

Abstract
The ankyrin repeat is one of the most common, modular, protein–protein interaction motifs in nature. To understand the structural determinants of this family of proteins and extract the consensus information that defines the architecture of this motif, we have designed a series of idealized ankyrin repeat proteins containing one, two, three, or four repeats by using statistical analysis of ≈4,000 ankyrin repeat sequences from the PFAM database. Biophysical and x-ray crystallographic studies of the three and four repeat constructs (3ANK and 4ANK) to 1.26 and 1.5 Å resolution, respectively, demonstrate that these proteins are well-folded, monomeric, display high thermostability, and adopt a very regular, tightly packed ankyrin repeat fold. Mapping the degree of amino acid conservation at each position on the 4ANK structure shows that most nonconserved residues are clustered on the surface of the molecule that has been designated as the binding site in naturally occurring ankyrin repeat proteins. Thus, the consensus amino acid sequence contains all information required to define the ankyrin repeat fold. Our results suggest that statistical analysis and the consensus sequence approach can be used as an effective method to design proteins with complex topologies. These generic ankyrin repeat proteins can serve as prototypes for dissecting the rules of molecular recognition mediated by ankyrin repeats and for engineering proteins with novel biological functions.
Understanding the complex relationship between amino acid sequence and protein structure remains a major challenge in structural biology. The number of naturally occurring protein sequences is much larger than the number of unique protein folds. Thus, many different proteins must fold into similar structures. For proteins with identical folds but unrelated functional properties, an effective way to extract the structural determinants and to delineate them from functional ones is to analyze the variability of amino acid residues at each position. The statistical properties of such a family can be described in a hierarchical manner by using multiple sequence alignments. At the simplest level, the variability is defined by the probability of each of the 20 amino acids occurring at each position in the sequence. At the next level of approximation, the covariation between residues at two different positions in the sequence family can be calculated. In theory, the statistical description can be expanded to cover the entire sequence space in terms of covariations of all positions with respect to one another.
Accurate statistical analysis requires the use of a large data set. Short repeating sequences, such as the leucine-rich repeat (1), the tetratricopeptide repeat (2, 3), the armadillo/HEAT repeat (4, 5), and the ankyrin repeat (6, 7), are extremely abundant in protein databases and can serve as an excellent source for a consensus-based design strategy (8). These modular units often form tandem arrays that function as molecular scaffolds to interact with a wide range of protein partners depending on the details of the repeating sequence. To date, few generalizations have been made regarding the structural properties of these repeats. In particular, it is not clear which residues determine the structure of the scaffold and which residues are specifically required for function.
One of the most commonly occurring repeats is the ankyrin repeat, a 33-residue sequence motif found in proteins with diverse functions, such as transcription initiation, cell cycle regulation, cytoskeletal integrity, ion transport, and cell–cell signaling (9). Each ankyrin repeat folds into a helix-loop-helix structure with a β-hairpin/loop region projecting outward from the helices at a 90° angle. Because the set of helices that face away from the β-hairpin/loop region are slightly longer, the repeats stack together to form a concave L-shaped structure (10). To date, 10 high-resolution structures of ankyrin repeat proteins have been solved. These structures closely resemble one another despite their different cellular functions, supporting the role of the ankyrin repeat as a versatile scaffold for protein–protein interactions. Indeed, it appears that the ankyrin repeat motif is defined by its fold rather than by its function, as there is no specific sequence or structural motif that is universally recognized by ankyrin repeat proteins. This situation stands in contrast to other protein–protein binding motifs such as SH2, SH3, or PTB domains (11).
Here, we report the design and biophysical characterization of a series of ankyrin repeat proteins containing one, two, three, or four consensus repeats based on statistical analysis of ≈4,000 ankyrin repeat sequences in the nonredundant protein database. The high-resolution x-ray structures of proteins containing three and four identical repeats demonstrate that these proteins contain the necessary structural elements to form the ankyrin repeat fold. The numerous contacts observed in the crystal structures combined with the high thermostability of our consensus ankyrin repeat proteins indicate that the statistical approach described here is an effective method to design well-folded proteins.
Materials and Methods
Database Analysis.
