

Abstract
Background: NV1066, a replication-competent oncolytic herpes simplex virus type 1 (HSV-1) attenuated by a deletion in the gene γ134.5, preferentially replicates in and kills malignant cells. γ134.5 encodes ICP34.5, a viral protein essential for productive replication, which has homology with mammalian stress response induced GADD34 (Growth Arrest and DNA Damage-Inducible Protein). We hypothesized that cisplatin upregulates GADD34 expression, which enhances NV1066 replication and oncolysis.
Methods: Ten human malignant pleural mesothelioma (MPM) cell lines were infected with NV1066 at multiplicities of infection (MOI; ratio of viral particles per tumor cell) 0.005 to 0.8 in vitro, with and without cisplatin (1 to 4 μM). In the MPM cell line VAMT, viral replication was determined by plaque assay, cell kill by lactate dehydrogenase assay, and GADD34 induction by quantitative RT-PCR and Western blot. Synergistic efficacy was confirmed by the isobologram and combination index methods of Chou-Talalay. GADD34 upregulation by cisplatin was inhibited with GADD34 siRNA to further confirm the synergistic efficacy dependence with GADD34.
Results: Combination therapy with NV1066 and cisplatin showed strong synergism in epithelioid (H-2452, H-Meso), sarcomatoid (H-2373, H-28), and biphasic (JMN, Meso-9, MSTO-211H) MPM cell lines, and an additive effect in others. In VAMT cells combination therapy enhanced viral replication 4 to 11-fold (p < 0.01) and cell kill 2 to 3-fold (p < 0.01). Significant dose reductions for both agents (2 to 600-fold) were achieved over a wide range of therapeutic-effect levels (LD50 – LD99) without compromising cell kill. Synergistic cytotoxicity correlated with GADD34 upregulation (2 to 4-fold, p < 0.01) and was eliminated following transfection with GADD34 siRNA.
Conclusion: Cisplatin-induced GADD34 expression selectively enhanced the cytotoxicity of the γ134.5-deficient oncolytic virus, NV1066. This provides a cellular basis for combination therapy with cisplatin and NV1066 to treat MPM and achieve synergistic efficacy, while minimizing dosage and toxicity.
Keywords: Chemotherapy, Gene therapy, HSV, Combination therapy
Introduction
Malignant pleural mesothelioma (MPM) is an aggressive cancer with a median survival of 4-12 months(1). Because of the long latency period between asbestos exposure and tumor development, the annual incidence of 2500 new cases is expected to increase by more than 50% in the coming decade(2;3). MPM is a diffuse disease and resistant to currently available therapeutic modalities. Cytotoxic monotherapy results in rates of tumor regression between 10-30%, with no significant impact on median survival(4).
Cisplatin has been studied as a single agent and in combined regimens for MPM. In phase II studies of high-dose cisplatin, response rates of 14-36% were achieved but significant discontinuations (34%) occurred due to toxicity(5;6). In a phase II trial of intrapleural and systemic cisplatin, the dosage of cisplatin had to be reduced because of renal toxicity(7). Therefore, therapies are necessary that may synergize with cisplatin to increase tumor response without increasing dosage, thereby minimizing toxicity.
Herpes simplex virus type 1 (HSV-1) mediated oncolysis and gene therapy have emerged as promising treatment modalities against cancer(8-11). Oncolysis results from the replicative life cycle of the virus, which lyses infected tumor cells and releases viral progeny for propagation of infection and resultant lysis of neighboring cancer cells. NV1066 is a replication-competent oncolytic HSV-1, which has mutations in the internal repeat sequence, resulting in deletions of one copy each of the genes encoding ICP0, ICP4, and ICP34.5(12). These deletions attenuate the virus and help to ensure that it preferentially replicates within cancer cells. The HSV gene γ134.5 encodes ICP34.5, which functions as a virulence factor by preventing the shutoff of protein synthesis in virus-infected cells(13), thereby allowing viral replication. ICP34.5 has significant homology at its carboxyl terminus to mammalian GADD34 (Growth Arrest and DNA Damage-Inducible Protein)(14). GADD34 is induced by various types of DNA damage, including ionizing radiation and chemotherapy(15;16). Previously, we have shown that mitomycin upregulates GADD34, which enhances replication and cytotoxicity of γ134.5-mutant HSV-1 in gastric and bladder cancer(17;18).
In the present study we show that cisplatin and NV1066 have synergistic cytotoxicity in MPM cell lines, allowing dosage reductions for both agents. Cisplatin upregulated GADD34 and increased viral replication. Inhibition of GADD34 with siRNA eliminated the synergistic cytotoxicity, suggesting that the mechanism of synergism is at least partly due to GADD34 substituting for the γ134.5 deletion in NV1066.
Methods
Cell culture: Human malignant mesothelioma cell lines of various histological subtypes were studied including: sarcomatoid (VAMT, H-2052, H-2373), epithelioid (H-2452, H-Meso), biphasic (JMN, H-Meso1A, MSTO-211H), and other pathological types (Meso-9, Meso-10). MSTO-211H and Vero cells (from the African green monkey kidney) were obtained from the American Type Culture Collection (ATCC® Rockville, MD). H-Meso and H-Meso1A cell lines were obtained from the National Cancer Institute (Bethesda, MD). JMN, VAMT, Meso-9, and Meso-10 cell lines were a kind donation from Dr. Sirotnik from Memorial Sloan-Kettering Cancer Center (New York, NY). H-2052, H-2452, and H-2373 cell lines were a kind donation from Dr. Pass from the Karmanos Cancer Institute, Wayne State University (Detroit, MI). All cell lines were maintained in appropriate media as recommended and incubated in a humidified incubator with 5% CO2.
