

Abstract
Over the last decade a variety of MS measurements, such as H/D exchange, collision cross sections, and electron capture dissociation (ECD), have been used to characterize protein folding in the gas phase, in the absence of solvent. To the extensive data already available on ubiquitin, here photofragmentation of its ECD-reduced (M + nH)(n−1)+• ions shows that only the 6+ to 9+, not the 10+ to 13+ ions, have tertiary noncovalent bonding; this is indicated as hydrogen bonding by the 3,050–3,775 cm−1 photofragment spectrum. ECD spectra and H/D exchange of the 13+ ions are consistent with an all α-helical secondary structure, with the 11+ and 10+ ions sufficiently destabilized to denature small bend regions near the helix termini. In the 8+ and 9+ ions these terminal helical regions are folded over to be antiparallel and noncovalently bonded to part of the central helix, whereas this overlap is extended in the 7+, 6+, and, presumably, 5+ ions to form a highly stable three-helix bundle. Thermal denaturing of the 7+ to 9+ conformers both peels and slides back the outer helices from the central one, but for the 6+ conformer, this instead extends the protein ends away to shrink the three-helix bundle. Thus removal of H2O from a native protein negates hydrophobic interactions, preferentially stabilizes the α-helical secondary structure with direct solvation of additional protons, and increases tertiary interhelix dipole-dipole and hydrogen bonding.
For many decades, understanding the in vivo folding of a protein into its biologically active form has attracted extensive research attention, both experimental (1–4) and theoretical (5–7). To define better the critical effects of aqueous solvation, a novel approach has been to study protein conformations in the gas phase. H/D exchange (8–12) and collision cross-section measurements (13–19) using MS have shown clearly that proteins such as cytochrome c (12.3 kDa) and ubiquitin (8.6 kDa) have a multiplicity of stable conformers of yet undefined structures in the gas phase, in sharp contrast to the single native conformer in solution whose structure and folding are well established (2–4). A new method, electron capture dissociation (ECD) (20–28), has recently provided structural details of the tertiary noncovalent bonding of gaseous cytochrome c (26) and ubiquitin (27), including thermal denaturation and refolding kinetics as a function of the number of protons (“acidity”) on the gaseous molecular ion. We report here photofragment spectra (3,050–3,775 cm−1) and further ECD and H/D exchange data that now lead to postulated conformational structures for the 5+ to 13+ ubiquitin ions in the gas phase. Ubiquitin has recently been recognized as a “superhero” of cell biology (29, 30).
For conformers in solution versus those formed in the gas phase by electrospray ionization, removal of water negates hydrophobic bonding while enhancing hydrogen bonding; however, reversible folding and unfolding between stable gaseous conformers is still observable (9, 11, 14, 19, 26, 27). In solution, basic side chains on protonation are solvated into the surrounding water to denature the protein; in the gas phase, these protonated sites are solvated internally, decreasing intramolecular H/D exchange through backbone protection by the newly protonated side chain (11, 12, 26, 27) (Fig. 1). Collisional cross-section measurements of 5+ to 15+ ubiquitin ions define multiple conformer types whose cross sections increase with increasing charge (13, 14, 16, 19). For simple gaseous peptide ions, conformational structures have been proposed from theoretical calculations of their expected cross-section values (15, 17), such as the α-helical structure of (Ala-35 + 3H)3+ (15).
Fig 1.

Number of nonproton deuterium atoms exchangeable in 5+ to 13+ ubiquitin ions: ▴, ref. 8; ▾, this study; □ and 5+, 6+ ions (image width indicates abundance), ref. 12.
Fast (<10−12 s) nonergodic ECD produces backbone dissociation of covalent bonds (Eq. 1) without significant dissociation of tertiary noncovalent bonds; a c, z⋅ product
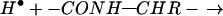 |
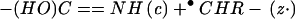 |
pair still held together by tertiary bonding exhibits the unchanged mass of the reduced molecular ion (M + nH)(n−1)+• (26, 27). Thus specific thermal denaturations of a charge-selected protein ion are indicated by the new ECD spectral cleavages that appear with increasing temperature. Destroying the noncovalent bonding of (M + nH)(n−1)+• with collisionally activated dissociation (31, 32) or IR multiphoton dissociation (IRMPD) (33) then releases and identifies the c, z⋅ products (26, 27). Such energetic dissociation of even-electron protein ions produces complementary b and y product ions (Eq. 2).