All sequence analysis was carried out by using in-house JAVA- or PERL-based programs. The source of the ≈4,400 ANK sequences (at the time of analysis) was the PFAM database (http://pfam.wustl.edu), which retrieves sequences from the nonredundant protein database by using a hidden Markov model (12). The multiply aligned sequences were downloaded from the Internet, and duplications were removed. Insertions in the repeat sequences (represented by lowercase letters) were disregarded from analysis. The probability of each of the 20 amino acids occurring at each of the 33 positions was calculated, as well as the average distribution of various types of amino acids per sequence such as charged (positive or negative), polar, hydrophobic, and structure breakers (proline and glycine).
Covariation analysis was carried out by calculating the covariation score Cij =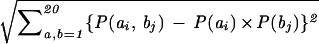 , where P(ai, bj) is the joint probability for amino acid a occurring at position i and amino acid b occurring at position j in one sequence, and P(ai) and P(bj) are the single-site probabilities derived from the amino acid conservation data. To estimate the background, the ankyrin repeat sequences were randomly shuffled and the covariation scores were recalculated and compared with that of the original database. Covarying positions with scores higher than 2 standard deviations from the average were evaluated.
, where P(ai, bj) is the joint probability for amino acid a occurring at position i and amino acid b occurring at position j in one sequence, and P(ai) and P(bj) are the single-site probabilities derived from the amino acid conservation data. To estimate the background, the ankyrin repeat sequences were randomly shuffled and the covariation scores were recalculated and compared with that of the original database. Covarying positions with scores higher than 2 standard deviations from the average were evaluated.
Gene Construction.
The genes encoding multiple consensus ankyrin repeats were constructed by using two rounds of recursive PCR (13, 14). The first round used two overlapping primers that, when annealed, created tandem repeats of the designed ANK sequence. The second round used primers that incorporated cloning sites, a stop codon, an N-terminal methionine and a C-terminal tyrosine (for concentration determination). Genes were cloned into the pAED-4 expression vector (15) downstream from a Trp-LE leader sequence which forces the protein into inclusion bodies (16). The designed repeat and the Trp-LE leader sequence contain no internal methionine therefore the N-terminal Met residue, introduced during the second round of PCR, is unique at the junction between the Trp-LE leader sequence and the designed gene. The genes encoding 1ANK, 2ANK, 3ANK, and 4ANK were first identified by restriction digestion, and then confirmed by automated DNA sequencing analysis.
Protein Expression and Purification.
The proteins were expressed in Escherichia coli BL21 (DE3) pLysS and purified from inclusion bodies. The inclusion bodies were resuspended in 5 ml of 6 M GuHCl overnight at 4°C and diluted to a final concentration of 0.6 M GuHCl with H2O, and the precipitate was collected and redissolved in 10 ml of 70% formic acid. The proteins were cleaved from the leader sequence with 0.6 g of CNBr for 3 h. The reaction was stopped by the addition of 2 g of glycine and dialyzed overnight in 5% acetic acid. The precipitate formed during dialysis was removed by centrifugation, whereas the supernatant was purified by reverse-phase HPLC on a Vydac C18 preparative column and finally lyophilized. The identities of all proteins were verified by electrospray mass spectrometry. Protein concentration was determined by measuring the absorbance at 280 nm in 6 M GuHCl, assuming a molar extinction coefficient of 1,280 for all constructs (17).
CD.
All CD experiments were carried out in 10 mM succinate, 1 mM EDTA, and 50 mM NaCl by using a JASCO J-715 spectropolarimeter with a thermoelectric temperature controller. The CD spectra were recorded at 10 μM protein concentration with a 0.1-cm pathlength cuvette at 10 nm/min scan rate, 2-nm bandwidth, and 8-s response time. The results shown are the average of three scans, except for 1ANK, which is the average of nine scans because of the low signal-to-noise ratio.
Thermal denaturation studies were performed in the same buffer as described above at 2 μM protein concentration in a 1-cm pathlength cuvette. The CD signal was monitored at 222 nm, with 2-nm bandwidth, 16-s response time, and temperature increasing at a rate of 25°C per hour. To determine the midpoint of transition (Tm), the thermal denaturation data were fit with an apparent two-state folding model.
Size Exclusion Chromatography–Multiple Angle Laser Light Scattering (SEC–MALLS).
SEC–MALLS studies were conducted by using a Varian HPLC system connected to a Pharmacia Superdex 75 size exclusion column with a miniDAWN light scattering detector and an Optilab refractive index detector (both from Wyatt Technology). Samples (200 μl) were injected at 100 μM concentration and eluted at 0.5 ml/min flow rate. Scattering data were collected and analyzed by using the astra software. The apparent molecular weights were calculated by using the Zimm method (18).