Virus: NV1066 is a replication-competent, attenuated HSV-1 oncolytic virus with deletions of one copy each of the genes encoding ICP0, ICP4, and ICP34.5 genes, as previously described(12). Viral stocks were propagated in Vero cells, harvested by freeze-thaw lysis and sonication, and titered by standard plaque assay.
Cisplatin: Cisplatin was obtained from Bristol Laboratories (Princeton, NJ) as a 1 mg/ml aqueous solution. Dilutions were made with respective cell culture media for each cell line to achieve concentrations from 1 to 4 μM. Fresh dilutions were made for each experiment. After a 6 h exposure to cisplatin, cells were washed with PBS and incubated in fresh cisplatin-free media. For combination therapy experiments virus was added 1-2 h after the end of cisplatin treatment.
In vitro cytotoxicity assay: VAMT cells were plated in 24-well flat-bottom plates (Becton Dickinson, Franklin Lake, NJ) in 1 ml of media. Cells were treated with media alone (control wells), cisplatin alone, NV1066 alone, or combination therapy using both cisplatin and NV1066. NV1066 infection was carried out at multiplicities of infection (MOI: ratio of viral plaque forming units (PFU) per tumor cell) of 0.03, 0.06, or 0.09 in a total volume of 100 μl of medium. Combination therapy was performed using serial dilutions of cisplatin (1, 2, and 4 μM) and NV1066 (MOI 0.03, 0.06, and 0.09) in a 1:20 ratio. This ratio was determined by estimating the LD50 for each therapy in initial experiments and by using these doses to determine the ratio of combination therapy. Typically, cells were plated overnight, treated with cisplatin in the morning, and infected with virus 1-2 h after the end of cisplatin treatment. Percent survival for each group was determined on each day for 6 days after treatment using a standard lactate dehydrogenase (LDH) release bioassay (Promega, Madison, WI). Results were expressed as surviving fraction, based on the measured absorbance of treated cellular lysates, compared to that of untreated control cellular lysates. All samples were tested in triplicate. Experiments were repeated in triplicate to ensure reproducibility. Cytotoxicity assays were also performed in nine other mesothelioma cell lines and NV1066 virus, with and without cisplatin. The combination ratio between both therapies was kept at 1:10 or 1:20.
Quantitative analysis of synergy between cisplatin and NV1066: The multiple drug effect analysis of Chou and Talalay was used to determine the pharmacologic interaction between cisplatin and NV1066. This method defines the expected additive effect of two (or more) agents and quantifies synergism or antagonism by determining how the combination effect differs from the expected additive effect. The equations and computer software used for data analysis have been described in detail elsewhere(19-22). The combination index (CI) equation, which takes into account both the potency (LD50 or Dm values) and the shapes of the dose-effect curves (m values), is used to precisely analyze two-drug combinations. Interpretation of CI values is defined such that CI=1 indicates an additive effect, and CI<1 and a CI>1 indicate synergism and antagonism, respectively.
Cytotoxicity data obtained from the experiments described above were used in the Chou-Talalay analysis(20). The CI values for each dose and corresponding effect level, referred to as the fraction affected (Fa), were generated. Based on the actual experimental data, computer software was used to calculate serial CI values over an entire range of effect levels (Fa) from 5% to 95%. These data were then used to generate Fa-CI plots, which is an effect-oriented means of presenting synergism or antagonism. Data were also analyzed by the isobologram technique, which is dose-oriented. The axes on an isobologram represent the doses of each drug. Two points on the x and y axes are chosen that correspond to the doses of each drug necessary to generate that given Fa value. The straight line (hypotenuse) drawn between these two points on the x and y axes corresponds to the possible combination of doses that would be required to generate the same Fa value, indicating that the interaction between the two drugs is strictly additive. If these drug combination points lie on the straight line, then the effect is additive at that Fa value. If the point lies to the lower left of the hypotenuse, then the effect is synergistic, and if the point lies to the upper right of the hypotenuse, then the effect is antagonistic at that Fa value. Another calculation available using the CI method is the dose-reduction index (DRI)(19;22). The DRI is a determination of the fold of dose reduction allowed for each drug when given in synergistic combination, as compared with the concentration of a single agent that is needed to achieve the same effect level. DRI>1 signifies a favorable reduction in toxicity while still maintaining therapeutic efficacy.
In vitro viral growth analysis: The ability of NV1066 to replicate within VAMT cells in the presence or absence of cisplatin was evaluated by viral growth analysis. 5 x 104 cells per well were plated into 6-well plates. Cells were then infected with either NV1066 (MOI 0.03 or 0.06) alone, or with NV1066 following cisplatin (1 or 2 μM). Cells and media were harvested at 48, 72, 96, 120, and 144 hours post-infection. After three cycles of freeze-thaw lysis, standard plaque assay was performed on Vero cells to evaluate viral titers. All samples were performed in triplicate.
