

Abstract
Oxidative stress has been widely implicated as an important factor in the aging process. Because mitochondrial respiration is the principal source of reactive oxygen within cells, the mitochondrially localized superoxide dismutase (SOD) 2 is thought to play an important front-line defensive role against aging-related oxidative stress. Although genetic studies with mutants deficient in SOD1, the predominantly cytosolic isoform of SOD, have been instrumental in elucidating the role of reactive oxygen metabolism in aging in Drosophila, the lack of available mutations in the Sod2 gene has hampered an equivalent analysis of the participation of this important antioxidant enzyme in the Drosophila aging model. Here we report that ablation of mitochondrial SOD2 through expression of a GAL4-regulated, inverted-repeat Sod2 RNA-interference transgene in an otherwise normal animal causes increased endogenous oxidative stress, resulting in loss of essential enzymatic components of the mitochondrial respiratory chain and the tricarboxylic acid cycle, enhances sensitivity to applied oxidative stress, and causes early-onset mortality in young adults. In sharp contrast, ablation of SOD2 has no overt effect on the development of larvae and pupae, which may reflect a fundamental transition in oxygen utilization and/or reactive oxygen metabolism that occurs during metamorphosis from larval to adult life.
The mitochondrial respiratory chain that reduces oxygen to water is the metabolic engine of high-energy aerobic metabolism. However, the escape of the univalently reduced reactive intermediate, superoxide (O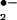 ), from the respiratory chain is an incipient threat to mitochondrial integrity. The superoxide dismutases (SODs) constitute a family of antioxidant enzymes that catalyze the disproportionation of superoxide to oxygen and hydrogen peroxide (H2O2) (1). Most eukaryotic cells contain a copper- and zinc-containing SOD (SOD1) that is located principally in the cytoplasm (2), with a small fraction located in the intermembrane space of mitochondria (3, 4), and a manganese-containing SOD (SOD2) that is strategically located in the mitochondrial matrix (2) to capture superoxide released during respiration.
), from the respiratory chain is an incipient threat to mitochondrial integrity. The superoxide dismutases (SODs) constitute a family of antioxidant enzymes that catalyze the disproportionation of superoxide to oxygen and hydrogen peroxide (H2O2) (1). Most eukaryotic cells contain a copper- and zinc-containing SOD (SOD1) that is located principally in the cytoplasm (2), with a small fraction located in the intermembrane space of mitochondria (3, 4), and a manganese-containing SOD (SOD2) that is strategically located in the mitochondrial matrix (2) to capture superoxide released during respiration.
In Drosophila melanogaster, SOD1 and SOD2 seem to comprise the total complement of SOD (5, 6); no other form of SOD, such as the extracellular SOD (SOD3) of mammals (7, 8), has been identified biochemically or by inference from the Drosophila genome-sequence database (9). Extensive genetic analysis of SOD1 has revealed the critical role of this enzyme in many diverse aspects of the biology of Drosophila. SOD1-null mutants of Drosophila, in sharp contrast to those of mice (10), are debilitated severely and exhibit male sterility and reduced female fertility, hypersensitivity to paraquat, ionizing radiation, transition metals, hyperoxia (5), and buthionine sulfoximine-mediated depletion of glutathione (11), retinal degeneration (12), premature aging (13), and reduction of adult life span by 85–90% (5). All these phenotypes can be restored virtually to WT by genomic Sod1 transgenes (14). Overexpression of SOD1 using a genomic transgene, under specific circumstances, can lead to apparent extension of adult life span (15), whereas overexpression of SOD1 using tissue-specific (16) and inducible (17) transgenes confers convincing and robust life-span extension.
In contrast, relatively little progress has been made in the genetic analysis of SOD2 function in Drosophila. Although the Sod2 gene has been cloned and characterized (6), no naturally occurring allelic variants have been identified yet as for Sod1 (18), nor have extensive attempts to recover induced mutations yet been fruitful. Interestingly, the Sod2 genetic locus lies within a small region in the Drosophila genome for which no chromosomal deletions have been reported yet (19). This observation, coupled with the difficulty encountered in generating loss-of-function alleles of Sod2, has kept alive the possibility that the Sod2 gene may be one of a small number of haplolethal genes in the D. melanogaster genome.
To circumvent the lack of mutants, we used RNA interference (RNAi) to investigate SOD2 function in Drosophila. RNAi occurs when introduced double-stranded RNA silences gene expression via specific degradation of the cognate mRNA. RNAi methodology has been applied to several animal models including Caenorhabditis elegans (20) and Drosophila (21–23). Here we show that expression of a GAL4-regulated, inverted-repeat Sod2 RNAi transgene virtually eliminates detectable SOD2, confers elevated oxidative stress in mitochondria, and causes rapidly progressing early adult-onset mortality with no apparent ill effects on preadult development.
Experimental Methods
Drosophila Stocks and Culture Methods.
