

Abstract
G proteins are critical for the regulation of membrane protein function and signal transduction. Nevertheless, coupling between G proteins and membrane proteins with multiple membrane-spanning domains has so far been observed only in higher organisms. Here we show that the polytopic membrane protein FeoB, which is essential for Fe(II) uptake in bacteria, contains a guanine-nucleotide-specific nucleotide binding site. We identify the G4-motif, NXXD, responsible for guanine nucleotide specificity, and show that GTP hydrolysis occurs very slowly. In contrast to typical G proteins, the association and dissociation of GDP were found to be faster than for GTP, suggesting that in the absence of additional factors, FeoB's G protein domain may exist mostly in the GTP-bound form. Furthermore, the binding of GTP is required for efficient Fe(II) uptake through the FeoB-dependent system. Notably, even in bacteria, this covalent linkage between a G protein and a polytopic membrane protein appears, to our knowledge, to be unique. These findings raise the intriguing question whether FeoB represents a primordial archetype of G protein-regulated membrane proteins.
The integration of extracellular signals through G protein-coupled membrane proteins is crucial for maintaining homeostasis in multicellular organisms. However, very little is known about the origins of G protein-coupled membrane processes, mainly because no suitable model systems seemed to exist in prokaryotes. In this study, we provide evidence that the polytopic bacterial membrane protein FeoB contains a G protein similar to small regulatory G proteins found in eukaryotes, and we demonstrate that Fe(II) uptake depends on the function of the G protein. The existence of a polytopic membrane protein that is covalently tethered to a G protein is, to our knowledge, unprecedented in bacteria and raises the intriguing question whether G protein-coupled signal transduction and solute transport in higher organisms are evolutionarily linked with the ancient process of Fe(II) uptake in prokaryotes.
Deletion of FeoB abolishes the ability of bacteria to accumulate Fe(II), a defect that can be rescued by the reintroduction of FeoB into a feoB-deficient strain (1–3). Furthermore, in vivo studies using the pathogen Helicobacter pylori showed that dissipation of the proton motive force across the inner membrane of bacteria and the ATPase inhibitor orthovanadate inhibited Fe(II) uptake. Based on these observations, it was postulated that FeoB functions as a transport ATPase for Fe(II) (1, 2). Whether this working model is correct remains an open question because previously published data can also be explained by alternative models. Notably, the amino acid sequences from FeoB proteins do not share any sequence homology with transport ATPases (4), ABC-type transporters (5, 6), or other Fe(II) transporters (7–11). The only sequence homology that exists links FeoB to GTP-binding proteins. Interestingly, the putative G protein is highly conserved among FeoB proteins from different bacterial species. This finding suggests that GTP binding is important for FeoB function, and at the same time, creates a puzzle because in vivo data suggest that Fe(II) uptake depends on ATP hydrolysis. To address this issue we determined the properties of FeoB's nucleotide binding domain. Our results indicate that the domain is indeed specific for GTP binding and that function of the G protein is required for Fe(II) uptake. Moreover, a detailed kinetic analysis of the G protein domain suggests that the molecular mechanism of FeoB function in Fe(II) uptake is more complex than was previously anticipated.
Materials and Methods
Plasmids.
pET24a-based expression vectors containing the hydrophilic N terminus of FeoB from Escherichia coli (NFeoB, amino acids 1–274) with and without a hexahistidine tag were generated (pET24a-NFeoBh6 and pET24a-NFeoB, respectively) starting from the original FeoB gene described by Kammler et al. (1). In the case of a histidine-tagged protein, the exact amino acid sequence following AA274 in NFeoB was KLAAALEHHHHHH. The expression plasmid pET24a-NFeoBh6 was used to generate two expression vectors coding for single amino acid mutations at position D94 and D123 in NFeoBh6 (NFeoBh6D94N and NFeoBh6D123N) by using Stratagene's QuikChange site-directed mutagenesis kit. For Fe(II) uptake measurements, plasmids containing a cDNA encoding C-terminally decahistidine-tagged (PGHHHHHHHHHH) WT FeoB and a point-mutated FeoBD123N were generated (ptac-FeoBh10 and ptac-FeoBh10D123N).
Overexpression and Purification of NFeoB, NFeoBh6, NFeoBh6D94N, and NFeoBh6D123N.
