). With 300 μM bromide, the IC50 was 0.3 μM (results not shown). The maximal inhibition achieved with either thiocyanate or bromide was approx. 80%.
Figure 2. Inhibition of eosinophil peroxidase by nitrite.
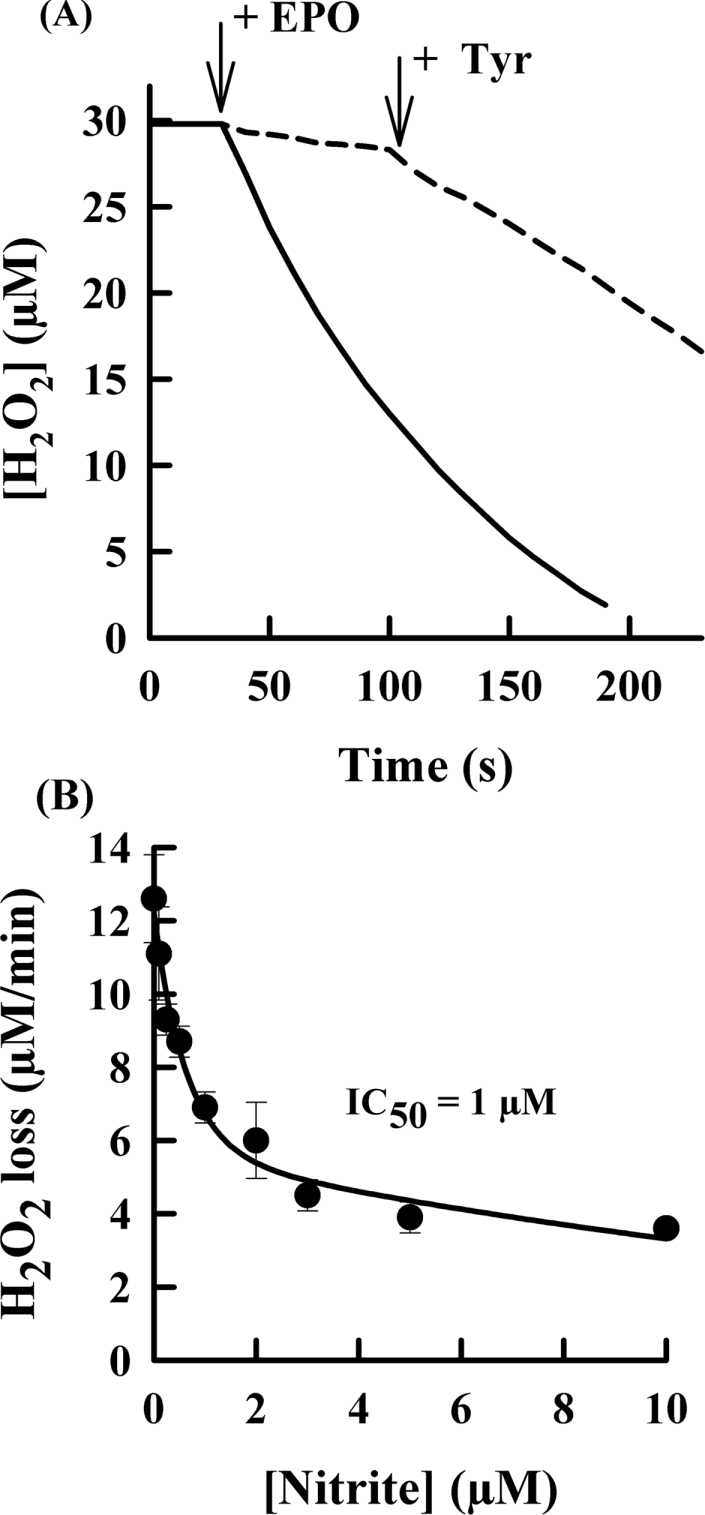
(A) Reactions were started by the addition of 2 nM eosinophil peroxidase (EPO) to 30 μM hydrogen peroxide in 100 mM sodium phosphate buffer (pH 7.4) containing 20 μM DTPA, 100 μM glutathione and 100 μM thiocyanate with (broken line) or without (solid line) 10 μM nitrite. Tyrosine (50 μM) was added where indicated by the arrow. Results are representative of at least duplicate experiments. (B) Reaction conditions were as for (A) except that the concentration of nitrite was varied. Initial rates were calculated over the first 10 s of each reaction. The IC50 was calculated by fitting a rectangular hyperbola to the data by using nonlinear regression. Results are means and ranges for at least duplicate experiments.