Real-time reverse transcription-PCR analysis for GADD34 in cells treated with cisplatin: 1 x 105 VAMT cells per well were plated in 6-well plates and incubated for 12 hours. Cells were treated with cisplatin doses of 1, 2, or 4 μM. Each sample was done in triplicate. After 24, 48, 72, and 96 hours of incubation, cells from each well of the plate were collected after washing with cold PBS and frozen for RNA collection. RNA from each sample was collected and isolated with an RNeasy protect kit (QIAGEN Inc., Valencia, CA) using the manufacturer's protocol. GADD34 in each sample was measured quantitatively by real-time RT-PCR using a SYBR green fluorophore with a Bio-Rad iCycler iQ detection system (Bio-Rad laboratories, Hercules, CA) and standardized to an 18S rRNA control. For GADD34 the following primers were applied: GADD34 forward 5′-GGA GGA AGA GAA TCA AGC CA-3′; GADD34 reverse 5′-TGG GGT CGG AGC CTG AAG AT-3′; for 18S: 18S forward 5′-GTA ACC CGT TGA ACC CCA TT-3′; 18S reverse 5′-CCA TCC AAT CGG TAG TAG CG-3′. A comparison between each treatment sample and the control group, which did not receive any cisplatin, was made to determine GADD34 upregulation. The results were represented as fold upregulation in the treatment sample compared to the control group. GADD34 siRNA transfection: Duplex siRNAs targeting human GADD34 outside the viral homology domain were designed and tested for the ability to decrease GADD34 expression. After preliminary experiments the following sequence targeting from codon 635 was chosen for further experiments: 5′-GUCAAUUUGCAGAUGGCCATTUGGCCAUCUGCAAAUUGACTT-3′. VAMT cells were plated at a concentration of 5x104 per well in 24-well plates 12 hours prior to transfection in appropriate medium without antibiotics. Standard siRNA transfection protocol as described before was used(17). Cells transfected with lacZ siRNA under a similar protocol were used as controls.
Western blot for GADD34 protein: VAMT cells (lacZ transfected and GADD34 siRNA-transfected) were incubated overnight and treated in the morning with 1 or 2 μM cisplatin. Untreated cells served as a control. Cells were lysed and collected with Cell Lysis Buffer (Cell Signaling Technology, Inc., Beverly, MD). Equal amounts of proteins were resolved on 10% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA) under reducing conditions and blotted on PVDF membrane (Schleicher & Schuell Bioscience, Keene, NH). Protein expression was determined by using primary rabbit polyclonal antihuman GADD34 (Santa Cruz Biotechnology, Santa Cruz, CA) and primary goat polyclonal antihuman actin (Santa Cruz Biotechnology). A secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) was used to visualize the expression level of GADD34 and actin on chemiluminescence film (Hyperfilm, Amersham Biosciences, Buckinghamshire, England) by application of an ECL Plus Western Blotting Detection System (Amersham Biosciences).
Statistical analysis: All data were expressed as mean ± standard error of the mean. Comparisons between groups were made using the two-tailed Student's t-test. To identify statistical significance for multiple comparisons, the ANOVA test was used where appropriate.
Results
In vitro cytotoxicity of cisplatin and NV1066: Cytotoxicity derived by LDH release assay on each day up to day 6 is represented in Figure 1. Both cisplatin (1μM) and NV1066 (MOI 0.03) demonstrated dose-dependent cytotoxicity against VAMT cells on each day up to day 6. Combination therapy killed more cells (92% ± 2%) than either agent alone (cisplatin killed 1 ± 3%, and NV1066 killed 55 ± 3%). Combination therapy showed greater efficacy than the expected additive effect by day 6 (p < 0.001). Synergistic cytotoxicity (p < 0.01) was demonstrated with combination therapy compared to single-agent therapy alone across a wide range of doses (cisplatin 1, 2 and 4 μM and NV1066 MOI 0.03, 0.06 and 0.09).
Figure 1.
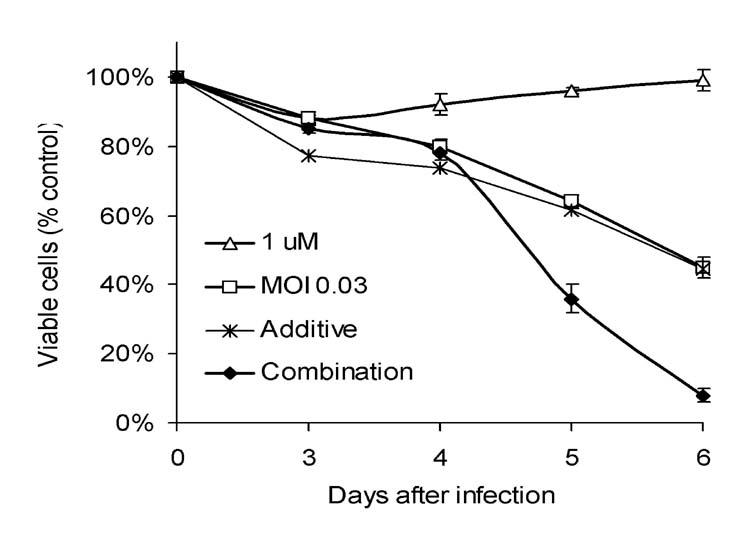
Cytotoxic effect of cisplatin, NV1066, or both on VAMT cells in vitro. VAMT cells were treated with cisplatin (1 μM), NV1066 (MOI 0.03), or the combination. Results for the treated groups are expressed as cell survival compared to untreated control cells grown under identical conditions. MOI: multiplicity of infection, ratio of viral particles to tumor cells.