The daG32Gal4 driver stock (FlyBase: P{GAL4-da.G32}) used for widespread expression of UAS-transgenes was obtained originally from G. Boulianne (University of Toronto, Toronto). Stocks were maintained on cornmeal agar medium at 25°C unless otherwise stated. CO2 anesthesia was used throughout, allowing at least 5 h of recovery at room temperature before the onset of experimental procedures to avoid potential latent effects of anesthesia (24).
Generation of Transformants.
P-vector transformants were generated by standard embryo-injection methods (25) in which the P vector, pPUAST (26), carrying the Sod2IR construct and the p(Δ2–3) helper plasmid were coinjected into w1 recipient embryos. Adult G0 transformants were identified by outcrossing to w1 and balanced over SM5 or TM3 balancer chromosomes (27). A total of 18 independent transformants was generated and screened for suppression of SOD2 activity with the GAL4 driver, daG32Gal4 (see below). Two transgenic strains with robust expression, SodIR15 and SodIR24, were selected for more thorough characterization as described here.
Construction of the Sod2 Inverted-Repeat Transgene (Sod2IR).
The complete Drosophila Sod2 cDNA was inserted into the EcoRI site of pBluescriptII SK(+/−). The entire cDNA less the 3′-polyadenylation site was amplified with the addition of a 3′-KpnI site by PCR using the T3 primer of pBluescriptII SK(+/−) and Sod2IR. The 841-bp PCR product was digested with KpnI and EcoRI, and the 757-bp product was self-ligated and digested with EcoRI to produce a 1,514-bp inverted-repeat product, which then was ligated into the EcoRI site of the P vector, pPUAST.
Northern Hybridization Analysis.
Total RNA was extracted from 25 adults aged 2–4 days by using TRIzol reagent (Invitrogen). After electrophoretic separation of denatured RNA (20 μg per lane) through 1.5% formaldehyde agarose gels, the RNA was transferred and UV-crosslinked to a nylon membrane (Roche Diagnostics). Sod2 and Rp49 (control) RNA was detected by using digoxigenin-labeled DNA probes made with a digoxigenin nonradioactive DNA-labeling kit (Roche Diagnostics).
Western Immunoblot Analysis of SOD2.
Adult males (1–2 days old) were homogenized in 1% Triton X-100, the extracts were centrifuged at 13,000 × g for 5 min at 4°C, and the supernatant was mixed with an equal volume of loading buffer (125 mM Tris⋅HCl, pH 7.2/4% SDS/10% mercaptoethanol/20%glycerol/0.01% bromophenol blue). Samples were boiled for 5 min, and 10 μg total protein was separated on an SDS polyacrylamide gel (4% stacking, 15% separating). The protein was transferred to Hybond-ECL nitrocellulose membrane (Amersham Pharmacia) and probed with rat anti-SOD2 polyclonal antibody (StressGen Biotechnologies, Victoria, Canada) and goat anti-rabbit IgG horseradish peroxidase conjugate (Stressgen). In addition to reacting strongly with Drosophila SOD2, this antibody preparation gives a very weak background signal common to all genotypes. Amersham Pharmacia ECL Western blotting detection reagents were used. For this and all other procedures, proteins were determined with the Bio-Rad protein assay.
SOD Activity Determination.
The GAL4/UAS strains were initially characterized for SOD1 and SOD2 activities using the 6-hydroxydopamine autooxidation method (28) as described (6, 14). Thereafter, the following method was used to determine activities of SOD1 and SOD2. Adult males, 1–2 days old, were homogenized in 50 mM sodium phosphate, pH 7.4/0.1 mM EDTA, the extract was sonicated for 15 sec to rupture mitochondria, and centrifuged at 13,000 × g. The supernatant was assayed for SOD1 and SOD2 by using an in-gel activity assay after separation of SOD1 and SOD2 by native protein gel electrophoresis (29). Qualitative estimation of SOD activity using the in-gel activity assay after serial dilution of extracts provides a lower detection limit of ≈0.1 fly equivalents for SOD2 and 0.05 fly equivalents for SOD1 activity of a single 1- to 2-day- old WT male.
Preparation of Crude Mitochondrial Extracts.
Mitochondria were prepared by using a procedure modified from that described by Van den Bergh (30). The final enriched mitochondrial pellet was resuspended in extraction buffer and sonicated for 15 sec.
Succinate Dehydrogenase (SDH) Assay.
SDH activity was assayed in crude mitochondrial extracts by using the dichlorophenolindophenol (DCIP) method as described in ref. 31.
Aconitase Assay.