To establish whether the inhibition of eosinophil peroxidase by nitrite was reversible, tyrosine was added to each reaction (Figure 2A). Tyrosine is a substrate for compound I and compound II of eosinophil peroxidase [16]. If the enzyme was inhibited through the accumulation of compound II, which is unable to oxidize thiocyanate or bromide, then tyrosine should reverse the inhibition by reducing the enzyme back to its active ferric form. Indeed, tyrosine restored almost all the enzyme's activity. A similar effect of tyrosine was observed when bromide was the reducing substrate for the enzyme (results not shown).
To confirm that nitrite inhibits eosinophil peroxidase by promoting the formation of compound II, the absorption spectrum of eosinophil peroxidase was monitored during oxidation of bromide (Figure 3A). With bromide alone as the substrate, there was no change in the absorption spectrum of eosinophil peroxidase upon addition of hydrogen peroxide. This is expected because the rate-determining step would be reaction of hydrogen peroxide with the native enzyme [15]. However, when 10 μM nitrite was added, there was a clear shift in the Soret maximum. The peak for the ferric enzyme at 415 nm shifted to 435 nm and there was an isosbestic point at 425 nm (Figure 3A). This peak subsequently decayed back to the native spectrum (results not shown). The new spectrum was the same as that previously identified for compound II of eosinophil peroxidase [14]. To check that the spectral changes were not due to the formation of a complex between nitrite and the haem group of the enzyme, the spectra obtained on addition of 10 μM and 10 mM nitrite to native eosinophil peroxidase were compared. Addition of 10 μM nitrite to the native enzyme caused minimal change in the native spectrum (results not shown). When 10 mM nitrite was added to the enzyme, there was a shift in the Soret maximum from 415 to 425 nm with the isosbestic point at 419 nm (Figure 3B). This new spectrum differs from that obtained during the oxidation of nitrite by eosinophil peroxidase. However, it resembles complexes between nitrite and the haem groups of other peroxidases [31,32].
Figure 3. Spectral changes of eosinophil peroxidase during the oxidation of nitrite.

(A) The spectrum of 840 nM native eosinophil peroxidase (EP3+) (grey line) in PBS containing 100 μM bromide, 10 μM nitrite and 1 mM methionine was recorded prior to the addition of 50 μM hydrogen peroxide (black line). (B) The spectrum of 600 nM native enzyme in 100 mM phosphate buffer (100 mM sodium dihydrogen phosphate and disodium hydrogen phosphate buffer, pH 7.4) was recorded (grey line) prior to adding 10 mM nitrite (black line).
We also determined how nitrite affects production of hypobromous acid by isolated human eosinophils. Cells were stimulated with PMA and hypobromous acid was trapped with taurine to form stable taurine bromamine [33]. Increasing the concentration of nitrite caused progressive inhibition of hypobromous acid production (Figure 4). The concentration of nitrite that inhibited by 50% was approx. 20 μM. At 200 μM nitrite, there was little production of hypobromous acid by eosinophils.
Figure 4. Effect of nitrite on hypobromous acid formation by human eosinophils.
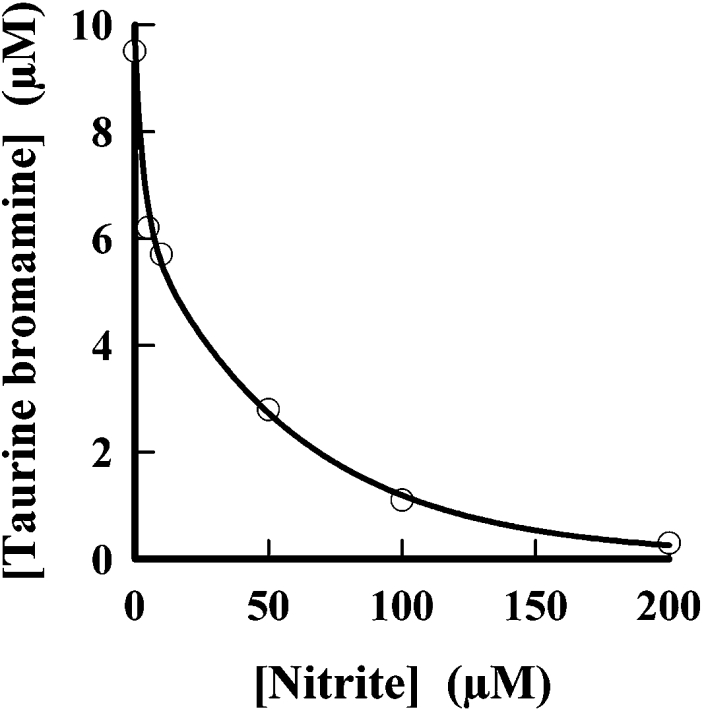
Human eosinophils (1×106 ml−1) were incubated at 37 °C in PBS containing 1 mM CaCl2, 0.5 mM MgCl2, 1 mg/ml glucose, 100 μM bromide, 10 mM taurine and various concentrations of nitrite. The cells were stimulated with 100 ng/ml PMA. Reactions were stopped after 30 min with 20 μg/ml catalase and accumulated taurine bromamine was measured with TNB. Results shown are representative of four independent experiments.