Pharmacological analysis of synergy between cisplatin and NV1066: Chou-Talalay analysis demonstrated that the combination index values remained < 1 over the entire range of Fa values for VAMT cells (Table 1). The DRI was calculated for each Fa value. DRI values > 1 indicated that a reduction in toxicity was achieved without loss of efficacy. The cisplatin dose could be lowered 13 – 21 -fold, and the NV1066 dose could be lowered 9 – 197 -fold when given as combination therapy. Synergism was present across the entire range of fractional cell kill from LD50 to LD99. Isobolograms were constructed for the doses of cisplatin and NV1066 necessary to kill 50% of cells (LD50), 75% of cells (LD75), and 95% of cells (LD95) (Figure 2). Experimental combination data points at drug and viral concentrations were well below the expected additive effect line for each of these Fa values (50, 75, and 95). These studies confirmed synergism between cisplatin and NV1066 across a wide range of therapeutic doses.
Table 1.
Cisplatin and viral doses needed to kill various fractions (Fa) of VAMT cells and fold-dose reduction possible when delivered in combination.
| Fraction Affected (Fa) | Cisplatin alone (μM) | NV1066 alone (MOI) | Cisplatin dose reduction index | NV1066 dose reduction index |
|---|---|---|---|---|
| Dose reduction index is the –fold of dose reduction possible to achieve the same cell kill if radiation and virus are used in combination. LDx = Lethal dosex, dose needed to kill x% of cells, MOI = multiplicity of infection, ratio of viral particles to tumor cells. | ||||
| LD70 | 8 | 0.2627 | 16 | 9 |
| LD87 | 13 | 1.0432 | 13 | 17 |
| LD95 | 20 | 4.0885 | 13 | 45 |
| LD99 | 41 | 35.472 | 21 | 197 |
Figure 2.
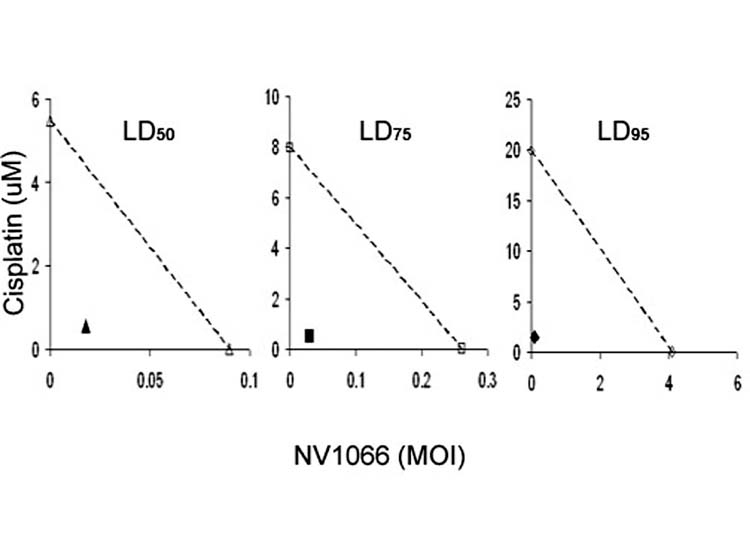
Isobolograms demonstrate dose-reductions achieved due to synergism of cisplatin with NV1066 in VAMT cells. The doses of cisplatin and NV1066 necessary to achieve 50% cell kill (open triangles), 75% cell kill (open squares), and 95% cell kill (open circles) are plotted on the axes, and the connecting solid lines represent the expected additive effects for combination therapy. Experimental combination therapy doses necessary to generate actual LD values of 50% (filled triangle), 75% (filled square), and 95% (filled circle) all lie to the lower left of the corresponding lines, indicating synergism.
Synergistic cytotoxicity in multiple malignant pleural mesothelioma (MPM) cell lines: To further confirm that the synergistic cytotoxicity was not isolated to one particular cell line or pathological type, cytotoxicity experiments were performed in multiple MPM cell lines. These cell lines had varied sensitivity to NV1066 (LD50 MOI 0.16 to 0.70) and cisplatin (2-20 μM). Combination therapy resulted in synergistic cytotoxicity in most of the cell lines (H-2452, H-Meso, H-2373, H-28, JMN, Meso-9 and MSTO-211H) and additive effect in others. With combination therapy strong synergism was present in H2373 and Meso9 cells, and moderate in JMN, H2452, Meso, and MSTO cells. Synergism and possible dose reductions were calculated by the isobologram and combination index methods of Chou-Talalay for 5 to 95% cell kill (LD5 - LD95) in all the cell lines. Table 2 shows representative data of treatment doses for single agent therapy, combination therapy, and fold dose reduction achieved for 50% cell kill (LD50).
Table 2.