Aconitase activity in mitochondrial extracts prepared as described above was assayed spectrophotometrically by determining the conversion of isocitrate to cis-aconitate at 240 nm (32). Alternatively, mitochondrial and cytoplasmic aconitase activities in whole-fly extracts were assayed jointly after electrophoretic separation (33). Thirty adult males were homogenized in 120 μl of extraction buffer (0.6 mM MnCl2/2 mM citric acid/50 mM Tris⋅HCl, pH 8.0) and centrifuged at 13,000 × g. Aliquots were electrophoresed on Sepraphore III membranes (Pall). Aconitase activity was detected chromogenically by incubating the membrane in 100 mM potassium phosphate, pH 6.5/1 mM NADPH/2 mM cis-aconitic acid/1.2 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide/0.3 mM phenazine methosulfate/25 mM MgCl2/5 units/ml isocitrate dehydrogenase.
Aconitase Reactivation.
Aconitase was reactivated in vitro by incubating 22.5 μl of whole-fly extract with 2.5 μl of 0.5 M DTT and 2.5 μl of 50 mM ferrous ammonium sulfate/50 mM Tris⋅HCl, pH 8.0, for 1 min at room temperature before electrophoresis (34).
Fumarase Assay.
Fumarase was assayed spectrophotometrically in mitochondrial extracts by following the conversion of malate to fumarate at 240 nm as described (35).
Applied Oxidative Stress, Eclosion, and Life-Span Determinations.
Paraquat.
One-day-old males were maintained on fresh cornmeal for 24 h after collection and then transferred to vials (10 flies per vial) containing Whatman 3M filter disks with 250 μl of freshly prepared 2.0 mM methyl viologen (Sigma) in 1% sucrose (5). Survival at 25°C in the dark was monitored at 48 h.
Eclosion rate.
Virgin w1;+/+;daG32/daG32 females were crossed to w1;Sod2IR/SM5;+/+ males and allowed to lay eggs for 4 days in cornmeal vials, and eclosing adult progeny were scored.
Life span.
Survival of 200 adult males on standard cornmeal medium (20 flies per vial) was followed at 25°C with transfer of survivors to fresh vials every 2–3 days.
Statistical Analysis.
Results are expressed as mean ± SD. The Student's unpaired t test was used to determine the significance of differences between means. P < 0.05 is considered significant.
Results
Sod2IR Transformants.
Eighteen independent UAS-Sod2IR transformants were generated and screened for efficacy in suppressing SOD2 activity by crossing to the daG32GAL4 driver (see below). Two transgenic strains with robust expression, Sod2IR15 and Sod2IR24, were selected for the experiments reported here.
Sod2IR Expression Diminishes Endogenous Sod2 Transcripts, SOD2 Protein, and SOD2 Activity.
We used the GAL4/UAS binary regulatory system to express the Sod2IR transgenes by crossing the Sod2IR lines to the daG32Gal4 driver line (36). The daG32Gal4 driver (FlyBase P{GAL4-da.G32}) expresses Gal4 under the control of regulatory sequences from the class I helix–loop–helix transcription factor gene, daughterless (da). This driver expresses widely throughout development and in most if not all adult tissues (37). To avoid potential recessive position effects associated with P-vector insertions, the experiments reported here were conducted with flies hemizygous for the daG32Gal4 driver and/or the UAS-Sod2IR expression vector. Expression of Sod2IR leads to the apparent degradation of the endogenous 800-kb Sod2 transcript (Fig. 1) and to high levels of a broad range of putative transcript fragments. This correlates with a nearly complete loss of immunodetectable SOD2 protein (Fig. 2) and with the virtually complete loss of detectable SOD2 enzymatic activity in Sod2IR-expressing adults (Fig. 3). Sod2IR expression by the daG32Gal4 driver also suppresses SOD2 activity in late third instar larvae and pharate adults (late pupae) (Fig. 4) but has no major effect on SOD2 activity in embryos, the latter being likely due to maternally transmitted SOD2 and/or Sod2 transcripts (6). RNAi-mediated silencing of Sod2 has no collateral effect on the level of SOD1 activity. These results suggest that the viability of late larvae and pupae has little if any direct dependence on SOD2.
Fig. 1.

Sod2IR promotes degradation of Sod2 mRNA. Total RNA (20 μg) isolated from 1- to 2-day-old adults of different genotypes was electrophoresed, transferred to nylon membranes, and hybridized with digoxigenin-labeled probes for Sod2 and the ubiquitously expressed marker, RP49. Genotypes are +/+;daG32Gal4/+ (lane 1), UASSod2IR15/+;daG32Gal4/+ (lane 2), and UASSod2IR24/+;daG32Gal4/+ (lane 3).
Fig. 2.

Sod2IR suppression of SOD2 protein. Protein (10 μg) isolated from adult males (1–2 days old) of different genotypes was electrophoresed through SDS polyacrylamide gels, transferred to nitrocellulose, and probed with SOD2 antibody. In addition to the strong signal for Drosophila SOD2, this heterologous rat polyclonal antibody interacts weakly and nonspecifically with a variety of unknown proteins in Drosophila extracts regardless of genotype. Genotypes are +/+;daG32Gal4/+ (lane 1), UASSod2IR15/+;daG32Gal4/+ (lane 2), and UASSod2IR24/+;daG32Gal4/+ (lane 3).