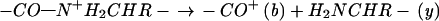 |
In contrast to tertiary bonding, secondary noncovalent bonding can enhance, not restrict, ECD; for the 13+ ion of ubiquitin, ECD produces extensive cleavages, yet there is persuasive evidence of strong secondary stabilization of these ions (16, 34), such as by an α-helix (27). In 60% MeOH solution at pH 2, the C-terminal half of the ubiquitin native state is converted to an α-helix (the A state) (2).
The nature of the noncovalent bonding is investigated here by using photofragmentation of (M + nH)(n−1)+• ions; such studies with tunable (3,050–3,775 cm−1) IR lasers have not been applied previously to peptides or proteins. Evidence from several such methods is used for the first prediction of gaseous conformational structures (Fig. 2).
Fig 2.

Proposed conformational structures for gaseous ubiquitin ions. Black bars, α-helices; shaded areas, H+ bound basic residues; dashed loops, possible salt bridges.
Materials and Methods
The 6 T Fourier transform ion cyclotron resonance mass spectrometer (Finnigan-MAT, San Jose, CA) has been described (11, 24, 26, 27). Bovine ubiquitin (Sigma) was electrosprayed from 10 μM solutions (90:8:2 H2O/CH3OH/CH3COOH for 7+ and 8+ ions; 83:15:2 for 9+ and 10+ ions; 49:49:2 for 10+ to 13+ ions) and trapped in the ion cell with N2 gas (10−6 Torr, gauge). After a 20-s thermalization delay, ions of interest were isolated by consecutive stored waveform inverse Fourier transform waveforms, in broadband and narrowband mode. ECD used electrons of ≈0.20–0.35 μA from a Rh ribbon filament for 0.6–2.0 s.
IRMPD used a 25-W continuous CO2 laser (Synrad, Mukilteo, WA, 10.6 μm) for 0.1–2 s. Photofragment spectroscopy used a single-stage optical parametric oscillator laser (IR OPO 2732, OPOtek, Carlsbad, CA) of unseeded cavity design with bulk potassium titanyl arsenate crystal as the nonlinear medium, pumped by 1,064 nm Nd:YAG laser (Brilliant, Quantel, Bozeman, MT), duration ≈4 ns, line width 3–4 cm−1, output power of 7–10 mJ per pulse at 10 Hz measured by using a Molectron (Sunnyvale, CA) PM 30 probe, and irradiation time controlled by a fast-response mechanical shutter (UNIBLITZ, Vincent Associate, Rochester, NY). Each point in a spectrum represents the depletion by 170 laser pulses, averaged over 20 measurements, corrected for variation in photon intensity as a function of wavelength. All mass spectra were obtained in broadband acquisition mode and interpreted by the automated thrash program (35).
Results and Discussion
α-Helicity Induced by Extensive Protonation.
High acidity (pH 2) in 60% methanol introduces an α-helix into residues 39–72 of ubiquitin (the A state), as found by Ernst and coworkers (2). For the gaseous 13+ ubiquitin ions whose charge corresponds to protonation at the 12 basic residues (Lys, Arg, His) plus the N-terminal amino group (Fig. 3), we have reported substantial evidence to support a complete α-helical secondary structure (25–27). The 13+ ions are stable to 125°C storage, to 0.25 s IRMPD (10.6 μm), and to 6 eV electron bombardment that cause dissociation of lower charge state ions. Only a highly compact conformer could restrict H/D exchange mainly to the 13 exposed protons (Fig. 1). The amide-H to carbonyl hydrogen bonding that stabilizes the α-helix in solution should be enhanced both by no competition from the aqueous solvent and by protonation from the solvated side chain (9, 11, 27). This H+ to carbonyl solvation also positions the proton for ECD (to form H⋅ in Eq. 1), with an α-helix favoring such intermediates on the first four residues on the N-terminal side of the protonated basic residue as well as itself (27), as shown in Fig. 3 and at 155°C in Fig. 4. Molecular modeling calculations on peptides reported by Jarrold and coworkers (17) and Clemmer and colleagues (15, 19) also provide persuasive evidence for gaseous α-helical conformers stabilized on the N-terminal side of protonated basic residues.
Fig 3.