NMR.
NMR experiments were performed by using a Varian Inova 500 MHz spectrometer with a triple resonance probe. Sample conditions consisted of 300 μM 3ANK, 10% D2O, 10 mM succinate (pH 4.0), 1 mM EDTA, and 100 mM NaCl. 15N–1H HSQC spectra were collected at 30°C, and data were processed by using nmrpipe (19).
Crystallization.
Crystals were grown by hanging drop vapor diffusion method at room temperature. 3ANK and 4ANK proteins were dissolved in water at a concentration of 20 mg/ml. 4ANK crystallized overnight in 2 to 8 μl drops consisting of 1:1 mixture of well solution [0.2 M MgBr2/0.1 M sodium cacodylate, pH 6.4/33% 2-methyl-2,4-pentanediol (MPD)/15% isopropanol] and protein solution. 3ANK crystals grew for 5 days in 2 μl drops consisting of a 1:1 mixture of well solution (0.1 M Hepes, pH 7.0/50% MPD) and protein solution with 0.2 μl of 30% MPD added to the hanging drop.
X-Ray Diffraction and Structural Refinement.
X-ray data were collected at beamline 8.3.1 (Advanced Light Source, Lawrence Berkeley Laboratory, Berkeley, CA) with an ADSC Quantum 210 CCD detector. Crystals were flash frozen in liquid nitrogen before data collection. Data were processed by using elves, an automated scripting program (J. Holton and T. Alber, personal communication). Reflections were indexed, integrated, and scaled with mosfilm and scala (20). For 4ANK crystals, single wavelength anomalous diffraction (SAD) data were collected at the bromine absorption edge (21). SAD phases were determined by using solve (22). Maps with experimentally determined phases were submitted to arp/warp for automated refinement and chain tracing (warpNtrace) (23) followed by manual rebuilding and refinement with o (24) and refmac (25). The structure of 3ANK was solved by molecular replacement using residues 1–92 of the 4ANK crystal structure as the search model and epmr (26). Diffraction data of 3ANK with resolution up to 2 Å were used in the search for molecular replacement. The model was refined by using arp/warp, o, and refmac with iterative rounds of automated and manual building. Stereochemistry of the 4ANK and 3ANK final models were assessed by procheck (27).
Results
Design of Consensus Ankyrin Repeat.
Our statistical analysis of the ≈4,000 ankyrin repeat sequences in the PFAM database calculated the probability of each of the 20 amino acid residues occurring at each of the 33 positions of the repeat (Fig. 1 a and b). We also determined, on average, how many amino acid residues of each type (e.g., polar, positively charged, or hydrophobic) were in an ankyrin repeat sequence (Fig. 1c). We used this information to classify each position into one of four categories. Positions where one amino acid was present >50% of the time were classified as well conserved and for the design the particular residue was automatically assigned to that position. Positions were classified as semiconserved if two to four amino acid residues occurred with a higher frequency than any others. The semiconserved positions were subdivided into two categories. If all high frequency residues had the same property (e.g., hydrophobic), the position was classified as semiconserved of the same type and the most frequently occurring residue was automatically assigned to that position. Positions were classified as semiconserved of different type if the high frequency residues belonged to different groups of amino acids. Finally, if a position had no preference for any amino acid, the position was classified as nonconserved. Positions falling into the last two categories were assigned based on an effort to satisfy the amino acid distribution data and their location in the proposed ankyrin repeat secondary structure. For example, position 24 (nonconserved) was assigned to alanine to satisfy the average number of hydrophobic residues (15) found in a single repeat and because of alanine's high α-helical propensity.
Fig 1.

Statistical analysis data used in the design of the consensus ankyrin repeat proteins. (a) Final sequence of the designed repeat and the corresponding secondary structure elements. Positions are colored according to the conservation level. (b) Representative histograms colored accordingly. (c) Examples of amino acid distribution data.
In addition to the amino acid conservation and distribution data, we also calculated the pairwise covariation between amino acid residues at two positions within the same repeat and between adjacent repeats (data not shown). The covariation analysis did not significantly influence the final sequence selection because the most frequently observed pairs were already well represented in our design. Our consensus ankyrin repeat sequence does not appear in the nonredundant protein database; the highest scoring match using blastp has 57% sequence identity and 87% sequence similarity (residues 188–220, NP_542421). The designed sequence uses only 12 of 20 amino acids and it does not contain cysteine, methionine, or aromatic residues.