Combination therapy with cisplatin and NV1066 to achieve LD50 (50% cell kill) in multiple malignant mesothelioma cells and fold dose reduction possible when delivered in combination.
| Treatment | Cell Line | |||||
|---|---|---|---|---|---|---|
| JMN | H2452 | H2373 | Meso | Meso9 | MSTO | |
| Dose reduction index is the –fold of dose reduction possible to achieve the same cell kill if cisplatin and virus are used in combination. LDx = Lethal dosex, dose needed to kill x% of cells, MOI = multiplicity of infection, ratio of viral particles to tumor cells. The combination index (CI) method was described by Chou and Talalay (20). CI < 1, = 1, and > 1 indicate synergism, additive effect, and antagonism, respectively. The symbols represent the degree of synergism (+++++, very strong synergism; ++++, strong synergism; +++, synergism; ++, moderate synergism; +, slight synergism). | ||||||
| Single therapy (LD50) | ||||||
| Virus (MOI) | 0.05 | 0.65 | 0.005 | 0.011 | 0.009 | 0.046 |
| Cisplatin (μM)24 | 11 | 53 | 1.4 | 66 | 23.1 | |
| Combination therapy (LD50) | ||||||
| Virus (MOI) | 0.036 | 0.325 | 0.00074 | 0.0048 0.0022 | ||
| 0.026 | ||||||
| + Cisplatin (μM)0.8 | 1.67 | 0.088 | 0.45 | 0.21 | 0.64 | |
| Dose Reduction Index (DRI) values | ||||||
| Virus | 1.4 | 2 | 6.8 | 2.3 | 4.1 | 1.8 |
| Cisplatin | 30 | 6.6 | 603 | 3.1 | 316 | 36 |
| Combination index (CI) value | ||||||
| 0.54 | 0.64 | 0.16 | 0.75 | 0.25 | 0.58 | |
| Synergism | ||||||
| +++ | +++ | ++++ | ++ | ++++ | +++ | |
Increased viral replication with cisplatin: After treatment with 1 μM of cisplatin, VAMT cells demonstrated an 11-fold increase in viral titers compared to NV1066 infection alone (MOI 0.03) 5 days post-infection (p < 0.01). At higher concentrations of virus (MOI 0.09), there was a 4-fold increase in viral yields in the presence of cisplatin (p = 0.02). This lower fold increase in viral replication is probably due to a higher loss of cellular substrates at an earlier time point with a higher MOI.
GADD34 induction with cisplatin: RNA extracted from VAMT cells treated with 1 μM cisplatin was analyzed for GADD34 levels, using real-time RT-PCR, standardized to an 18S rRNA control. RNA extracted from cells not treated with cisplatin served as negative controls. As shown in Figure 3, in VAMT cells 1 μM cisplatin increased GADD34 levels 4.3-fold at 48 hours, compared to untreated control cells. Western blot analysis confirmed GADD34 upregulation at the protein level.
Figure 3.
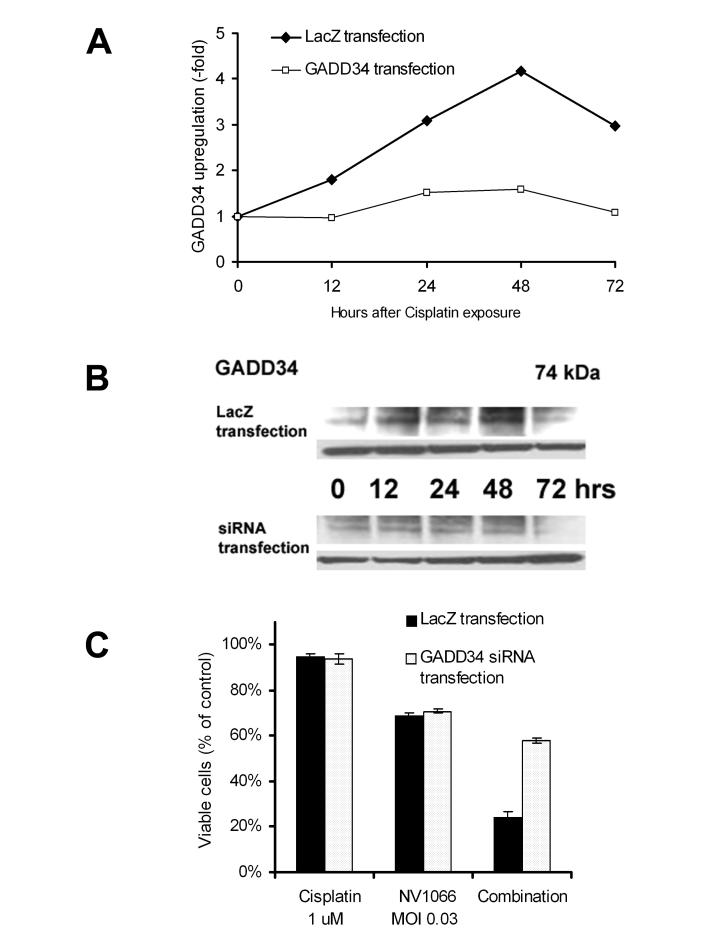
(A) GADD34 upregulation following cisplatin therapy and inhibition by GADD34 siRNA. LacZ transfected (control) and GADD34 siRNA-transfected VAMT cells were treated with cisplatin. Untreated cells served as control. GADD34 expression was measured by real-time RT-PCR at 0 to 72 h, standardized by an 18S control, and was upregulated in cisplatin-treated cells. This upregulation was inhibited in GADD34 siRNA-transfected cells. GADD34 upregulation was expressed as fold upregulation compared to untreated control cells. (B) GADD34 upregulation and its inhibition by siRNA transfection were confirmed at the protein level with Western blot analysis. (C) GADD34 upregulation inhibition by siRNA eliminated synergistic cytotoxicity. VAMT cells were treated with cisplatin (1 μM), NV1066 (MOI 0.06), or the combination, with or without GADD34 siRNA transfection. MOI = multiplicity of infection, ratio of viral particles to tumor cells. GADD34, Growth Arrest and DNA Damage-Inducible Protein 34, siRNA: small inhibitory RNA.