Fig. 3.

Sod2IR ablation of SOD2 activity. Extracts of adult males (1–2 days old) of different genotypes were electrophoresed on native protein gels, and SOD activity was determined by interference with superoxide-dependent reduction of nitroblue tetrazolium. Genotypes are +/+;daG32Gal4/+ (lane 1), UASSod2IR15/+;daG32Gal4/+ (lane 2), and UASSod2IR24/+;daG32Gal4/+ (lane 3). Each lane was loaded with 1.2 fly equivalents of extract.
Fig. 4.

Sod2IR expression during development. Extracts were prepared, electrophoresed, and assayed for SOD activity as described for Fig. 3. Embryos, 0–16 h, ≈100 embryo equivalents per lane; larvae, wandering third instar, 3 larvae per lane; pharate adults, pupal stages P12–P15, 3 pupae per lane; adults, 0- to 24-h males, 1 fly per lane. Embryos, larvae, and pupae were of mixed sex. Lanes: +/+;daG32Gal4/+/+;daG32Gal4/+ (−) and UASSod2IR24/+;daG32Gal4/+ (+).
Silencing of Sod2 Impairs Mitochondrial Iron-Sulfur Enzymes.
SOD2 mutants in yeast are sensitive to oxidative stress and show diminished mitochondrial aconitase and impaired cell-cycle progression arising from reduced tricarboxylic acid (TCA) cycle function (38–40). Mitochondrial aconitase, a constituent of the TCA cycle, can act as an indicator of altered superoxide flux in mitochondria through its susceptibility to inactivation by superoxide attack on its solvent-exposed [4Fe-4S] cluster (41, 42). Accordingly, we assessed the impact of Sod2IR expression on specific targets of mitochondrial oxidative stress by assaying enzymes of the respiratory chain and TCA cycle. As shown in Fig. 5, Sod2IR expression reduces mitochondrial aconitase to 66% and 61%, respectively, of control activity in the two Sod2IR strains. SDH, an Fe-S-containing constituent of respiratory complex II, is impacted to an even greater extent by Sod2IR expression, with activity reduced to 33% and 31%, respectively, in the two Sod2IR strains. Fumarase, an enzyme of the TCA cycle that is not susceptible to superoxide inactivation, remains unaffected by expression of Sod2IR.
Fig. 5.

RNAi suppression of SOD2 confers loss of mitochondrial aconitase (m-Aconitase) and SDH but not fumarase. Genotypes are +/+;daG32Gal4/+ (A), UASSod2IR15/+;daG32Gal4/+ (B), and UASSod2IR24/+;daG32Gal4/+ (C). The data points represent the mean ± SD of at least six independent determinations. **, Significantly different from +/+;daG32Gal4/+ control, P < 0.005.
To determine whether the loss of aconitase in SOD2-deficient mitochondria was due to reversible, superoxide-mediated loss of the [4Fe-4S] center, extracts were subjected to in vitro reactivation (Fig. 6). Treatment of extracts with iron and a thiol reductant (34) fully restores the activity of mitochondrial aconitase. The aconitase-assay procedure we used also shows that, in contrast to mitochondrial aconitase, cytoplasmic aconitase activity remains relatively unaffected under conditions of SOD2 depletion, suggesting that the immediate consequences of the loss of SOD2 remain confined to the mitochondrial compartment.
Fig. 6.

In vitro reactivation of mitochondrial aconitase inactivated by RNAi suppression of SOD2. Mitochondrial and cytoplasmic aconitase activities in unfractionated adult extracts were detected after reactivation and electrophoretic separation. Genotypes are UASSod2IR24/+;daG32Gal4/+ (1 and 2) and +/+;daG32Gal4/+ (3 and 4). The extent of reactivation was estimated by densitometric analysis of the ratio of mitochondrial/cytoplasmic aconitase activity for each genotype and treatment: +/+;daG32Gal4/+ (control, lane 3), 1.74; +/+;daG32Gal4/+ (reactivated control, lane 4), 2.26 (1.3-fold increase); UASSod2IR24/+;daG32Gal4/+ (RNAi, lane 1) 1.06; UASSod2IR24/+;daG32Gal4/+ (reactivated RNAi, lane 2), 2.55 (2.4-fold increase). These values derive specifically from the figure shown, which is representative of two independent experiments.
RNAi-Mediated Ablation of SOD2 Confers Hypersensitivity to Paraquat.