Ankyrin repeats arranged in tandem arrays invariably result in the beginning of the first repeat and the termination of the last repeat with the β-hairpin/loop region. The structure of this region is thought to depend on the stabilizing interactions of both neighboring repeats. In fact, in the 10 high-resolution structures of ankyrin repeat proteins, the terminal β-hairpin/loop region is either disordered or altogether absent in the protein. We reasoned that our designed constructs would be more soluble and less prone to aggregation if the terminal β-hairpin/loop was omitted. Thus, the N-terminal repeat started at position 1, and the C-terminal repeat ended at position 26. All internal repeats were identical and consisted of 33 residues corresponding to the consensus ankyrin repeat sequence shown in Fig. 1a. These proteins are referred to as 1ANK, 2ANK, 3ANK, and 4ANK based on the total number of repeats present in the construct.
Solution Characterization.
The folding and oligomerization state of the designed ankyrin repeat proteins were characterized by far-UV CD spectroscopy, 2D NMR spectroscopy, and SEC–MALLS. In general, the ANK proteins are more soluble, though somewhat less stable, under acidic conditions. Fig. 2a shows the CD spectra of 1ANK and 2ANK recorded at pH 5, 3ANK at pH 4.5, and 4ANK at pH 4.25. 1ANK is unfolded under all conditions tested (pH 3–8). 2ANK is partially folded under the conditions described for Fig. 2a, though it is not strictly a monomer based on SEC–MALLS studies (see below). 3ANK and 4ANK are well folded with mean residue ellipticity at 222 nm in agreement with other biophysically characterized ankyrin repeat proteins. NMR studies confirm that 3ANK is well folded in solution. The 15N–1H HSQC spectra consist of well-resolved peaks with the chemical shift dispersion range indicative of a protein containing mostly α-helical secondary structure (Fig. 2b).
Fig 2.

Solution characterization of the consensus ankyrin repeat proteins. (a) Far-UV CD spectra of 1ANK (⋄), 2ANK (□), (•), and (○) at 20°C. The pH values are described in the text. (b) 1H–15N HSQC spectra of recorded at 30°C, pH 4. (c) SEC–MALLS analysis of (○ and □) and (• and ▪). The detector values are the average of the 90° and 138° light scattering signals. The molar masses were calculated by using Astra software (Wyatt Technology, Santa Barbara, CA). (d) Thermal denaturation of (○) and (•) monitored by CD signals at 222 nm. The data were fit with an apparent two-state folding model (line).
Fig. 2c shows the SEC–MALLS data for 3ANK and 4ANK at the conditions used for CD analysis. These experiments can be used to determine the molecular weight of proteins in solution independently of the elution time on the size exclusion column (18). The apparent molecular weights calculated were both within 8.5% of the expected value. The polydispersity factor was 1.000 and 1.001, respectively. This analysis demonstrates that both 3ANK and 4ANK are homogeneous monomers in solution and agrees well with independent sedimentation equilibrium studies (data not shown). In contrast, we could not find any condition at which 2ANK was fully monomeric, despite reproducible CD spectra showing a fully folded protein at pH 6 and 7 (data not shown).
The thermal denaturation of 3ANK and 4ANK was monitored by CD at 222 nm (Fig. 2d), showing reversible, cooperative thermal transitions for both proteins. The calculated midpoint of transition, or Tm value, is equal to 69.4°C for 3ANK and 81.3°C for 4ANK. This type of transition is the hallmark of a well-packed, highly ordered protein (28).
X-Ray Crystal Structures of 3ANK and 4ANK.
The crystal structures of 3ANK and 4ANK were solved to 1.26 and 1.5 Å resolution, respectively (Table 1). The structure of 4ANK was solved by using SAD data generated from bromide ions present in the crystal (21). The structure of 3ANK was solved by molecular replacement by using a truncated version of 4ANK as the search model. Fig. 3a shows a portion of the experimental SAD-phased electron density map for the 4ANK structure. The structure of the designed ankyrin repeat is extremely similar to that of naturally occurring ones. For example, the backbone rms deviation of residues 5–120 of 4ANK and residues 41–156 of GA-binding protein β (GABPβ) is 0.79 Å. The structures of 3ANK and 4ANK overlay to 0.24 Å backbone rms deviation and 1.03 Å backbone and side chain rms deviation (Fig. 3b). The region with maximum structural discrepancy is located just after the second α-helices with an average rms deviation of 1.06 Å. This region is involved in crystal contacts in the 3ANK structure.