Inhibition of GADD34 induction with siRNA: Experiments were performed in VAMT cells to determine whether siRNA directed against GADD34 inhibited its upregulation by cisplatin. Transfection with siRNA inhibited GADD34 upregulation (60 – 80% knockdown) compared to lacZ transfected VAMT cells, as confirmed by real-time RT-PCR (Figure 3A). Western blot analysis confirmed GADD34 protein upregulation by cisplatin and inhibition by GADD34 siRNA (Figure 3B).
GADD34 inhibition with siRNA eliminated synergism: GADD34 inhibition eliminated synergistic cytotoxicity as demonstrated in Figure 3C. In GADD34 siRNA-transfected cells, 42 ± 1% of cells were killed with combination therapy, compared with 76 ± 2% of lacZ transfected cells (p < 0.01) (Figure 3C).
Discussion
Malignant pleural mesothelioma (MPM) is an aggressive neoplasm with a dismal prognosis. Combined surgery, radiation, and chemotherapy have limited success in a highly select group of patients. Cisplatin is one of the most effective drugs in MPM and is commonly used in combination regimens(23;24). Its cytotoxicity is mediated through platinum-DNA adducts, resulting in apoptosis and cell cycle arrest. However, this apoptosis is responsible for the characteristic nephrotoxicity, ototoxicity, and neurotoxicity(25). In MPM the diffuse tumor growth pattern and large surface area require high doses of cisplatin, with prohibitive toxicity(5-7). In addition, the clinical application of cisplatin is limited by the development of tumor resistance.
Oncolytic herpes simplex virus type 1 (HSV-1) has been shown to be effective in many experimental cancers including malignant pleural mesothelioma(8-11;17;26). Combining oncolytic HSV-1 with cisplatin may decrease cisplatin dosage and toxicity and may overcome cisplatin resistance. Previously, we showed that upregulation of DNA repair genes was beneficial for oncolytic viral activity(17). In this study we show that low-dose cisplatin was used to induce the cellular stress response, with minimal activation of apoptotic pathways. High-dose cisplatin causes not only toxicity but also a high apoptotic cell fraction, which hinders HSV-1 oncolysis by limiting viral replication(26). In addition, cisplatin and oncolytic viruses exert cytotoxicity via independent mechanisms, thereby circumventing the evolution of treatment-resistant cancer cells. Also, a major mechanism of cisplatin resistance in cancer cells is enhanced DNA repair(27), which may aid in enhanced viral replication and cytotoxicity when therapies are combined in these patients.
Oncolytic HSV-1 have strategic deletions in their genome to improve safety. Phase I trials of oncolytic HSV-1 demonstrated no adverse effects related to the virus(28). NV1066 has a deletion in the gene γ134.5, encoding the virulence factor ICP34.5, which precludes shutoff of protein synthesis and prevents apoptosis of virus-infected cells. ICP34.5 bears significant homology with the carboxyl terminus of mammalian GADD34 (Growth Arrest and DNA Damage-Inducible Protein), which is induced by stressful growth arrest conditions and treatment with DNA-damaging agents(14-16). In this study cisplatin enhanced viral replication and upregulated GADD34, which increased antitumor efficacy. Furthermore, inhibition of GADD34 upregulation with GADD34 siRNA abolished synergistic activity.
Previous studies showed that the carboxyl terminus of GADD34 substitutes for ICP34.5 in precluding premature shutoff of protein synthesis in neuroblastoma cells(29). Thus, HSV-1 γ134.5 mutants can appropriate the host cell GADD34 for their own use without increasing virulence. Recent attempts have been made to restore the γ134.5 gene into viral mutants since deletion of this gene markedly reduces cytotoxicity(30). In particular, investigators have attempted to insert the γ134.5 genes under control of a transcriptional-regulated promoter in order to facilitate selective gene expression in rapidly dividing cells. Although this approach is promising, insertion of the entire γ134.5 gene may theoretically restore neurovirulence and virulence in other non-malignant cells, in addition to providing any improvements in tumor cell kill. Our approach of inducing GADD34 function to replace the ICP34.5 function may provide a superior safety profile. Furthermore, such combination therapy may prove efficacious for tumor cells that may be resistant to either virus or cisplatin therapy alone.
Synergy was examined in this study using the CI and isobologram methods of Chou and Talalay(19-22). This type of analysis is one of the few methods available that determines synergy based on an extrapolated equation. The possibility of predicting false-positive synergistic interactions, a problem inherent in many other methods, is minimized as the analysis takes into account both the potency and shapes of the dose-effect curves in precisely analyzing two therapeutic combinations. Synergistic tumoricidal activity was demonstrated in multiple MPM cell lines, allowing dosage reductions for both agents while achieving the same therapeutic efficacy. Synergism was demonstrated in MPM cell lines, irrespective of their cisplatin sensitivity and histologic type. Sarcomatoid MPM is especially resistant to chemotherapy and associated with a poorer prognosis. In the H2373 sarcomatoid cell line combination therapy allowed a 603-fold reduction in cisplatin dose, while achieving the same cytotoxicity. Ultimately, the dose reduction is the most important parameter in determining the clinical applicability of combination therapy because potential toxicity can be reduced without sacrificing any therapeutic effect.