Toxic hypersensitivity to the redox-cycling agent paraquat is a robust indicator of oxygen defense status in Drosophila (5). As shown in Fig. 7, RNAi-mediated ablation of SOD2 confers exquisite hypersensitivity to paraquat toxicity, with strains Sod2IR15 and Sod2IR24 reduced to 17% and 2% survival, respectively, compared with the control. In terms of hypersensitivity to paraquat, RNAi-mediated suppression of SOD2 using the daG32Gal4 driver is roughly equivalent to loss of SOD1 by germ-line mutation (5).
Fig. 7.

RNAi suppression of SOD2 enhances sensitivity to paraquat toxicity. Young adult males (at least 200 flies per genotype, 10 flies per vial) were exposed to 0 or 2 mM paraquat as described in Experimental Methods. Survivors were enumerated after 48 h of exposure. Genotypes are +/+;daG32Gal4/+ (A), UASSod2IR15/+;daG32Gal4/+ (B), and UASSod2IR24/+;daG32Gal4/+ (C). The data points represent the mean ± SD of at least six independent determinations. **, Significantly different from +/+;daG32Gal4/+ control, P < 0.005. Note that the mean and SD for strain UASSod2IR24/+;daG32Gal4/+ at 0 mM paraquat reflects an accidental loss of flies in one experiment.
RNAi-Mediated Ablation of SOD2 Has No Major Impact on Preadult Viability.
Silencing of Sod2 by Sod2IR exerts no overt effect on preadult development, and developmental time from egg to adult is not altered (data not shown). Moreover, expression of Sod2IR confers no increased preadult mortality or difficulty in eclosion. A cross of [w;+/+;daG32/daG32] females × [w;Sod2IR15/SM5;+/+] males yielded 873 [w1;Sod2IR15/+;daG32/+]/840 [w1;SM5/+;daG32/+] progeny (873/840 = 1.04), whereas a cross of [w;+/+;daG32/daG32] females × [w;Sod2IR24/SM5;+/+] males yielded 780 [w1;Sod2IR24/+;daG32/+]/813 [w1;SM5/+;daG32/+] progeny (780/813 = 0.96). A progeny ratio of 1.0 indicates equivalent viabilities. The absence of detectable SOD2 in Sod2IR-expressing late larvae and pupae (above) excludes the possibility that preadult viability depends on perdurance of maternal SOD2 at levels detectable by this method.
RNAi-Mediated Silencing of Sod2 Causes Rapid Adult-Onset Mortality.
Silencing of Sod2 by daG32Gal4-driven expression of Sod2IR transgenes has a profound effect on adult life span with reductions of 86% and 76% for Sod2IR15 and Sod2IR24, respectively (Fig. 8). This level of life-span reduction is roughly equivalent to that seen in SOD1-null mutants (5).
Fig. 8.

RNAi suppression of SOD2 causes rapid mortality in young adults. Survival of 200 adult males of each genotype on standard cornmeal medium was followed (20 flies per vial) at 25°C with enumeration and transfer of survivors to fresh vials every 2–3 days.
Discussion
It is instructive to compare our results to SOD2-deficient mouse models (43, 44). These mouse models present severe phenotypic syndromes characterized in part by neonatal lethality, tissue-specific loss of the respiratory chain enzymes, NADH dehydrogenase and SDH, and the TCA-cycle enzyme, aconitase (45, 46). Notably, even the partial loss of SOD2 in Sod2−/+ heterozygous mice confers measurable oxidative damage to mitochondrial macromolecules that correlates with diminished mitochondrial function in these animals despite the absence of a gross phenotype (47, 48).
Because no mutant alleles of Sod2 have been reported in Drosophila, RNAi was envisioned to provide a surrogate tool for investigating the biological function of SOD2 in Drosophila. We used the binary GAL4/UAS expression system to broadly express inverted-repeat Sod2 RNAi transgenes in many tissues throughout development. Expression of Sod2IR with the daG32Gal4 driver virtually eliminates detectable SOD2 in late larvae, pupae, and adults, generates increased endogenous oxidant stress that results in diminished activity of critical mitochondrial enzymes, confers adult hypersensitivity to applied oxidant stress, and causes early mortality of newly eclosed adults under normoxic conditions. Although SOD2 activity in Sod2IR Drosophila extracts is below the limits of detection, the nature of RNAi silencing (49, 50) makes it unlikely that Sod2IR expression completely abolishes SOD2 synthesis. Thus, the phenotype of a true SOD2-null mutant might be more severe than observed here. In addition, because preadult viability is not obviously impaired when SOD2 levels are reduced to well below the 50% expected in a hypothetical Sod2-null mutant heterozygote, our results could be taken as evidence that the Sod2 gene is not the origin of the haplolethality of the chromosomal region in which the Sod2 locus resides in Drosophila, with the caveat that the daG32Gal4 driver might be insufficiently active to drive the level of SOD2 below the critical 50% threshold in a cell type that is the lethal focus in such a hypothetical SOD2-null mutant heterozygote.