Table 1.
Data collection and refinement statistics
| Data collection/phasing | ||
| Wavelength, Å | 0.9199 | 1.0000 |
| Resolution, Å | 46.14–1.50 | 19.96–1.26 |
| No. of reflections (free) | 16,966 (913) | 41,302 (2,246) |
| Rsym (highest shell) | 0.07 (0.436) | 0.087 (0.080) |
| Space group | P212121 | P212121 |
| Unit cell (a, b, c), Å | 28.429, 46.115, 81.588 | 39.636, 43.159, 105.574 |
| Completeness (highest shell) | 99.92 (5.8) | 99.52 (5.0) |
| I/σ (highest shell) | 10.1 (1.7) | 11.8 (1.7) |
| Number of bromide sites | 2 | 0 |
| Mean figure of merit | 82–1.5 Å 0.322 (0.604 after solvent flattening) | |
| Refinement | ||
| Rcryst/Rfree (resolution range), % (Å) | 21.6/24.9 (19.65–1.50) | 17.2/18.7 (19.96–1.26) |
| rms bonds, angles, Å, ° | 0.011, 1.49 | 0.011, 1.41 |
| No. of protein molecules per asymmetric unit | 1 | 2 |
| No. of water molecules | 86 | 183 (1 trifluoroacetic acid) |
| Ramachandran (most favored/ additional allowed/ generously allowed/disallowed), % | 92.6/7.4/0/0 | 92.4/7.6/0/0 |
Rsym = ∑|I − 〈I〉|/ΣI, I = intensity.
Mean figure of merit = ∥∑Pαeiα/∑Pα∥, α = phase, Pα = phase probability distribution.
Rcryst = ∑|Fobs − Fcalc|/∑Fobs.
Fig 3.

X-ray crystal structures of and . (a) Stereo representation showing the initial electron density map of residues 37–40 of , including the TPLH motif, contoured at 1.5 σ. The figure was made with O and rendered by using MOLRAY (43). (b) Overlay of (cyan) and (orange) backbone structures. This figure and all following structural representations were made with MOLMOL (44). (c) Hydrogen bonding network in β-hairpin/loop region of . Each repeat is colored differently and hydrogen bonds are represented by letters: a, D32(OD1) and N34(HN); b, D32(O) and G35(HN); c, D32(OD1) and R36(HN); d, D32(HN) and R36(O); e, R36(O) and H7(HNE2); f, A30(O) and H7(HNE2); g, A30(HN) and D27(O); h, D27(OD1) and N29(HN); i, N29(OD1) and D60(HN); j, N29(ND2) and G58(O); k, N34(O) and K66(HN); and l, R36(HNE) and D65(OD2). Equivalent hydrogen bonds in adjacent repeats are represented by the letter followed by ′ or " and for all residue numbers i > 33, the canonical ankyrin repeat position = i − 33. (d) Comparison of intramolecular packing of ankyrin repeat proteins based on the occluded surface area analysis. For each protein, the average ray length of all residues was plotted against the average occluded surface packing (OSP) value of all buried residues with exposed surface area of <5%. The average values of 152 high-resolution structures in PDB are shown by the dashed lines on the graph (31).
Examination of the hydrogen-bonding pattern in both structures shows a network of interactions with remarkable regularity. Hydrogen bonding in the β-hairpin/loop portion of the repeats is contributed by a combination of side chain and backbone interactions (Fig. 3c). For example, D32 forms backbone hydrogen bonds with the backbone of G2 and R3 in the second repeat, and side chain hydrogen bonds with the backbone of N1 and R3 in the second repeat. It is interesting to note that K33 appears to have varied H-bonding interactions depending on the repeat in which it exists. K33 in the first repeat forms a side chain hydrogen bond with the side chain of N1 and a backbone hydrogen bond with the side chain of K33 in the second repeat, but K33 in the third repeat forms a side chain hydrogen bond with the side chain of N1 in the same repeat. On the opposite side of the proteins, in the loop at the junction of the two α-helices, G13 forms a backbone hydrogen bond to L15.