Nitration of tyrosyl residues by eosinophils and neutrophils
Previously, nitration of HPA (hydroxyphenylpropanoic acid) by eosinophils and neutrophils was compared [10]. It was shown that eosinophils were approx 4-fold more efficient at nitration of this tyrosine analogue than neutrophils at physiological concentrations of nitrite. However, HPA is a tyrosine analogue that should be oxidized to radicals which would drive nitration by reacting with nitrogen dioxide. We wanted to assess how efficient eosinophils were at nitrating tyrosyl residues in the absence of a flux of tyrosyl radicals. Nitration was investigated by using heptapeptides that contained a tyrosyl residue. The tyrosyl residues of these peptides are not a substrate for eosinophil peroxidase because they did not affect the rate of hydrogen peroxide utilization by eosinophil peroxidase during the oxidation of bromide or thiocyanate (results not shown). Any nitration of the heptapeptides should depend on the direct oxidation of nitrite by eosinophil peroxidase. Hence, the nitration mechanism should resemble that for a large protein rather than free tyrosine.
Eosinophil peroxidase readily nitrated a heptapeptide at physiological concentrations of nitrite (Table 1). Nitration was modestly enhanced by tyrosine, unaffected by bromide but substantially inhibited by thiocyanate. These results are in good agreement with those obtained by Wu et al. [6] for the nitration of albumin by eosinophil peroxidase and confirm that nitration of the heptapeptide was a good model for protein nitration.
Table 1. Effects of tyrosine and bromide on peptide nitration by eosinophil peroxidase.
Nitration of the peptide acetyl-Ser-Gln-Asn-Tyr-Pro-Val-Val (100 μM) was started by adding hydrogen peroxide (100 μM) to 20 nM eosinophil peroxidase and 100 μM nitrite, with or without additions of 10 μM tyrosine, 100 μM bromide, 100 μM thiocyanate or 1 mM methionine at 21 °C in 50 mM phosphate buffer (pH 7.4). Reactions were stopped after 1 h with 20 μg/ml catalase. Nitration of the peptide was measured by HPLC as outlined in the Experimental section. Results shown are means±S.D. for n experiments. EPO, eosinophil peroxidase; SCN−, thiocyanate.
| Reaction system | [Nitrated peptide] (μM) |
|---|---|
| Peptide alone | 0 |
| +EPO/H2O2/NO2− | 9.3±1.1 (n=6) |
| +Tyr | 14.1±0.4 (n=6) |
| +Br−/Met | 7.7±0.3 (n=4) |
| +Br−/Met/Tyr | 10.0±0.1 (n=4) |
| +SCN− | 2.2±1.3 (n=4) |
We measured nitration of a heptapeptide by eosinophils and neutrophils isolated from the same individual. When these granulocytes were stimulated with phorbol ester, they both promoted nitration in a reaction that was dependent on the concentration of nitrite (Figure 5). Eosinophils were much more responsive to the concentration of nitrite. At 100 μM nitrite, the level of peptide nitration observed with eosinophils (0.57±0.07 μM 3-nitrotyrosine) was approx. four times greater than that for neutrophils (0.13±0.02 μM 3-nitrotyrosine). The extent of nitration was increased 3-fold by the presence of superoxide dismutase, which indicates that superoxide inhibits nitration. Tyrosine increased the level of nitration either in the presence or absence of superoxide dismutase. Superoxide production by eosinophils was approx. 3-fold greater than that for neutrophils (results not shown). With eosinophils, there was minimal nitration when azide or catalase was added to the cells (Figure 6). These results indicate that nitration was dependent on an active respiratory burst, hydrogen peroxide and eosinophil peroxidase.
Figure 5. Comparison of tyrosyl residue nitration by human neutrophils and eosinophils.