How then can we account for the high level of SOD2 activity in Sod2IR-expressing embryos? The parentage of the Sod2IR-expressing flies produced for these experiments (+/+;daG32Gal4/TM3 females × Sod2IR/SM5;+/+ males), in which the maternal parent carries only one of the two components of the binary Gal4 system, suggests that the SOD2 seen in embryos is likely maternally derived because both maternally transmitted and zygotic Sod2 transcripts in the embryo (6) presumably would be degraded by embryonic RNAi. However, we believe it unlikely that the preadult viability of Sod2IR-expressing animals relies solely on this maternal SOD2, because daG32Gal4/Sod2IR-expressing females do produce viable, fertile offspring, and a stock doubly homozygous for the daG32Gal4 and Sod2IR transgenes has been established (data not shown). Further experiments will be required to determine whether Sod2IR is actually expressed in embryos and whether it triggers the degradation of Sod2 transcript in embryos as it does in late larvae, pupae, and adults.
It has been suggested that mitochondrial aconitase is a preferential target of oxidative inactivation during aging in Drosophila and in the house fly, Musca domestica (51, 52). Our results show that the reduced mitochondrial aconitase activity conferred by RNAi suppression of SOD2 arises from reversible inactivation, a finding consistent with the mechanism of superoxide-mediated attack on the solvent-exposed [4Fe-4S] cluster (41, 42). Interestingly, the absence of a parallel reduction of cytoplasmic aconitase activity suggests that this superoxide-mediated effect does not directly extend beyond the confines of the mitochondria. Cytoplasmic aconitase is a dual-function protein that exhibits aconitase activity when its [4FE-4S] cluster is intact, but if this cluster becomes lost through cellular iron depletion or superoxide attack, the apoprotein, also known as the iron-regulatory protein 1 (IRP-1) (53), acquires a translational regulatory function by binding to the stem–loop structure of iron-regulatory elements found in several mRNAs related to iron metabolism (54). In Drosophila, such iron-regulatory elements have been identified in several transcripts including SDH-Ip (54) and ferritin (55). Whether IRP-1 of Drosophila (56) regulates the synthesis of mitochondrial aconitase remains to be determined.
The impact of SOD2 deficiency on SDH surpasses the effect on mitochondrial aconitase. Although the structure of Drosophila SDH has not been determined, mammalian SDH possesses an iron-containing subunit (Ip) carrying three Fe-S clusters, one of which is tetranuclear. However, the internal location of the critical [4Fe-4S] cluster likely shields it from inactivation by superoxide. The presence of an iron-regulatory element in the 5′ UTR of the mRNA for the Ip subunit of Drosophila SDH (54) raises the more likely possibility that the reduction in SDH activity conferred by depletion of SOD2 arises through diminished translation of Ip transcripts. Regardless of the mechanism, the striking loss of complex II function suggests that mitochondrial oxidative damage arising from RNAi ablation of SOD2 in Drosophila is widespread and includes important protein targets other than mitochondrial aconitase.
In contrast to the striking and widespread effects of SOD2 deficiency presented by adults, SOD2 deficiency confers no overtly harmful effects on larval and pupal development. Although the reasons for this are unclear, several possibilities could be considered: (i) larvae may use some unknown SOD2-independent mechanism for metabolizing respiration-derived reactive oxygen, (ii) the specific rate of reactive oxygen generation in larvae may be substantially lower than in adults, and (iii) metabolic demands on the TCA cycle may be greater in adults than in larvae. If energy metabolism in larvae is more highly glycolytic than adults, then the latter possibilities would be favored. Although energetically less efficient, glycolysis would generate a lower flux of reactive oxygen by-products than the more highly aerobic respiration of the adult and possibly provide a metabolic refuge for successful development in the absence of SOD2. Further experimentation will be required to resolve this issue.
The data presented here demonstrate that selective diminution of mitochondrial SOD2 in an otherwise normal animal leads in turn to the loss of enzymatic activities critical to the function of the mitochondrial respiratory chain and the TCA cycle and to catastrophic early adult mortality. Although the implications of these results to the mechanisms of aging remain unclear, the results are consistent with the recent observation that FLP transgene-mediated overexpression of SOD2 can extend the life span of Drosophila adults significantly (57).
Acknowledgments
We thank Fanis Missirlis and Aubry deGray for critical reading of the manuscript. This work was supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada (to J.P.P. and A.J.H.).