Studies suggest that recombinant HSV-1 lacking ICP34.5 is capable of killing ovarian cancer cells that lack p53 function, resist apoptosis, and/or are chemotherapy resistant(31). Other investigators have shown that cisplatin did not inhibit the efficacy of replication-competent HSV in the treatment of head and neck squamous cell carcinoma and in non-small cell lung cancer(32;33). Studies from our laboratory have shown the synergistic interaction of HSV with chemotherapy in the treatment of gastric and bladder cancer(17;18). In this particular study, we further extend those observations to a relatively chemoresistant tumor, malignant pleural mesothelioma, and further elicited the mechanism of such synergistic interaction.
In a study of cisplatin's effects on HSV infection in mice, cisplatin caused significant reductions in HSV infection rates(34). In our experiments high-dose cisplatin resulted in an inhibitory effect on HSV replication and efficacy when cells were exposed to both agents simultaneously. When cells were exposed to cisplatin alone, the stress response enhanced the efficacy of subsequent HSV therapy. This observation is beneficial in clinical translation, as it prevents the administration of both therapies simultaneously and avoids combination toxicity.
In summary, we show that cisplatin and NV1066 have synergistic cytotoxicity in MPM cell lines, allowing dosage reductions for both agents. Cisplatin results in increased viral replication and GADD34 upregulation. Inhibition of GADD34 with siRNA abrogates the synergistic activity, demonstrating that one mechanism of synergistic activity is GADD34 substituting for the γ134.5 deletion in NV1066. Such synergistic activity is shown across multiple different pathological types of malignant pleural mesothelioma cell lines. These data support future clinical investigation of such combined therapy for malignant pleural mesothelioma that aims to increase efficacy while minimizing toxicity.
Acknowledgments
The authors thank Brian Horsburgh, Ph.D. and Medigene, Inc. for constructing and providing us with the NV1066 virus. We thank Liza Marsh of the Department of Surgery at Memorial Sloan-Kettering Cancer Center for her editorial assistance.
Footnotes
Meeting presentation: 8th Annual meeting of American Society of Gene Therapy, St. Louis, June 2005.
Supported in part by AACR-Astra Zeneca Cancer Research and Prevention Foundation Fellowship (P.S.A), grants RO1 CA 75416 and RO1 CA/DK80982 (Y.F.) from the National Institutes of Health, grant MBC-99366 (Y.F.) from the American Cancer Society, grant BC024118 from the US Army (Y.F.), grant IMG0402501 from the Susan G. Komen Cancer Foundation (Y.F. and P.S.A.) and grant 032047 from Flight Attendant Medical Research Institute (Y.F. and P.S.A.)
Reference List
- 1.Flores RM. Induction chemotherapy, extrapleural pneumonectomy, and radiotherapy in the treatment of malignant pleural mesothelioma: the Memorial Sloan-Kettering experience. Lung Cancer. 2005;49(Suppl 1):S71–S74. doi: 10.1016/j.lungcan.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Connelly RR, Spirtas R, Myers MH, Percy CL, Fraumeni JF., Jr. Demographic patterns for mesothelioma in the United States. J Natl Cancer Inst. 1987;78(6):1053–60. [PubMed] [Google Scholar]
- 3.Walker AM, Loughlin JE, Friedlander ER, Rothman KJ, Dreyer NA. Projections of asbestos-related disease 1980-2009. J Occup Med. 1983;25(5):409–25. [PubMed] [Google Scholar]
- 4.Tomek S, Manegold C. Chemotherapy for malignant pleural mesothelioma: past results and recent developments. Lung Cancer. 2004;45(Suppl 1):S103–S119. doi: 10.1016/j.lungcan.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 5.Planting AS, Schellens JH, Goey SH, van der Burg ME, de Boer-Dennert M, Stoter G, et al. Weekly high-dose cisplatin in malignant pleural mesothelioma. Annals of Oncology. 1994;5(4):373–4. doi: 10.1093/oxfordjournals.annonc.a058846. [DOI] [PubMed] [Google Scholar]
- 6.Mintzer DM, Kelsen D, Frimmer D, Heelan R, Gralla R. Phase II trial of high-dose cisplatin in patients with malignant mesothelioma. Cancer Treatment Reports. 1985;69(6):711–2. [PubMed] [Google Scholar]
- 7.Rusch V, Saltz L, Venkatraman E, Ginsberg R, McCormack P, Burt M, et al. A phase II trial of pleurectomy/decortication followed by intrapleural and systemic chemotherapy for malignant pleural mesothelioma. J Clin Oncol. 1994;12(6):1156–63. doi: 10.1200/JCO.1994.12.6.1156. [DOI] [PubMed] [Google Scholar]
- 8.Bennett JJ, Delman KA, Burt BM, Mariotti A, Malhotra S, Zager J, et al. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Therapy. 2002;9(11):935–45. doi: 10.1038/sj.cgt.7700510. [DOI] [PubMed] [Google Scholar]
- 9.Delman KA, Zager JS, Bennett JJ, Malhotra S, Ebright MI, McAuliffe PF, et al. Efficacy of multiagent herpes simplex virus amplicon-mediated immunotherapy as adjuvant treatment for experimental hepatic cancer. Annals Of Surgery. 2002;236(3):337–42. doi: 10.1097/00000658-200209000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong RJ, Chan MK, Yu Z, Ghossein RA, Ngai I, Adusumilli PS, et al. Angiogenesis inhibition by an oncolytic herpes virus expressing interleukin 12. Clinical Cancer Research. 2004;10(13):4509–16. doi: 10.1158/1078-0432.CCR-04-0081. [DOI] [PubMed] [Google Scholar]