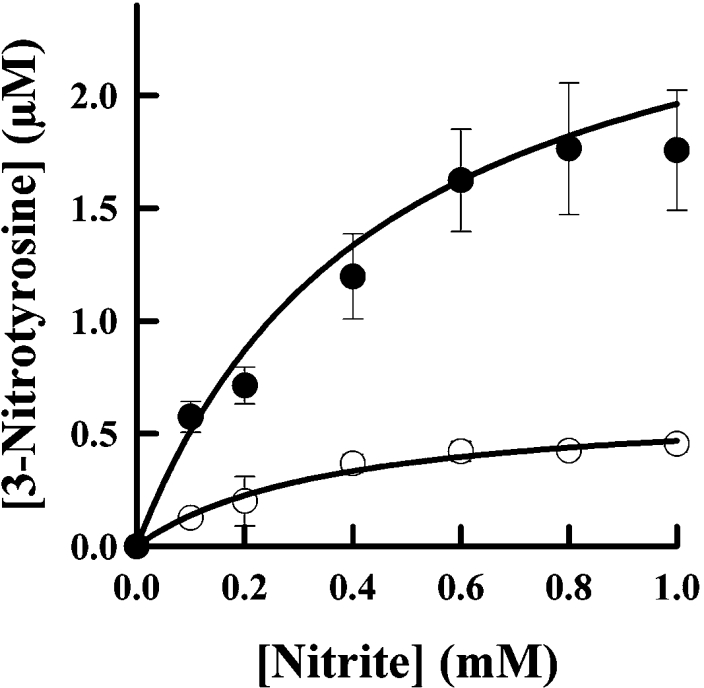
Eosinophils (●) and neutrophils (○) (1×106 cells/ml) were isolated from the same individual, and incubated at 37 °C in PBS containing 1 mM CaCl2, 0.5 mM MgCl2, 1 mg/ml glucose, 100 μM Ala-Pro-Arg-Leu-Arg-Phe-Tyr-Ser and increasing concentrations of nitrite. Cells were stimulated with 100 ng/ml PMA and the reactions were stopped after 1 h by adding 40 μg/ml catalase. Concentrations of the nitrated peptide in the supernatant were determined by HPLC. Results shown are means and S.D. for triplicate experiments and are representative of two separate experiments.
Figure 6. Nitration of tyrosyl residues by human eosinophils.

Eosinophils (1×106 cells/ml) were incubated with 100 μM acetyl-Ser-Gln-Asn-Tyr-Pro-Val-Val, 100 μM nitrite and either 10 μg/ml superoxide dismutase (SOD), 100 μM tyrosine (Tyr), 100 μM azide (Az) or 20 μg/ml catalase (CAT). They were either unstimulated (US) or stimulated with 100 ng/ml PMA, and the reactions were stopped after 1 h. Other reaction conditions were as described in Figure 5. Results shown are means and S.E.M. for three experiments.
DISCUSSION
In the present study we have extended the earlier work of Hazen and co-workers [6,7,10,34] on eosinophil peroxidase and now provide kinetic and spectral evidence that this enzyme oxidizes nitrite by single one-electron reactions involving compound I and compound II (see Scheme 1). The product of these reactions will be nitrogen dioxide as demonstrated previously [7]. There are two important consequences that arise from establishing the mechanism and kinetics of nitrite oxidation. First, nitrite will inhibit hypohalous acid production by eosinophil peroxidase under most physiological concentrations of nitrite, thiocyanate and bromide. Secondly, nitrite is a comparatively good substrate for compound II of eosinophil peroxidase. This reaction allows for efficient production of nitrogen dioxide, such that nitrosative stress should be considered as a likely outcome of eosinophilic inflammation.
Scheme 1. The reaction mechanism for oxidation of substrates by eosinophil peroxidase.

Two lines of evidence clearly pointed to the one-electron oxidation of nitrite by eosinophil peroxidase (Scheme 1). We found that nitrite inhibited the oxidation of thiocyanate and bromide, and that this could be reversed by tyrosine. These results are best explained by nitrite reducing compound I to compound II. Under these conditions, consumption of hydrogen peroxide would be inhibited because turnover of compound II by nitrite is much slower than reaction of compound I with either thiocyanate or bromide [15]. Tyrosine would have reversed inhibition by readily reducing compound II back to the ferric enzyme [16]. This mode of inhibition was confirmed by showing spectrally that nitrite converted eosinophil peroxidase into compound II during oxidation of bromide. There was no spectral evidence that nitrite bound to the enzyme (reaction 6) at the low concentrations of nitrite that were effective at inhibiting oxidation of thiocyanate and bromide. Hence, it is unlikely that nitrite acts by preventing hydrogen peroxide from reacting with the ferric enzyme (reaction 1).