Abbreviations
SOD, superoxide dismutase
RNAi, RNA interference
SDH, succinate dehydrogenase
TCA, tricarboxylic acid
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.McCord J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 256-275. [PubMed] [Google Scholar]
- 2.Weisiger R. A. & Fridovich, I. (1973) J. Biol. Chem. 248, 4793-4696. [PubMed] [Google Scholar]
- 3.Okado-Matsumoto A. & Fridovich, I. (2001) J. Biol. Chem. 276, 38388-38393. [DOI] [PubMed] [Google Scholar]
- 4.Sturtz L. A., Diekert, K., Jensen, L. T., Lill, R. & Culotta, V. C. (2001) J. Biol. Chem. 276, 38084-38089. [DOI] [PubMed] [Google Scholar]
- 5.Phillips J. P., Campbell, S. D., Michaud, D., Charbonneau, M. & Hilliker, A. J. (1989) Proc. Natl. Acad. Sci. USA 86, 2761-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duttaroy A., Parkes, T., Emtage, P., Kirby, K., Boulianne, G. L., Wang, X. D., Hilliker, A. J. & Phillips, J. P. (1997) DNA Cell Biol. 16, 391-399. [DOI] [PubMed] [Google Scholar]
- 7.Marklund S. L. (1982) Proc. Natl. Acad. Sci. USA 79, 7634-7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hjalmarsson K., Marklund, S., Engstrom, A. & Edlund, T. (1987) Proc. Natl. Acad. Sci. USA 84, 6340-6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., Amanatides, P. G., Scherer, S. E., Li, P. W., Hoskins, R. A., Galle, R. F., et al. (2000) Science 287, 2185-2195. [DOI] [PubMed] [Google Scholar]
- 10.Reaume A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., Wilcox, H. M., Flood, D. G., Beal, M. F., Brown, R. H., et al. (1996) Nat. Genet. 13, 43-47. [DOI] [PubMed] [Google Scholar]
- 11.Parkes T. L., Hilliker, A. J. & Phillips, J. P. (1993) Genome 36, 1007-1014. [DOI] [PubMed] [Google Scholar]
- 12.Phillips J. P., Tainer, J. A., Getzoff, E. D., Boulianne, G. L., Kirby, K. & Hilliker, A. J. (1995) Proc. Natl. Acad. Sci. USA 92, 8574-8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rogina B. & Helfand, S. L. (2000) Biogerontology 1, 163-169. [DOI] [PubMed] [Google Scholar]
- 14.Parkes T. L., Kirby, K., Phillips, J. P. & Hilliker, A. J. (1998) Genome 41, 642-651. [PubMed] [Google Scholar]
- 15.Orr W. C. & Sohal, R. S. (1994) Science 263, 1128-1130. [DOI] [PubMed] [Google Scholar]
- 16.Parkes T. L., Elia, A. J., Dickinson, D., Hilliker, A. J., Phillips, J. P. & Boulianne, G. L. (1998) Nat. Genet. 19, 171-174. [DOI] [PubMed] [Google Scholar]
- 17.Sun J. & Tower, J. (1999) Mol. Cell. Biol. 19, 216-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee Y. M., Misra, H. P. & Ayala, F. J. (1981) Proc. Natl. Acad. Sci. USA 78, 2052-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.FlyBase (1999) Nucleic Acids Res. 27, 85-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fire A., Xu, S., Montgomery, M. K., Kostas, S. A., Driver, S. E. & Mello, C. C. (1998) Nature 391, 806-811. [DOI] [PubMed] [Google Scholar]
- 21.Misquitta L. & Paterson, B. M. (1999) Proc. Natl. Acad. Sci. USA 96, 1451-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennerdell J. R. & Carthew, R. W. (2000) Nat. Biotechnol. 18, 896-898. [DOI] [PubMed] [Google Scholar]
- 23.Lam G. & Thummel, C. S. (2000) Curr. Biol. 10, 957-963. [DOI] [PubMed] [Google Scholar]
- 24.Ashburner M., (1989) Drosophila: A Laboratory Handbook (Cold Spring Harbor Lab. Press, Plainview, NY).
- 25.Rubin G. M. & Spradling, A. C. (1981) Science 218, 348-352. [DOI] [PubMed] [Google Scholar]
- 26.Brand A. H. & Perrimon, N. (1993) Development (Cambridge, U.K.) 118, 401-415. [DOI] [PubMed] [Google Scholar]
- 27.Lindsley D. L. & Zimm, G., (1992) The Genome of Drosophila melanogaster (Academic, New York).