For overexpression of NFeoB, NFeoBh6, and the D123N mutant, the respective plasmids were transformed into BL21(DE3) cells (Novagen) under kanamycin selection (30 μg/ml); the D94N mutant was overexpressed in BL21(DE3) pLysS cells (Novagen) in the presence of kanamycin (30 μg/ml) and chloramphenicol (12.5 μg/ml). The induction of protein expression was carried out according to the manufacturer's protocol (Novagen). Cells were harvested, resuspended in 20 mM Tris⋅HCl/100 mM NaCl, pH 7.5, and lysed by passing the cells twice through a French press. Cell debris was removed by centrifugation (in a TLA-110 rotor, at 110,000 rpm, for 18 min, at 4°C). The histidine-tagged proteins were purified on a Ni-NTA Superflow agarose column (Qiagen, Valencia, CA) by using 0.5 M imidazole in 20 mM Mops/100 mM NaCl, pH 8.0 (25°C), as the elution buffer. The nontagged NFeoB was purified by ion-exchange chromatography. Cleared cell lysate was applied onto a Q Sepharose (Pharmacia) column and washed with at least 10 column volumes of 20 mM bis-Tris⋅HCl/5 mM NaCl, pH 6.5 (25°C). Bound proteins were eluted by using a linear salt gradient from 5 to 500 mM NaCl in 20 mM bis-Tris⋅HCl, pH 6.5 (25°C). Aliquots (1.5 ml) were collected and analyzed by SDS/15% PAGE. To increase purity, NFeoB-containing aliquots were pooled, diluted 3-fold, and rechromatographed using the same protocol.
Stopped-Flow Measurements.
Stopped-flow measurements were used to determine fast binding and the release of fluorescent nucleotides as described (12). Fluorescent mant-GDP and the nonhydrolyzable nucleoside 5′-[β,γ-imido]triphosphate derivatives mant-GMPPNP, mant-AMPPNP (all from Molecular Probes), and mant-XMPPNP (JenaBioScience, Jena, Germany) were used as probes. Measurements were carried out in 20 mM Mops/100 mM NaCl, pH 7.0, by using an Applied Photophysics stopped-flow instrument (Leatherhead, U.K.). The mant group (N-methylanthraniloyl) was excited at 360 nm and the fluorescence was monitored through a 405-nm cut-off filter. In addition, off-rate constants were also measured directly in an independent set of experiments. The protein (10 μM) was first preincubated with mant-nucleotide (0.5 μM) for 15 min. Thereafter, the fluorescent nucleotide was displaced from the protein by rapidly mixing the incubation mixture in the stopped-flow instrument with a large excess of unlabeled GTP (0.5 mM final concentration; Calbiochem). The off-rate constant can be directly obtained by fitting a single-exponential function to the data points during the first 100 ms. The equilibrium binding constants were calculated according to Kd = koff (direct)/kon.
GTPase Assays.
NFeoB was dialyzed against 20 mM Mops/100 mM NaCl, pH 7.0. Protein (20 μM) and nucleotides (1 mM GTP or ATP final concentrations) were added in a total volume of 100 μl at 25 or 37°C. At several times after starting the hydrolysis reaction, samples were taken and the nucleotide composition of the reaction mixture was analyzed by reversed-phase chromatography with optical detection (254 nm), essentially as described in ref. 13.
Fe(II) Uptake Measurements.
Iron uptake was monitored essentially as described (1) except that LB plates were used in combination with the chromogenic substrate 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal, American Bioanalytical, Natick, MA; see Fig. 3 for the principle of the complementation assay) instead of MacConkey plates. All measurements were done in a feoB-deletion strain background [H1771, kindly provided by K. Hantke, University of Tübingen, Tübingen, Germany (14)]. Plasmids harboring FeoB (ptac-FeoBh10), and FeoB (D123N) (ptac-FeoBh10D123N), as well as the “vector-only” control (pMALc2x, maltose-binding protein under control of the Ptac promoter, New England Biolabs), were transformed into H1771 and grown at 37°C for 16 h under the appropriate antibiotic selection (30 μg/ml kanamycin and/or 100 μg/ml ampicillin) on LB-agar supplemented with different concentrations of Fe(II) ammonium sulfate (4 μM, 100 μM, and 400 μM) or the Fe(II)-specific chelator Ferrozine (0.5 mM). β-Galactosidase activity was monitored by adding 0.2 mg/ml X-Gal to the LB-agar.
Fig 3.

Ferrous iron uptake requires a functional G protein domain and is abolished by a single point mutation. Intracellular iron levels were monitored in vivo by using a fhuF-lacZ reporter construct (A). At normal intracellular levels of Fe(II), the binding of the Fur/Fe(II) complex to the Fur-binding site, fhuF (“Fur-BS”), shuts off lacZ expression. Under these conditions, colonies will appear white. Depletion of intracellular Fe(II) stores and insufficient Fe(II) uptake lead to derepression and will manifest themselves in the formation of blue colonies. (B) Fe(II)-specific uptake was measured in a feoB-deletion strain (Δ) and in the feoB-deletion strain complemented with plasmids harboring histidine-tagged WT FeoB (FeoBh10), histidine-tagged D123N (D123N) mutant unable to bind guanine nucleotides, or a vector-only control (vect. only). Only complementation with FeoB rescued the Fe(II)-uptake defect of the feoB-deletion strain. In this case, complete rescue could be achieved at Fe(II) concentrations >40 μM. Only partial rescue was observed at Fe(II) concentrations <40 μM (not shown), and no rescue was observed at low Fe(II) concentrations (≈4 μM) or in the presence of the Fe(II)-specific chelator Ferrozine. The introduction of the single point mutation D123N, interfering with GTP binding, resulted in derepression (blue colonies) and thus failed to support uptake of sufficient Fe(II). This result established that Fe(II) uptake is functionally coupled to the G protein domain present in the N-terminal domain of FeoB.