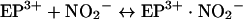 |
(6) |
The observed rate constant for the turnover of eosinophil peroxidase during oxidation of nitrite, which mainly reflects reduction of compound II, was determined to be 5.6×103 M−1·s−1. It is an observed rate constant because under the conditions of the assay, where the nitrite concentration was as high as 10 mM, nitrite would have also affected hydrogen peroxide consumption by binding to ferric eosinophil peroxidase (reaction 6). This was seen spectrally and indicates that nitrite should inhibit the reaction of hydrogen peroxide with the native enzyme. It was not possible to calculate the actual rate constant for reaction of compound II with nitrite. Previously, we undertook the same kinetic experiments for peroxidation of nitrite by myeloperoxidase [17]. A replot of the data obtained in that investigation gives a value of 3.0×102 M−1·s−1 for the observed rate constant for reaction of nitrite with compound II (results not shown). This compares favourably with the value of 5.5×102 M−1·s−1 obtained by stopped-flow techniques [18].
These rate constants indicate that nitrite is approx. 20-fold more favoured as a substrate for compound II of eosinophil peroxidase than myeloperoxidase. Consequently, eosinophil peroxidase is a much better catalyst for the production of nitrogen dioxide than myeloperoxidase. This result explains why eosinophils were better at nitrating tyrosyl residues than neutrophils. The greater capacity of eosinophils to produce hydrogen peroxide should not affect the extent of nitration because the rate-determining step in nitration does not involve hydrogen peroxide. Based on these in vitro findings, we propose that nitration should be more evident in eosinophilic inflammation, as occurs in asthma [35], than in neutrophilic inflammation. However, other factors such as the presence of competing substrates and pH are likely to impact on the efficiency of nitration in vivo [7]. Rate constants for reaction of tyrosine and ascorbate with compound II of eosinophil peroxidase are 2.7×104 and 6.7×103 M−1·s−1 respectively [16]. Hence, at sites of inflammation where the concentration of nitrite is likely to be high, nitrite can be expected to be a major substrate for compound II.
A value for the rate constant for the reaction of nitrite with compound I of eosinophil peroxidase would give an indication of how effectively nitrite should compete with other substrates of the enzyme that are present at inflammatory sites. To get an estimate of this rate constant, we constructed a kinetic model of the reactions given in Scheme 1. In this model, we used published values for the rate constants for thiocyanate (1×108 M−1·s−1) and bromide (1.9×107 M−1·s−1) reacting with compound I, and hydrogen peroxide reacting with the native enzyme (4.3×107 M−1·s−1) [15]. To calculate the rate constant for reaction of nitrite with compound II, we used the reaction conditions described in Figure 2 for hydrogen peroxide consumption by eosinophil peroxidase in the presence of thiocyanate or bromide. The concentration of nitrite was set at its experimentally determined IC50 value. Rate constants that gave 50% inhibition of the rate of loss of hydrogen peroxide were then calculated. Values of 2×106 and 4×106 M−1·s−1 were obtained for the thiocyanate and bromide data respectively. Thus our estimate for the rate constant for reaction of nitrite with compound I of eosinophil peroxidase is approx. 3×106 M−1·s−1. This gives a relative substrate preference for compound I of 1:7:33 for nitrite, bromide and thiocyanate respectively. On this basis, we conclude that nitrite reacts rapidly with compound I but is not as good a substrate for eosinophil peroxidase as bromide and thiocyanate.
From our kinetic and mechanistic investigations into the oxidation of nitrite, we conclude that nitrite will modulate oxidant production by eosinophils. At low concentrations, it will inhibit hypohalous acid production by converting eosinophil peroxidase into compound II, which does not oxidize bromide or thiocyanite [14,15]. Unless compound II can be recycled by more abundant reductants, such as tyrosine and ascorbate [16], the enzyme will be trapped in this form and unable to produce oxidants. At higher concentrations, nitrite will compete with bromide and thiocyanate, and efficiently reduce compound II. Under these conditions, nitrogen dioxide will be a major oxidant produced by eosinophil peroxidase.
Nitration of proteins occurs by the following series of reactions where P-Tyr is a tyrosyl residue.
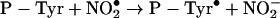 |
(7) |
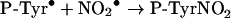 |
(8) |
If tyrosine is present, the following reaction will also occur.