- 28.Heikkila R. E. & Cabbat, F. (1976) Anal. Biochem. 75, 356-362. [DOI] [PubMed] [Google Scholar]
- 29.Beauchamp C. O. & Fridovich, I. (1971) Anal. Biochem. 44, 276-287. [DOI] [PubMed] [Google Scholar]
- 30.Van den Bergh S. G. (1967) Methods Enzymol. 10, 117-122. [Google Scholar]
- 31.Ackrell B. A., Kearney, E. B. & Singer, T. P. (1997) Methods Enzymol. 53, 466-483. [DOI] [PubMed] [Google Scholar]
- 32.Henson C. P. & Cleland, W. W. (1967) J. Biol. Chem. 242, 3833-3838. [PubMed] [Google Scholar]
- 33.Huang T. T., Raineri, I., Eggerding, F. & Epstein, C. J. (2002) Methods Enzymol. 349, 191-213. [DOI] [PubMed] [Google Scholar]
- 34.Hausladen A. & Fridovich, I. (1996) in Nitric Oxide Part B: Physiological and Pathological Processes, ed. Packer, L. (Academic, New York), Vol. 269, pp. 37–41. [Google Scholar]
- 35.Racker E. (1950) Biochim. Biophys. Acta 4, 211-214. [DOI] [PubMed] [Google Scholar]
- 36.Wodarz A., Hinz, U., Engelbert, M. & Knust, E. (1995) Cell 82, 67-76. [DOI] [PubMed] [Google Scholar]
- 37.Smith J. E. & Cronmiller, C. (2001) Development (Cambridge, U.K.) 128, 4705-4714. [DOI] [PubMed] [Google Scholar]
- 38.van Loon A. P. G. M., Pesold-Hurt, B. & Shatz, G. (1986) Proc. Natl. Acad. Sci. USA 83, 3820-3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strain J., Lorenz, C. R., Bode, J., Garland, S., Smolen, G. A., Ta, D. T., Vickery, L. E. & Culotta, V. C. (1998) J. Biol. Chem. 273, 31138-31144. [DOI] [PubMed] [Google Scholar]
- 40.Guidot D. M., McCord, J. M., Wright, R. M. & Repine, J. E. (1993) J. Biol. Chem. 268, 26699-26703. [PubMed] [Google Scholar]
- 41.Gardner P. R. & Fridovich, I. (1991) J. Biol. Chem. 266, 19328-19333. [PubMed] [Google Scholar]
- 42.Gardner P. R., Nguyen, D.-D. & White, C. W. (1994) Proc. Natl. Acad. Sci. USA 91, 12248-12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y., Huang, T. T., Carlson, E. J., Melov, S., Ursell, P. C., Olson, J. L., Noble, L. J., Yoshimura, M. P., Berger, C., Chan, P. H., et al. (1995) Nat. Genet. 11, 376-381. [DOI] [PubMed] [Google Scholar]
- 44.Lebovitz R. M., Zhang, H., Vogel, H., Cartwright, J., Jr., Dionne, L., Lu, N., Huang, S. & Matzuk, M. M. (1996) Proc. Natl. Acad. Sci. USA 93, 9782-9787.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melov S., Schneider, J. A., Day, B. J., Hinerfeld, D., Coskun, P., Mirra, S. S., Crapo, J. D. & Wallace, D. C. (1998) Nat. Genet. 18, 159-163. [DOI] [PubMed] [Google Scholar]
- 46.Melov S., Coskun, P., Patel, M., Tuinstra, R., Cottrell, B., Jun, A. S., Zastawny, T. H., Dizdaroglu, M., Goodman, S. I., Huang, T. T., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 846-851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams M. D., Van Remmen, H., Conrad, C. C., Huang, T. T., Epstein, C. J. & Richardson, A. (1998) J. Biol. Chem. 273, 28510-28505. [DOI] [PubMed] [Google Scholar]
- 48.Van Remmen H., Williams, M. D., Guo, Z., Estlack, L., Yang, H., Carlson, E. J., Epstein, C. J., Huang, T. T. & Richardson, A. (2001) Am. J. Physiol. 281, H1422-H1432. [DOI] [PubMed] [Google Scholar]
- 49.Matzke M., Matzke, A. J. & Kooter, J. M. (2001) Science 293, 1080-1083. [DOI] [PubMed] [Google Scholar]
- 50.Sharp P. A. (2001) Genes Dev. 15, 485-490. [DOI] [PubMed] [Google Scholar]
- 51.Yan L. J., Levine, R. L. & Sohal, R. S. (1997) Proc. Natl. Acad. Sci. USA 94, 11168-11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das N., Levine, R. L., Orr, W. C. & Sohal, R. S. (2001) Biochem. J. 360, 209-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klausner R. D. & Rouault, T. A. (1993) Mol. Biol. Cell 4, 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gray N. K., Pantopoulous, K., Dandekar, T., Ackrell, B. A. & Hentze, M. W. (1996) Proc. Natl. Acad. Sci. USA 93, 4925-4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charlesworth A., Georgieva, T., Gospodov, I., Law, J. H., Dunkov, B. C., Ralcheva, N., Barillas-Mury, C., Ralchev, K. & Kafatos, F. C. (1997) Eur. J. Biochem. 247, 470-475. [DOI] [PubMed] [Google Scholar]
- 56.Muckenthaler M., Gunkel, N., Frishman, D., Cyrklaff, A., Tomancak, P. & Hentze, M. W. (1998) Eur. J. Biochem. 254, 230-237. [DOI] [PubMed] [Google Scholar]
- 57.Sun J., Folk, D., Bradley, T. J. & Tower, J. (2002) Genetics 161, 661-672. [DOI] [PMC free article] [PubMed] [Google Scholar]