ECD spectra of ubiquitin ions at 25°C. Vertical bars: black segment, c ions; open, z⋅ ions; gray filled and open, c and z⋅, respectively, from IRMPD of (M + nH)(n-1)+• ions. Vertical lines: basic residues of highest protonation probability; estimated intrinsic gas phase basicities of an amino acid in a protein: Arg, 251; His, 237; Lys, 237; and Pro, 227 kcal/mol (38). Some of these data are reprinted with permission from figure 3 of ref. 28 [Copyright (2002) American Chemical Society].
Fig 4.

ECD spectra of 6+ to 9+ ions at 155°C. Dashed and dotted vertical lines: higher and lower protonation probability, respectively.
Effect of Decreased Protonation.
Cassady and Carr (10) first showed that 12+ ions are of two conformer types. The ECD spectrum of the 12+ ions corresponds to a 1:1 mixture of the 13+ and 11+ ECD spectra (27), which show substantial differences in the regions of residues 5–11 and 56–68 (Fig. 3). Thus one type of 12+ ion conformer should also represent an α-helix that, like the 13+ ion, yields ECD cleavages next to protonated sites under the assumption of multiple protonated site isomers (16) or facile proton transfer to the vacant site (32). This removal of a proton allows the exchange of an additional 5–10 H atoms with D2O (Fig. 1), consistent with less stabilization of the α-helix.
Nonhelical Sites from Lowered Proton Density.
For the 13+ ions, cleavages on the N- and C-terminal sides of Lys-6 produce c ions of 1+ and 2+ charges, respectively, indicating that Lys-6 is charged. However, those for the 11+ ions produce 1+ and 1+ charges, indicating no protonation between these cleavage sites (27). Similarly, reduced ECD cleavages at bonds 4–9 and bonds 63–68 (Fig. 3) indicate that a major fraction of the 11+ ions are formed by removing the Lys-6 and His-68 protons from the helical 13+ ubiquitin ions, consistent with calculations of protonation frequency (36, 37). The loss of α-helicity in these regions, not just the charge removal, must negate ECD, as the same ECD cleavages are also absent in the corresponding 12+ conformer type that has lost only one H+. These 12+ ions exchange ≈23 more hydrogens, and at a 4-fold faster initial rate (data not shown), than the all-helix type 12+ ions (Fig. 1), in close correspondence to complete H/D exchange of the 10 amide backbone and side-chain H atoms in each of these six residue “bend” regions near Lys-6 and His-68 (Fig. 2). Five conformers of gaseous cytochrome c characterized by H/D exchange show 10 ± 1 D differences (11), suggesting similar bend regions. For the 9+ and 11+ ions, but not the 12+, using CH3OD instead of D2O gave H/D values corresponding to exchange at only a single bend (Fig. 1). For (Ala-36 + 3H)3+ peptide ions, collision cross-section data and molecular modeling calculations indicate that shifting proton density allows conversion of a portion of an extended helical conformation to a “folded hinged helix-coil” state (15). The unusually high values of activation energy (Ea), 1.6 eV, and Arrhenius pre-exponential factor (A), 1016-1017 s−1, found by Williams and coworkers (34) for blackbody IR dissociation of 10+ and 11+ ions is consistent with a strong helical bonding that also places an incipient proton near the amide N for the Eq. 2 mechanism (38).
Helicity loss has less influence on collision cross section, with values of 11+, 1,798, 1,814, 1,829, and 1,849; 12+, 1,875, 1,888, 1,900, and 1,929; and 13+, 1,942 and 1,970 Å2 (16). The four cross-section values for the 11+ ubiquitin ions suggest conformers with other bend locations. After removal of the His-68 proton from the 13+ ions, the next most favored loss is the H+ at Lys-6 or, somewhat less, at Pro-19; this would form a second conformer bent instead at His-68 and Pro-19 (Fig. 3 shows such evidence in ECD of the 10+, 11+ ions for reduced cleavages at bonds 13–18). Similarly, a bend could be formed by H+ loss at Arg-72 instead of His-68. Thus each of these four 11+ conformers (Fig. 2) would exchange ≈23 additional D atoms.
Two-Bend Three-Helix Conformers.