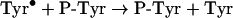 |
(9) |
From this investigation, it is apparent that superoxide will influence protein nitration at sites of inflammation. We found that superoxide dismutase increased peptide nitration by approx. 3-fold. Its effect can be attributed to inhibition of the diffusion-controlled reaction of nitrogen dioxide with superoxide (kNO2/O2−=4.5×109 M−1·s−1) [36]. This reaction would lower the concentration of nitrogen dioxide, thereby inhibiting the rate-determining step in nitration, which is the oxidation of tyrosyl residues by nitrogen dioxide (reaction 7; k7<1×105 M−1·s−1) [37]. Our results are in contrast with those of MacPherson et al. [10] who found that superoxide dismutase had no effect on nitration of HPA by eosinophils. HPA is an analogue of tyrosine and should react with eosinophil peroxidase at similar rates to tyrosine. Hence, under the conditions of their experiment, the steady-state concentration of phenoxyl radicals would have been sufficiently high to trap most of the nitrogen dioxide (reaction 8; k8=3×109 M−1·s−1) [37]. If the steady-state concentration of phenoxyl radicals was very much greater than that of superoxide, then reactions of superoxide with nitrogen dioxide and phenoxyl radicals (k=1.5×109 M−1·s−1 [38]) would not have been expected to affect nitration. This would explain the lack of effect of superoxide dismutase on nitration of HPA. In vivo, generation of tyrosyl radicals would promote the nitration through radical exchange with tyrosyl residues (reaction 9) and eliminate the need for reaction 7. Tyrosyl radical generation would also attenuate the effect of superoxide on nitration as we observed with eosinophils. However, in the absence of coincident generation of tyrosyl radicals, superoxide should be expected to block nitration of proteins by nitrogen dioxide.
Superoxide will also limit nitration by reacting with tyrosyl radicals on proteins [39]. These reactions of superoxide with nitrogen dioxide and tyrosyl radicals should not be considered protective against damaging reactions of nitrogen dioxide. Rather, both give rise to additional oxidants. Superoxide adds to tyrosyl radicals to produce tyrosine peroxide, which may inflict site-specific damage to proteins [39,40]. Nitrogen dioxide reacts with superoxide to generate peroxynitrate (O2NOO−) [36]. Peroxynitrate exists in equilibrium with peroxynitric acid (O2NOOH) and has a pKa of 5.85. At physiological pH, peroxynitrate is the dominant species. It has a half-life of approx. 1 s decomposing to nitrite and molecular oxygen [41]. It does not nitrate tyrosine [42]. However, peroxynitric acid is stable and is a strong oxidant that oxidizes aromatics, halides, azide, NADH [41] and methionione [43]. Inflammation is associated with a decline in pH, so it is possible that appreciable peroxynitric acid is formed and contributes to inflammatory tissue damage.
In conclusion, we have shown that nitrite is oxidized by eosinophil peroxidase by two successive one-electron oxidations to produce nitrogen dioxide. Low concentrations of nitrite will limit oxidant production by eosinophil peroxidase by converting the enzyme into compound II. At high concentrations of nitrite, nitrogen dioxide will be a major oxidant generated by eosinophil peroxidase. Its ability to nitrate proteins will be influenced by the presence of both superoxide and tyrosine radicals.
Acknowledgments
This work was supported by the Health Research Council of New Zealand.
References
- 1.Eiserich J. P., Hristova M., Cross C. E., Jones A. D., Freeman B. A., Halliwell B., van der Vliet A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature (London) 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 2.van der Vliet A., Eiserich J. P., Halliwell B., Cross C. E. Formation of reactive nitrogen species during peroxidase-catalyzed oxidation of nitrite: a potential additional mechanism of nitric oxide-dependent toxicity. J. Biol. Chem. 1997;272:7617–7625. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 3.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dedon P. C., Tannenbaum S. R. Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Klebanoff S. J. Reactive nitrogen intermediates and antimicrobial activity: role of nitrite. Free Radical Biol. Med. 1993;14:351–360. doi: 10.1016/0891-5849(93)90084-8. [DOI] [PubMed] [Google Scholar]