One of the most prominent features of the ankyrin repeat is the conserved TPLH sequence at positions 4–7 (10). Inspection of this sequence in the crystal structure uncovers an intricate network of hydrogen bonds with H7, located in the first α-helix, forming a backbone hydrogen bond to the side chain of T4 in the upper loop and a side chain hydrogen bond to the backbone of A30. H7 also forms a side chain hydrogen bond with the backbone of R3 in the second repeat, thereby supporting the β-hairpin/loop region. Proline at position 5 is located at the 90° junction formed by the β-hairpin/loops and helix-loop-helices. Proline residues C-terminal to threonine are among the most commonly occurring residues found in N-capping motifs of α-helices (29), and are likely to be responsible for the regular L shaped configuration adopted by the ankyrin repeat.
In an effort to compare the intramolecular packing interactions among ankyrin repeat proteins, the program os (occluded surface) (30, 31) was used to evaluate 3ANK, 4ANK, GABPβ, Bcl-3, p18, and the ankyrin repeat domain of 53bp2 (Fig. 3d). All structures had <2.5 Å resolution and contained three or more contiguous repeats. The analysis was based on surface normals extended outward from each atom until they intersect with the van der Waals surface of a neighboring atom. According to the original report, a lower value of the average ray length and a higher occluded surface packing value obtained from the buried residues indicate a protein with tighter packing (31). Fig. 3d shows a comparison of six ankyrin repeat proteins. Based on this criterion, GABPβ, 3ANK, and 4ANK are the most well packed proteins in this group.
Discussion
Our results demonstrate that the structure of the ankyrin repeat fold can be defined statistically by the probability of amino acid usage at each position in this motif. The consensus ankyrin repeat proteins reported here are the first de novo designed multiple repeat proteins with identical repeats. A similar approach has been used to design coiled coils (32, 33), a monomeric 33-residue WW domain using 65 WW domain sequences (34), and a 444-residue phytase with enhanced stability using 13 fungal phytase sequences (35). Together, these studies support the application of multiple sequence alignments in protein structure prediction and design. Our design was based on a large database of ≈4,000 sequences that allowed us to calculate the pairwise covariation between two sequence positions in addition to the conservation level at each position. The result of this analysis, however, did not provide significant input to the design strategy. Generally, it appears that higher-order correlations between positions are more important for the intricate details of protein structure such as stability (36) or long-range energetic coupling within the molecule (37). In contrast, the zero-order approximation, as defined by the amino acid conservation and distribution data, may be sufficient for determining the overall fold.
The 3ANK and 4ANK proteins exhibit high thermostability exceeding the level observed for p16 (38), Drosophila notch (39), p19 (40), myotrophin (41), and GABPβ (L.K.M. and Z.-y.P., unpublished results). In addition, 3ANK and 4ANK form highly ordered crystals that diffract to high resolution. These properties may relate to the regularity of the designed sequence in fostering the simultaneous formation of many favorable hydrogen bonding and side chain packing interactions both within a single repeat and between adjacent repeats. Indeed, the occluded surface packing analysis indicates that the most tightly packed proteins are 3ANK, 4ANK, and GABPβ. These three proteins also display the most ordered topology of the six proteins evaluated.
Mapping the conservation level of each residue on the structure of 4ANK supports its classification into either a structural or functional role (Fig. 4). The β-hairpin/loop region and the short α-helices comprising the concave face have been previously characterized as the recognition surface. This is due to the observation that this face often participates in intermolecular interactions in crystal structures of ankyrin repeat proteins in complex with binding partners. Positions classified as nonconserved and semiconserved of different type are mainly present on the recognition surface (Fig. 4a), whereas the opposite face (Fig. 4b) shows mostly semiconserved positions of the same type. This distribution is consistent with the notion that positions on the binding side of the molecule should have the highest variability to accommodate a diverse group of potential binding partners. Conversely, a conservation level mapping of the interior residues of 4ANK reveals mainly positions classified as well conserved with a few positions classified as semiconserved of the same type (Fig. 4c). In fact, essentially all well-conserved positions are located in the interior of the ankyrin repeat, implicating the role of these residues in structure formation rather than binding specificity.
Fig 4.