Our 1993 data (8) for complete H/D exchange (Fig. 1) show values for charge states 7+ to 10+ that correlate linearly (lower line) with these 11+ and 12+ values, evidence that the 7+ to 10+ ions also can have a helical conformation interrupted by two bends. Removal of one charge increases their exchange by ≈4 D atoms, whereas a value of ≈2 D per charge was found for five gaseous conformers of cytochrome c, presumably from the change in side-chain solvation (11). Although Marshall and coworkers (12) find H/D exchange values in general agreement with each of our 1993 values (8), for the 6+ to 10+ ions they also find a second value corresponding to a more dominant conformer that exchanges an additional ≈20 D atoms (Fig. 1), suggesting four-bend conformers. H/D exchange conditions causing isomerization between our four postulated conformers, which in total have four bends, would give values on the upper correlation line (Fig. 1), whereas the broad distributions for the 5+ and 6+ ions (12) are consistent with even slower conformer isomerizations during their exchange. Under our conditions, however, collisionally activated dissociation of 9+, 11+, and 13+ ions after 60 min of D2O exchange that caused 25% dissociation gave recovered molecular ions that showed negligible H/D exchange in an additional 60 min exposure to D2O (Fig. 1), showing no conformer isomerization or H/D scrambling (11, 39).
Tertiary Noncovalent Bonding.
The (M + nH)(n−1)+• molecular ions in ECD spectra that had captured an electron without change in mass were isolated in the Fourier transform MS and subjected to 10.6 μm IRMPD. Photodissociation of n = 10–13 differs greatly from that of n = 6–9. For example, n = 12 (Fig. 5) only produces b, y ions (Eq. 2) and requires far more fluence than n = 6–9 that instead produce c, z⋅ products (27). The n = 12 ions apparently (25) undergo initial H⋅ loss followed by the higher energy dissociation expected for the covalent bonds of the resulting even-electron (M + 11H)11+ ions; direct IRMPD of (M + 11H)11+ ions yields a similar spectrum. However, IRMPD (10.6 μm) of (M + nH)(n−1)+• ions for n = 6–9 instead gave c,z⋅ products (gray vertical bars, Fig. 3) with far less laser irradiation (Fig. 5), consistent with noncovalent bond dissociation. Thus these c, z⋅ products (Fig. 3) pinpoint the backbone cleavage that occurred during ECD, but for which tertiary noncovalent bonding across that region prevented product separation.
Fig 5.

Dissociation of (M + 7H)6+• and (M + 12H)11+• ions versus laser irradiation times using 3.0- and 10.6-μm photons, with the 3.0-μm data corrected to be energy equivalent.
Photofragmentation of (M + 7H)6+•.
To characterize these noncovalent bonds, the effect of wavelength on depletion was measured for (M + 7H)6+• and (M + 8H)7+• ubiquitin ions (Fig. 6). The spectrum shows a broad absorption envelope beginning before 3,100 cm−1, peaking at ≈3,350 cm−1, and approaching baseline at ≈3,500 cm−1. Denaturation at 75°C is indicated by reduced photodissociation up to ≈3,200 cm−1, whereas the spectrum of the (M + 8H)7+• ions at 27°C (of poor signal/noise) is also reduced from 3,200 to 3,400 cm−1. No peptide or protein reference spectra for proton-bound species appear to have been measured previously. Our preliminary data (Fig. 6) for the proton bound dimers of (Ac-Lys-OMe)2 H+ and (Ac-Asp-OH)2 H+ show mainly narrow absorptions at ≈3,450 cm−1 (N-H?) and 3,600 cm−1 (COO-H?), but their heterodimer does show some absorptions below 3,400 cm−1. These are greatly increased in their (Ac-Lys-OMe)2(Ac-Asp-OH)H+ trimer in which salt bridge stabilization is possible. Hydrogen-bonded stretching frequencies are usually several hundred wavenumbers less than the corresponding free stretching frequencies (40, 41), consistent with hydrogen bonding as a major factor in the tertiary structure of the 6+ to 9+ ions. This photodissociation, apparently a multiphoton process, requires further study.
Fig 6.

Photofragment spectra of (M + 7H)6+•, (M + 8H)7+•, and reference ions.
Noncovalently Bound Antiparallel Helices.