- 6.Wu W., Chen Y., Hazen S. L. Eosinophil peroxidase nitrates protein tyrosyl residues. Implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J. Biol. Chem. 1999;274:25933–25944. doi: 10.1074/jbc.274.36.25933. [DOI] [PubMed] [Google Scholar]
- 7.Brennan M. L., Wu W., Fu X., Shen Z., Song W., Frost H., Vadseth C., Narine L., Lenkiewicz E., Borchers M. T., et al. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- 8.Duguet A., Iijima H., Eum S. Y., Hamid Q., Eidelman D. H. Eosinophil peroxidase mediates protein nitration in allergic airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2001;164:1119–1126. doi: 10.1164/ajrccm.164.7.2010085. [DOI] [PubMed] [Google Scholar]
- 9.Iijima H., Duguet A., Eum S. Y., Hamid Q., Eidelman D. H. Nitric oxide and protein nitration are eosinophil dependent in allergen-challenged mice. Am. J. Respir. Crit. Care Med. 2001;163:1233–1240. doi: 10.1164/ajrccm.163.5.2003145. [DOI] [PubMed] [Google Scholar]
- 10.MacPherson J. C., Comhair S. A., Erzurum S. C., Klein D. F., Lipscomb M. F., Kavuru M. S., Samoszuk M. K., Hazen S. L. Eosinophils are a major source of nitric oxide-derived oxidants in severe asthma: characterization of pathways available to eosinophils for generating reactive nitrogen species. J. Immunol. 2001;166:5763–5772. doi: 10.4049/jimmunol.166.9.5763. [DOI] [PubMed] [Google Scholar]
- 11.Klebanoff S. J. Oxygen metabolites from phagocytes. In: Gallin J. I., Snyderman R., editors. Inflammation: Basic Principles and Clinical Correlates. Philadelphia, PA: Lippincott Williams & Wilkins; 1999. pp. 721–768. [Google Scholar]
- 12.Weiss S. J., Test S. T., Eckmann C. M., Roos D., Regiani S. Brominating oxidants generated by human eosinophils. Science. 1986;234:200–203. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 13.Slungaard A., Mahoney J. R. Thiocyanate is the major substrate for eosinophil peroxidase in physiologic fluids. J. Biol. Chem. 1991;266:4903–4910. [PubMed] [Google Scholar]
- 14.Van Dalen C. J., Kettle A. J. Substrates and products of eosinophil peroxidase. Biochem. J. 2001;358:233–239. doi: 10.1042/0264-6021:3580233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furtmüller P. G., Burner U., Regelsberger G., Obinger C. Spectral and kinetic studies on the formation of eosinophil peroxidase compound I and its reaction with halides and thiocyanate. Biochemistry. 2000;39:15578–15584. doi: 10.1021/bi0020271. [DOI] [PubMed] [Google Scholar]
- 16.Furtmüller P. G., Jantschko W., Regelsberger G., Obinger C. Spectral and kinetic studies on eosinophil peroxidase compounds I and II and their reaction with ascorbate and tyrosine. Biochim. Biophys. Acta. 2001;1548:121–128. doi: 10.1016/s0167-4838(01)00230-8. [DOI] [PubMed] [Google Scholar]
- 17.Van Dalen C. J., Winterbourn C. C., Senthilmohan R., Kettle A. J. Nitrite as a substrate and inhibitor of myeloperoxidase: implications for nitration and hypochlorous acid production at sites of inflammation. J. Biol. Chem. 2000;275:11638–11644. doi: 10.1074/jbc.275.16.11638. [DOI] [PubMed] [Google Scholar]
- 18.Burner U., Furtmüller P. G., Kettle A. J., Koppenol W. H., Obinger C. Mechanism of reaction of myeloperoxidase with nitrite. J. Biol. Chem. 2000;275:20597–20601. doi: 10.1074/jbc.M000181200. [DOI] [PubMed] [Google Scholar]
- 19.Beers R. J., Sizer I. W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 20.Jorg A., Portmann G., Fellay G., Dreyer J. L., Meyer J. A rapid and simple method for the isolation of pure eosinophilic leucocytes from horse blood. Experientia. 1978;34:1654–1655. doi: 10.1007/BF02034734. [DOI] [PubMed] [Google Scholar]
- 21.Maurer H. R. Berlin: Walter de Gruyter; 1971. Disc Electrophoresis and Related Techniques of Poly-acrylamide Gel Electrophoresis. [Google Scholar]
- 22.Kettle A. J., Winterbourn C. C. Assays for the chlorination activity of myeloperoxidase. Methods Enzymol. 1994;233:502–512. doi: 10.1016/s0076-6879(94)33056-5. [DOI] [PubMed] [Google Scholar]
- 23.Thomas E. L. Products of lactoperoxidase-catalysed oxidation of thiocyanate and halides. In: Pruitt K. M., Tenovuo J. V., editors. The Lactoperoxidase System: Chemistry and Biological Significance. New York: Marcel Dekker; 1985. pp. 31–53. [Google Scholar]