Conservation level of residues mapped onto the structure of . Colors represent the conservation level: blue, well-conserved; pink, semiconserved of the same type; green, semiconserved of different type; and yellow, nonconserved. The C-terminal tyrosine is colored in gray. (a) Front (concave) surface of . (b) Back surface of . The ribbon diagrams on the left show orientations. (c) Longitudinal cross-section of the protein displaying the interior residues. Protein is in the same orientation as in b.
We used statistical analysis to set up a guideline for the design of ankyrin repeat proteins. The ability of our designed sequence to fold into a unique, well-defined structure indicates that the consensus sequence carries all of the necessary structural determinants, most likely in the conserved residues, to form the ankyrin repeat fold. Less conserved residues, located on the surface, are amenable to mutation and therefore are good candidates for directing functional intermolecular interactions. The consensus ankyrin repeat proteins described here will be useful model systems for ankyrin repeat-related studies. The identical nature of the repeats within the protein makes these constructs ideal for studying the general topological characteristics of ankyrin repeats (42) as well as for protein folding studies. The biophysical properties of ankyrin repeats can be assessed without the complications of sequence variation between repeats because all interactions between repeats are identical. The designed consensus ankyrin repeat sequence can also serve as a blueprint for protein engineering, complete with a map of possible residues that are acceptable at each position. Beginning with the general scaffold as a foundation, it may be possible to engineer binding specificities into 3ANK or 4ANK to create novel biological functions.
Acknowledgments
We thank Mark Maciejewski for help with NMR and computational support, James Holton for beamline support, Ping Bai and Scott Robson for technical assistance, James Berger and Katjuša Brejc for help with x-ray crystallography, and members of the Peng lab for critical reading of the manuscript. The Advanced Light Source Beamline 8.3.1 was funded by the National Science Foundation, the University of California, and Henry Wheeler. This work was supported by National Institutes of Health Grant GM-54533 and American Cancer Society Grant GMC-103045 (to Z.-y.P.).
Abbreviations
SAD, single wavelength anomalous diffraction
SEC–MALLS, size exclusion chromatography–multiple angle laser light scattering
GABPβ, GA-binding protein β
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Coordinates for the structures of and have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes and , respectively).
References
- 1.Takahashi N., Takahashi, Y. & Putnam, F. W. (1985) Proc. Natl. Acad. Sci. USA 82, 1906-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sikorski R. S., Boguski, M. S., Goebl, M. & Hieter, P. (1990) Cell 60, 307-317. [DOI] [PubMed] [Google Scholar]
- 3.Hirano T., Kinoshita, N., Morikawa, K. & Yanagida, M. (1990) Cell 60, 319-328. [DOI] [PubMed] [Google Scholar]
- 4.Riggleman B., Weieschaus, E. & Schedl, P. (1989) Genes Dev. 3, 96-113. [DOI] [PubMed] [Google Scholar]
- 5.Andrade M. A. & Bork, P. (1995) Nat. Genet. 11, 115-116. [DOI] [PubMed] [Google Scholar]
- 6.Breeden L. & Nasmyth, K. (1987) Nature 329, 651-654. [DOI] [PubMed] [Google Scholar]
- 7.Lux S. E., John, K. M. & Bennett, V. (1990) Nature 344, 36-42. [DOI] [PubMed] [Google Scholar]
- 8.Marcotte E. M., Pellegrini, M., Yeates, T. O. & Eisenberg, D. (1999) J. Mol. Biol. 293, 151-160. [DOI] [PubMed] [Google Scholar]
- 9.Bork P. (1993) Proteins 17, 363-374. [DOI] [PubMed] [Google Scholar]
- 10.Sedgwick S. G. & Smerdon, S. J. (1999) Trends Biochem. Sci. 24, 311-316. [DOI] [PubMed] [Google Scholar]
- 11.Kuriyan J. & Cowburn, D. (1997) Annu. Rev. Biophys. Biomol. Struct. 26, 259-288. [DOI] [PubMed] [Google Scholar]
- 12.Bateman A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., Griffiths-Jones, S., Howe, K. L., Marshall, M. & Sonnhammer, E. L. (2002) Nucleic Acids Res. 30, 276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prodromou C. & Pearl, L. H. (1992) Protein Eng. 5, 827-829. [DOI] [PubMed] [Google Scholar]
- 14.Chen G.-q., Choi, I., Ramachandran, B. & Gouaux, J. E. (1994) J. Am. Chem. Soc. 116, 8799-8800. [Google Scholar]
- 15.Doering D. S., (1992) Ph.D. thesis (Massachusetts Institute of Technology, Cambridge, MA).