As argued above, 7+ through 9+ ions are also two bend conformers stabilized by tertiary noncovalent bonding. This is most probably between the folded-over α-helical regions; the further proton removal has reduced their Coulombic repulsion sufficiently to allow antiparallel dipole-dipole stabilization (1) and tertiary bonding rather than to disrupt (“bend”) further regions of the α-helical structure. The strongest hydrogen bonds should involve a basic-site proton of one helix bonded to an adjacent helix at a nonprotonated Arg, His, or Lys side chain, or at an amide carbonyl, although the latter should be less favored sterically; a salt bridge of an acidic residue between two basic residues can form even stronger proton binding (42). Lowering the number of charges will increase a site's proton affinity (36, 37, 42), strengthening this tertiary bonding. To rationalize the ECD data, Fig. 2 shows possible two-bend conformers with the proton bound between basic residues; salt bridges are also indicated, but could be sterically unfavorable.
Compared with the 11+ ECD spectrum, that of the 9+ ions (Fig. 3) has lost most cleavages induced by Lys-48 and Arg-54 (Eq. 1), suggesting that these are H+-bonded (Fig. 2); consistent with this finding, the IRMPD spectrum of (M + 9H)8+• ions shows c48. Its z⋅54 and the decreased cleavages at bonds 15–20 suggest interhelix bonding from Pro-19 to adjacent carbonyls, or just that the Pro-19 bend is now more favored than it is in the 11+ conformers. The c71 and c75 (Fig. 3) from the (M + 9H)8+• ions suggest an additional conformer with H+ noncovalent binding from Lys-29 to Arg-74. Dissociation of the 9+ and 8+ ions exhibit lower Ea values of 1.2 eV (34), consistent with the substantially reduced proton stabilization of the α-helical regions.
For ECD of (M + 8H)8+, the diminished 12–29 and 43–67 ECD cleavages suggest the salt bridge Pro-19/Glu-18/Lys-29 and the noncovalent bonds Lys-48 to His-68 and Arg-54 to Lys-63 (Fig. 2). The z⋅70,71,73 and c71 ions in the IRMPD (M + 8H)7+• spectrum indicate that additional terminally bound conformers with Lys-6 to Arg-42 or Arg-42 to Arg-74 can be present. ECD spectra of 9+ ions stored for 10 min showed substantial additional folding (27) suggestive of such 8+ conformer features (27).
In the 7+ ions, the prominent ECD cleavages at bonds 10–72 have mostly disappeared with the further reduction in Coulombic repulsion. The Fig. 2 overlap between the three helical regions would release products from cleavages at bonds 38 and 39, but these would not appear for a 7+ conformer that has additional salt bridge stabilization from Lys-6.
For the 6+ ions, nearly all ECD cleavages are eliminated, suggesting overlap extension to form an unusually stable three-helix bundle (1). The importance of the multiple salt bridging possibilities shown will depend heavily on steric factors that as yet have not been considered. This 6+ conformer should give little H/D exchange; slow isomerization between the more denatured structures could lead to the near continuum of reported (12) H/D data (Fig. 1). Dissociations (Eq. 2) of 6+ and 7+ ions have Ea values of only 0.9–1.0 eV (34), consistent with low proton stabilization of the helices. ECD achieves no cleavages of the 5+ ubiquitin ions, and collisionally activated dissociation or IRMPD (Ea = 1.2 eV) (34) causes mainly H2O and NH3 loss (31, 32, 34), indicating further strengthening of the noncovalent binding between the multiple helical regions.
Thermal Unfolding of 6+ to 9+ Ions.
Similar to the addition of protons, heating the 6+ to 9+ ions also increases their ECD fragmentation (155°C, Fig. 4). The charge states and masses of their fragment ions also suggest conversion to more helical structures (see above) with destruction of the noncovalent tertiary structure.
However, “denaturation” of the 6+ ions by heating differs from that by proton addition. Heating reduces the no-cleavage region to bonds 21–58 at 115°C and 32–50 at 155°C (Fig. 4) and 175°C (27). This denaturation could occur (Fig. 2) by the top and bottom helices sliding outward, shortening the three-helix bundle. At 155°C, this conformation could convert to a single bend structure.