- 24.Böyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of mononuclear cells by one centrifugation; and of granulocytes by combined centrifugation and sedimentation at 1 g. Scand. J. Clin. Lab. Invest. 1968;21:77–89. [PubMed] [Google Scholar]
- 25.Cramer R., Dri P., Zabucchi G., Patriarca P. A simple and rapid method for isolation of eosinophilic granulocytes from human blood. J. Leukoc. Biol. 1992;52:331–336. doi: 10.1002/jlb.52.3.331. [DOI] [PubMed] [Google Scholar]
- 26.Hansel T. T., De Vries I. J., Iff T., Rihs S., Wandzilak M., Betz S., Blaser K., Walker C. An improved immunomagnetic procedure for the isolation of highly purified human blood eosinophils. J. Immunol. Methods. 1991;145:105–110. doi: 10.1016/0022-1759(91)90315-7. [DOI] [PubMed] [Google Scholar]
- 27.Babior B. M., Kipnes R. S., Curnutte J. T. Biological defense mechanisms: the production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Invest. 1973;52:741–744. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fridovich I. Cytochrome c. In: Greenwald R. A., editor. Handbook of Methods for Oxygen Radical Research. Boca Raton, FL: CRC Press; 1985. pp. 213–215. [Google Scholar]
- 29.Van Dalen C. J., Whitehouse M. W., Winterbourn C. C., Kettle A. J. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Vliet A., Eiserich J. P., Shigenaga M. K., Cross C. E. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am. J. Respir. Crit. Care Med. 1999;160:1–9. doi: 10.1164/ajrccm.160.1.9807044. [DOI] [PubMed] [Google Scholar]
- 31.Cooper C. E., Odell E. Interaction of human myeloperoxidase with nitrite. FEBS Lett. 1992;314:58–60. doi: 10.1016/0014-5793(92)81461-t. [DOI] [PubMed] [Google Scholar]
- 32.Floris R., Piersma S. R., Yang G., Jones P., Wever R. Interaction of myeloperoxidase with peroxynitrite: comparison with lactoperoxidase, horseradish peroxidase and catalase. Eur. J. Biochem. 1993;215:767–775. doi: 10.1111/j.1432-1033.1993.tb18091.x. [DOI] [PubMed] [Google Scholar]
- 33.Thomas E. L., Bozeman P. M., Jefferson M. M., King C. C. Oxidation of bromide by the human leukocyte enzymes myeloperoxidase and eosinophil peroxidase: formation of bromamines. J. Biol. Chem. 1995;270:2906–2913. doi: 10.1074/jbc.270.7.2906. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Soud H. M., Hazen S. L. Nitric oxide is a physiological substrate for mammalian peroxidases. J. Biol. Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 35.Andreadis A. A., Hazen S. L., Comhair S. A., Erzurum S. C. Oxidative and nitrosative events in asthma. Free Radical Biol. Med. 2003;35:213–225. doi: 10.1016/s0891-5849(03)00278-8. [DOI] [PubMed] [Google Scholar]
- 36.Logager T., Sehested K. Formation and decay of peroxynitric acid – a pulse radiolysis study. J. Phys. Chem. 1993;97:10047–10052. [Google Scholar]
- 37.Prutz W. A., Monig H., Butler J., Land E. J. Reactions of nitrogen dioxide in aqueous model systems: oxidation of tyrosine units in peptides and proteins. Arch. Biochem. Biophys. 1985;243:125–134. doi: 10.1016/0003-9861(85)90780-5. [DOI] [PubMed] [Google Scholar]
- 38.Jin F., Leitich J., von Sonntag C. The superoxide radical reacts with tyrosine-derived phenoxyl radicals by addition rather than by electron transfer. J. Chem. Soc. Perkin Trans. 1993;II:1583–1588. [Google Scholar]
- 39.Winterbourn C. C., Kettle A. J. Radical-radical reactions of superoxide: a potential route to toxicity. Biochem. Biophys. Res. Commun. 2003;305:729–736. doi: 10.1016/s0006-291x(03)00810-6. [DOI] [PubMed] [Google Scholar]
- 40.Winterbourn C. C., Parsons-Mair H. N., Gebicki S., Gebicki J. M., Davies M. J. Requirements for superoxide-dependent tyrosine hydroperoxide formation in peptides. Biochem. J. 2004;381:241–248. doi: 10.1042/BJ20040259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirsch M., Korth H. G., Sustmann R., de Groot H. The pathobiochemistry of nitrogen dioxide. Biol. Chem. 2002;383:389–399. doi: 10.1515/BC.2002.043. [DOI] [PubMed] [Google Scholar]
- 42.Kirsch M., Lehnig M., Korth H. G., Sustmann R., de Groot H. Inhibition of peroxynitrite-induced nitration of tyrosine by glutathione in the presence of carbon dioxide through both radical repair and peroxynitrate formation. Chemistry. 2001;7:3313–3320. doi: 10.1002/1521-3765(20010803)7:15<3313::aid-chem3313>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 43.Squadrito G. L., Pryor W. A. Biological chemistry of peroxynitrate. Free Radical Biol. Med. 2002;33(Suppl. 2):s241–s381. [Google Scholar]