- 11.Kucharczuk JC, Randazzo B, Chang MY, Amin KM, Elshami AA, Sterman DH, et al. Use of a “replication-restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Research. 1997;57(3):466–71. [PubMed] [Google Scholar]
- 12.Wong RJ, Joe JK, Kim SH, Shah JP, Horsburgh B, Fong Y. Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Human Gene Therapy. 2002;13(10):1213–23. doi: 10.1089/104303402320138998. [DOI] [PubMed] [Google Scholar]
- 13.Chou J, Roizman B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(8):3266–70. doi: 10.1073/pnas.89.8.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou J, Roizman B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(12):5247–51. doi: 10.1073/pnas.91.12.5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hollander MC, Zhan Q, Bae I, Fornace AJ., Jr. Mammalian GADD34, an apoptosis- and DNA damage-inducible gene. Journal Of Biological Chemistry. 1997;272(21):13731–7. doi: 10.1074/jbc.272.21.13731. [DOI] [PubMed] [Google Scholar]
- 16.Advani SJ, Chung SM, Yan SY, Gillespie GY, Markert JM, Whitley RJ, et al. Replication-competent, nonneuroinvasive genetically engineered herpes virus is highly effective in the treatment of therapy-resistant experimental human tumors. Cancer Res. 1999;59(9):2055–8. [PubMed] [Google Scholar]
- 17.Bennett JJ, Adusumilli P, Petrowsky H, Burt BM, Roberts G, Delman KA, et al. Up-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin C and a gamma134.5 deleted oncolytic herpes virus (G207) FASEB J. 2004;18(9):1001–3. doi: 10.1096/fj.02-1080fje. [DOI] [PubMed] [Google Scholar]
- 18.Mullerad M, Bochner BH, Adusumilli PS, Bhargava A, Kikuchi E, Hui-Ni C, et al. Herpes simplex virus based gene therapy enhances the efficacy of mitomycin C for the treatment of human bladder transitional cell carcinoma. Journal Of Urology. 2005;174(2):741–6. doi: 10.1097/01.ju.0000164730.38431.5c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou TC. The median-effect principle and the combination index for quantitation of synergism and antagonism. In: Chou TCR, editor. Synergism and antagonism in chemotherapy. Academic Press; New York: 1991. pp. 61–102. [Google Scholar]
- 20.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in Enzyme Regulation. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 21.Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. European Journal of Biochemistry. 1981;115(1):207–16. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- 22.Chou J, Chou T. Manual and Software. Biosoft; Cambridge UK: 1987. Dose-effect analysis with microcomputers: quantitation of ED50, LD50, synergism, antagonism, low-dose risk, receptor-ligand binding and enzyme kinetics. [Google Scholar]
- 23.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol. 2003;21(14):2636–44. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 24.Krarup-Hansen A, Hansen HH. Chemotherapy in malignant mesothelioma: a review. Cancer Chemotherapy & Pharmacology. 1991;28(5):319–30. doi: 10.1007/BF00685684. [DOI] [PubMed] [Google Scholar]
- 25.Boulikas T, Vougiouka M.Cisplatin and platinum drugs at the molecular level Oncology Reports 20031061663–82.Review [PubMed] [Google Scholar]
- 26.Stanziale SF, Petrowsky H, Adusumilli PS, Ben Porat L, Gonen M, Fong Y. Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clinical Cancer Research. 2004;10(9):3225–32. doi: 10.1158/1078-0432.ccr-1083-3. [DOI] [PubMed] [Google Scholar]
- 27.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22(47):7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 28.Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9(12):967–78. doi: 10.1038/sj.cgt.7700537. [DOI] [PubMed] [Google Scholar]
- 29.He B, Chou J, Liebermann DA, Hoffman B, Roizman B. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the gamma(1)34.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. Journal of Virology. 1996;70(1):84–90. doi: 10.1128/jvi.70.1.84-90.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullen JT, Kasuya H, Yoon SS, Carroll NM, Pawlik TM, Chandrasekhar S, et al. Regulation of herpes simplex virus 1 replication using tumor-associated promoters. Annals Of Surgery. 2002;236(4):502–12. doi: 10.1097/00000658-200210000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coukos G, Makrigiannakis A, Kang EH, Rubin SC, Albelda SM, Molnar-Kimber KL. Oncolytic herpes simplex virus-1 lacking ICP34.5 induces p53-independent death and is efficacious against chemotherapy-resistant ovarian cancer. Clin Cancer Res. 2000;6(8):3342–53. [PubMed] [Google Scholar]
- 32.Chahlavi A, Todo T, Martuza RL, Rabkin SD. Replication-competent herpes simplex virus vector G207 and cisplatin combination therapy for head and neck squamous cell carcinoma. Neoplasia. 1999;1(2):162–9. doi: 10.1038/sj.neo.7900016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber KL. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum Gene Ther. 1999;10(18):3013–29. doi: 10.1089/10430349950016410. [DOI] [PubMed] [Google Scholar]
- 34.Snyder MB, Saravolatz LD, Markowitz N, Pohlod D, Taylor RC, Ward SG. The in-vitro and in-vivo efficacy of cisplatin and analogues in the treatment of herpes simplex virus-II infections. Journal of Antimicrobial Chemotherapy. 1987;19(6):815–22. doi: 10.1093/jac/19.6.815. [DOI] [PubMed] [Google Scholar]