In contrast, thermal denaturation of the 7+ ions peels back the terminal helices (27), resembling proton addition. At 55°C, ECD cleavages are mainly missing from bonds 18–29 (denaturing Lys-11 to Lys-33 in Fig. 2) and 40–57 (bonding Arg-42 to Arg-54). Although the Glu-16 to Lys-27, 29 bonding is gone by 95°C, at 155°C (Fig. 4) extensive Arg-42 to Lys-48 bonding is preserved in the 7+ to 9+ ions. After an 0.25-s IR laser pulse unfolded the 7+ ions, refolding at bonds such as 14, 24, 51, and 54 showed nearly identical rate constants (27), so that these were apparently concerted, but refolding rates at other sites indicate additional conformers.
Heating the 8+ ions adds ECD cleavages that indicate that Pro-19 to Lys-29 is broken at ≈75°C, and that Arg-42 to His-68 has become Arg-42 to Arg-54 at 55°C. For the 155°C 9+ ECD spectrum, product abundances vary much less, indicating a decreased importance of both the secondary and tertiary bonding. Heating 11+ ions to 100°C gives some increase in cleavages at bonds 6 and 65, but not at 8 and 68, indicating only partial restoration of helicity in the 11+ ion bend regions (Fig. 2).
The 7+ ions unfolding below 100°C give a value of ΔH = 32 ± 2 kJ/mol (27), in agreement with ΔH = 30 ± 2 kJ/mol for the 6+ ion unfolding that also changes the triple noncovalently bound helix bundle into two doubles. At ≈100°C the ΔH(unfolding) for the 7+ ions becomes much lower, consistent with the denaturing of only two adjacent helices and similar to the values for the 8+ and 9+ ions, 11 and 6 ± 2 kJ/mol, respectively (27), that are postulated to have two and one adjacent helices.
Thermal unfolding of ubiquitin ions starting below room temperature may be indicated by the recent data of Clemmer and coworkers (19). Ions stored for 20 ms showed only compact conformers for 6+ and 7+ ions and partially folded for 8+ ions. “Solvent evaporation during the electrospray ionization process should cool the internal temperature of these ions” (19) can explain why our 7+ and 8+ ions (Fig. 2) are more unfolded. However, storage of their cooled 8+ ions for 35 ms gave dominant formation of the extended conformer, whereas 40 ms to 30 s storage of 7+ ions gave far more extensive denaturation than indicated by our Figs. 2 and 3. Collisions in their trap and drift tube can produce somewhat higher ion temperatures; substantially increased ion injection voltages produce only extended conformers (13, 18).
Conclusions
Removal of the aqueous solvent from the protein ubiquitin gives an extensive variety of conformational structures, in contrast to the singular native form in solution. The basic anhydrous structure appears to be an α-helix whose stability is enhanced by protonation, and whose extent is also enhanced by thermal denaturation of the tertiary structure. The 13+ ions, in which essentially all basic sites are protonated, are fully α-helical at 25°C, which is nearly true for the 7+ to 13+ ions at 155°C. At 25°C for the 10+ and 11+ ions, and half of the 12+ ions, the α-helix is broken near the termini to yield conformers with combinations of two different bend locations at each end.
Tertiary noncovalent bonding is found only for ions of charge state 9+ and lower. Preliminary 3,050–3,775 cm−1 photodissociation studies of (M + 7H)6+• and (M + 8H)7+• ions are consistent with hydrogen bonding, which increases in strength with decreased charge by increasing the proton affinity of basic residues. In the 9+ and 8+ ions, apparently one or both terminal α-helical regions can fold over for antiparallel dipole interaction with the central helix, whereas for the 7+ and 6+ ions this overlap increases to form a highly stable three-helix bundle. In heating these 6+ ions, the bundle actually becomes shorter by increasing the length of the denatured terminal α-helical regions. This finding is opposite to the effect of increased protonation or heating the 7+ to 9+ ions.
To the extent that these structural predictions (Fig. 2) are accurate and applicable to other gaseous proteins, this finding indicates that gas-phase protein folding is significantly different from that in solution. This finding underscores precautions (9, 11) that characterization of solution conformers from their gaseous data requires great care. Gaseous noncovalent structure is also far simpler; the α-helix appears to be the dominant secondary structure, and side-by-side helices stabilized by dipole-dipole and proton-bound hydrogen bonding form the common tertiary structure. This finding also means that many conformers of nearly identical stabilities will be possible by minor overlap repositioning, with conformer structures affected differently by changes in charge state versus temperature (e.g., the 6+ ions). Theoretical calculations are necessary to support these general results and provide further details.