- 16.Staley J. P. & Kim, P. S. (1994) Protein Sci. 3, 1822-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edelhoch H. (1967) Biochemistry 6, 1948-1954. [DOI] [PubMed] [Google Scholar]
- 18.Folta-Stogniew E. & Williams, K. R. (1999) J. Biomol. Tech. 10, 51-63. [PMC free article] [PubMed] [Google Scholar]
- 19.Delaglio F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277-293. [DOI] [PubMed] [Google Scholar]
- 20.Kabsch W. (1988) J. Appl. Crystallogr. 21, 916-924. [Google Scholar]
- 21.Dauter Z. & Dauter, M. (1999) J. Mol. Biol. 289, 93-101. [DOI] [PubMed] [Google Scholar]
- 22.Terwilliger T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perrakis A., Morris, R. & Lamzin, V. S. (1999) Nat. Struct. Biol. 6, 458-463. [DOI] [PubMed] [Google Scholar]
- 24.Jones T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 25.Murshudov G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999) Acta Crystallogr. D 55, 247-255. [DOI] [PubMed] [Google Scholar]
- 26.Kissinger C. R., Gehlhaar, D. K. & Fogel, D. B. (1999) Acta Crystallogr. D 55, 484-491. [DOI] [PubMed] [Google Scholar]
- 27.Laskowski R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 28.Privalov P. L. (1989) Annu. Rev. Biophys. Biophys. Chem. 18, 47-69. [DOI] [PubMed] [Google Scholar]
- 29.Aurora R. & Rose, G. D. (1998) Protein Sci. 7, 21-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pattabiraman N., Ward, K. B. & Fleming, P. J. (1995) J. Mol. Recognit. 8, 334-344. [DOI] [PubMed] [Google Scholar]
- 31.Fleming P. J. & Richards, F. M. (2000) J. Mol. Biol. 299, 487-498. [DOI] [PubMed] [Google Scholar]
- 32.Harbury P. B., Zhang, T., Kim, P. S. & Alber, T. (1993) Science 262, 1401-1407. [DOI] [PubMed] [Google Scholar]
- 33.Zhou N. E., Kay, C. M. & Hodges, R. S. (1994) J. Mol. Biol. 237, 500-512. [DOI] [PubMed] [Google Scholar]
- 34.Macias M. J., Gervais, V., Civera, C. & Oschkinat, H. (2000) Nat. Struct. Biol. 7, 375-359. [DOI] [PubMed] [Google Scholar]
- 35.Lehmann M., Kostrewa, D., Wyss, M., Brugger, R., D'Arcy, A., Pasamontes, L. & van Loon, A. P. G. M. (2000) Protein Eng. 13, 49-57. [DOI] [PubMed] [Google Scholar]
- 36.Larson S. M., Di Nardo, A. A. & Davidson, A. R. (2000) J. Mol. Biol. 303, 433-446. [DOI] [PubMed] [Google Scholar]
- 37.Lockless S. W. & Ranganathan, R. (1999) Science 286, 295-299. [DOI] [PubMed] [Google Scholar]
- 38.Zhang B. & Peng, Z.-y. (1996) J. Biol. Chem. 271, 28734-28737. [PubMed] [Google Scholar]
- 39.Zweifel M. E. & Barrick, D. (2001) Biochemistry 40, 14357-14367. [DOI] [PubMed] [Google Scholar]
- 40.Zeeb M., Rosner, H., Zeslawski, W., Canet, D., Holak, T. A. & Balbach, J. (2002) J. Mol. Biol. 315, 447-457. [DOI] [PubMed] [Google Scholar]
- 41.Mosavi L. M., Williams, S. & Peng, Z.-y. (2002) J. Mol. Biol. 320, 5-11. [DOI] [PubMed] [Google Scholar]
- 42.Groves M. R. & Barford, D. (1999) Curr. Opin. Struct. Biol. 9, 383-389. [DOI] [PubMed] [Google Scholar]
- 43.Harris M. & Jones, T. A. (2001) Acta Crystallogr. D D57, 1201-1203. [DOI] [PubMed] [Google Scholar]
- 44.Koradi R., Billeter, M. & Wuthrich, K. (1996) J. Mol. Graphics 14, 51-55. [DOI] [PubMed] [Google Scholar]