Acknowledgments
We thank Floyd Davis, Ron Elber, Mariam ElNaggar, Harold Hwang, Young Kee Kang, Sang-Youn Park, Harold Scheraga, Charles Wilcox, Evan Williams, and Roman Zubarev for helpful discussions and David Clemmer for preprints of unpublished research. The National Institutes of Health (Grant GM16609 to F.W.M.) and Austrian Science Fund (FWF Project P15767 to K.B.) provided generous financial support.
Abbreviations
ECD, electron capture dissociation
IRMPD, IR multiphoton dissociation
References
- 1.Chou K.-C., Maggiora, G. M. & Scheraga, H. A. (1992) Proc. Natl. Acad. Sci. USA 89, 7315-7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brutscher B., Bruschweiler, R. & Ernst, R. R. (1997) Biochemistry 36, 13043-13053. [DOI] [PubMed] [Google Scholar]
- 3.Rumbley J., Hoang, L., Mayne, L. & Englander, S. W. (2000) Proc. Natl. Acad. Sci. USA 98, 105-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jourdan M. & Searle, M. S. (2001) Biochemistry 40, 10317-10325. [DOI] [PubMed] [Google Scholar]
- 5.Bryngelson J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. (1995) Proteins Struct. Funct. Genet. 21, 167-195. [DOI] [PubMed] [Google Scholar]
- 6.Dill D. A. (1999) Protein Sci. 8, 1166-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinner A. R., Sali, A., Smith, L. J., Dobson, C. M. & Karplus, M. (2000) Trends Biochem. Sci. 25, 331-339. [DOI] [PubMed] [Google Scholar]
- 8.Suckau D., Shi, Y., Beu, S. C., Senko, M. W., Quinn, J. P., Wampler, F. M., III & McLafferty, F. W. (1993) Proc. Natl. Acad. Sci. USA 90, 790-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood T. D., Chorush, R. A., Wampler, F., III, Little, D. P., O'Connor, P. B. & McLafferty, F. W. (1995) Proc. Natl. Acad. Sci. USA 92, 2451-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassady C. J. & Carr, S. R. (1996) J. Mass Spectrom. 31, 247-254. [DOI] [PubMed] [Google Scholar]
- 11.McLafferty F. W., Guan, Z., Haupts, U., Wood, T. D. & Kelleher, N. L. (1998) J. Am. Chem. Soc. 120, 4732-4740. [Google Scholar]
- 12.Freitas M. A., Hendrickson, C. L., Emmett, M. R. & Marshall, A. G. (1999) Int. J. Mass Spectrom. 185/186/187, 565-575. [Google Scholar]
- 13.Valentine S. J., Counterman, A. E. & Clemmer, D. E. (1997) J. Am. Soc. Mass Spectrom. 8, 954-961. [DOI] [PubMed] [Google Scholar]
- 14.Li J., Taraszka, J. A., Counterman, A. E. & Clemmer, D. E. (1999) Int. J. Mass Spectrom. 185/186/187, 37-47. [Google Scholar]
- 15.Counterman A. E. & Clemmer, D. E. (2001) J. Am. Chem. Soc. 123, 1490-1498. [DOI] [PubMed] [Google Scholar]
- 16.Purves R. W., Barnett, D. A., Ells, B. & Guevremont, R. (2001) J. Am. Soc. Mass Spectrom. 12, 894-901. [DOI] [PubMed] [Google Scholar]
- 17.Kinnear B. S., Hartings, M. R. & Jarrold, M. F. (2002) J. Am. Chem. Soc. 124, 4422-4431. [DOI] [PubMed] [Google Scholar]
- 18.Wyttenbach T., Kemper, P. R. & Bowers, M. T. (2002) Int. J. Mass Spectrom. 212, 13-23. [Google Scholar]
- 19.Myung S., Badman, E. R., Lee, Y. J. & Clemmer, D. E. (2002) J. Phys. Chem. A 106, 9976-9982. [Google Scholar]
- 20.Zubarev R. A., Kelleher, N. L. & McLafferty, F. W. (1998) J. Am. Chem. Soc. 120, 3265-3266. [Google Scholar]
- 21.Zubarev R. A., Kruger, N. A., Fridriksson, E. K., Lewis, M. A., Horn, D. M., Carpenter, B. K. & McLafferty, F. W. (1999) J. Am. Chem. Soc. 121, 2857-2862. [Google Scholar]
- 22.Zubarev R. A., Horn, D. M., Fridriksson, E. K., Kelleher, N. L., Kruger, N. A., Lewis, M. A., Carpenter, B. K. & McLafferty, F. W. (2000) Anal. Chem. 72, 563-573. [DOI] [PubMed] [Google Scholar]
- 23.Horn D. M., Ge, Y. & McLafferty, F. W. (2000) Anal. Chem. 72, 4778-4784. [DOI] [PubMed] [Google Scholar]
- 24.Sze S. K., Ge, Y., Oh, H.-B. & McLafferty, F. W. (2002) Proc. Natl. Acad. Sci. USA 99, 1774-1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breuker K., Oh, H.-B., Cerda, B. A., Horn, D. M. & McLafferty, F. W. (2002) Eur. J. Mass Spectrom. 8, 177-180. [Google Scholar]
- 26.Horn D. M., Breuker, K., Frank, A. J. & McLafferty, F. W. (2001) J. Am. Chem. Soc. 123, 9792-9799. [DOI] [PubMed] [Google Scholar]
- 27.Breuker K., Oh, H.-B., Horn, D. M., Cerda, B. A. & McLafferty, F. W. (2002) J. Am. Chem. Soc. 124, 6407-6420. [DOI] [PubMed] [Google Scholar]
- 28.Sze, S. K., Ge, Y., Oh, H.-B. & McLafferty, F. W. (2002) Anal. Chem., in press. [DOI] [PubMed]
- 29.Conaway R. C., Brower, C. S. & Conaway, J. W. (2002) Science 296, 1254-1258. [DOI] [PubMed] [Google Scholar]
- 30.Marx J. (2002) Science 297, 1792-1794. [DOI] [PubMed] [Google Scholar]
- 31.Senko M. W., Speir, J. P. & McLafferty, F. W. (1994) Anal. Chem. 66, 2801-2808. [DOI] [PubMed] [Google Scholar]
- 32.Reid G. E., Wu, J., Chrisman, P. A., Wells, J. M. & McLuckey, S. A. (2001) Anal. Chem. 73, 3274-3281. [DOI] [PubMed] [Google Scholar]
- 33.Little D. P., Speir, J. P., Senko, M. W., O'Connor, P. B. & McLafferty, F. W. (1994) Anal. Chem. 66, 2809-2815. [DOI] [PubMed] [Google Scholar]
- 34.Jockusch R. A., Schnier, P. D., Price, W. D., Strittmatter, E. F., Demerev, D. A. & Williams, E. R. (1997) Anal. Chem. 69, 1119-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horn D. M., Zubarev, R. A. & McLafferty, F. W. (2000) J. Am. Soc. Mass Spectrom. 11, 320-332. [DOI] [PubMed] [Google Scholar]
- 36.Schnier P. D., Gross, D. S. & Williams, E. R. (1995) J. Am. Soc. Mass Spectrom. 6, 1086-1097. [DOI] [PubMed] [Google Scholar]
- 37.Peschke M., Blades, A. & Kebarle, P. (2002) J. Am. Chem. Soc. 124, 11519-11530. [DOI] [PubMed] [Google Scholar]
- 38.Yu W., Vath, J. E., Huberty, M. C. & Martin, S. A. (1993) Anal. Chem. 65, 3015-3023. [DOI] [PubMed] [Google Scholar]
- 39.Demmers J. A. A., Rijkers, D. T. S., Haverkamp, J., Killian, J. A. & Heck, A. J. R. (2002) J. Am. Chem. Soc. 124, 11191-11198. [DOI] [PubMed] [Google Scholar]
- 40.Wu C. C., Jiang, J. C., Boo, D. W., Lin, S. H., Lee, Y. T. & Chang, H. C. (2000) J. Chem. Phys. 112, 176-188. [Google Scholar]
- 41.Wu C. C., Jiang, J. C., Hahndorf, I., Chaudhuri, C., Lee, Y. T. & Chang, H. C. (2000) J. Phys. Chem. A 104, 9556-9565. [Google Scholar]
- 42.Price W. D., Schnier, P. D. & Williams, E. R. (1997) J. Phys. Chem. B 101, 664-673. [DOI] [PMC free article] [PubMed] [Google Scholar]