Results
FeoB Contains Four of Five G Protein Signature Sequences.
FeoB was originally identified, in E. coli by Kammler et al. (1). Since then, FeoB homologs have been identified in >95 species, which are distributed over all bacterial kingdoms. At the protein level, FeoBs share the same overall design, in which a hydrophilic domain at the N terminus (NFeoB), representing about one-third of the protein, is followed by a hydrophobic C-terminal domain. Within the hydrophobic domain, 7–12 α-helical membrane-spanning segments were predicted, depending on both the bacterial species and the algorithm used. Localization of the predicted transmembrane α-helices in multiple sequence alignments (not shown) suggested that seven of the membrane-spanning domains are shared by all FeoB proteins. Furthermore, based on the FeoB amino acid sequence, no homologies exist to hydrophobic domains of other membrane proteins.
In contrast to the membrane-embedded part, a highly conserved guanine nucleotide binding site is evident within the first ≈160 aa of FeoB (15). Structurally, this site is formed by five loop regions that contain the GTPase signature motifs G1–G5 (ref. 16; see Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). In FeoB, the G1 motif [GXXXXGK(S/T)], is located at position 10–17 (all positions refer to the E. coli FeoB amino acid sequence). The G2 motif is observed at position 37, and the G3 motif (DXXG) maps to residues 56–59. Two potential copies of the NXXD sequence (G4 motif) were identified at positions 91–94 and 120–123 in the E. coli FeoB. Finally, the least conserved consensus sequence within G proteins, the G5 motif (consensus [(T/G)(C/S)A]), could not be located in the various FeoB sequences. Because it had been suggested that FeoB itself is an ATP-dependent transporter for Fe(II) (1, 2) the question arose as to whether the G protein signatures are relevant and/or whether the N-terminal domain of FeoB contains an additional cryptic binding site for ATP.
The N-Terminal Domain of FeoB Specifically Binds GTP but Not ATP.
Nucleotide binding specificity was determined by using a purified, recombinant N-terminal domain of FeoB, NFeoB (=FeoB-1–274) in conjunction with the fluorescent, nonhydrolyzable nucleotide analogs of GTP and ATP, mant-GMPPNP and mant-AMPPNP (12). Association of the nucleotide analogs with the binding site increases fluorescence and can be monitored by measuring emission of the mant group at 450 nm (or >405 nm in stopped-flow experiments). Initial experiments indicated that nucleotide association with the protein occurred instantaneously. Therefore, we used a stopped-flow setup for determining the time dependence of nucleotide binding. These experiments revealed that NFeoB itself bound only to mant-GMPPNP (Fig. 1).
Fig 1.

The N-terminal domain of FeoB (NFeoB) specifically binds guanine nucleotides. Fluorescent, nonhydrolyzable GTP and ATP analogs, mant-GMPPNP and mant-AMPPNP, were rapidly mixed with histidine-tagged NFeoB (20 μM final) in a stopped-flow instrument and the nucleotide-binding reaction was followed by changes in fluorescence (a.u., arbitrary units). Of the two analogs, only the reaction with mant-GMPPNP resulted in the increase of fluorescence that is associated with nucleotide binding.
Identification of the G4 Motif and Change of Substrate Specificity.
To further confirm FeoB's specificity for guanine nucleotides, and to determine which of the two putative G4 motifs at positions 94 and 123 was responsible for nucleotide recognition, we exploited the fact that the NXXD motif engages in the formation of two hydrogen bonds to the guanine base (16) (Fig. 2). Replacement of the aspartic acid residue by asparagine interferes with hydrogen bond formation and changes ligand specificity to xanthine nucleotides (17–19). In binding experiments using mant-GMPPNP, the D94N mutant behaved like WT NFeoB, whereas the D123N mutant failed to recognize the guanine nucleotide (Fig. 2A). However, in the reciprocal experiment using the xanthosine analog mant-XMPPNP, only the D123N mutant was able to bind the ligand (Fig. 2B). This result documented that the mutant protein was properly folded and that the G4 motif, NXXD, responsible for guanine nucleotide specificity, mapped to position 120–123 within the E. coli protein.
Fig 2.

The fourth G protein consensus motif is located at amino acids 120–123 in FeoB (E. coli). Nonhydrolyzable mant-GMPPNP and mant-XMPPNP were used to monitor binding to WT (w.t.) NFeoBh6, and point-mutated proteins D94N and D123N. WT protein and D94N mutant NFeoBh6 bound mant-GMPPNP, whereas the D123N mutant showed no mant-GMPPNP binding (A), indicating that D123 was responsible for the specific interaction with the base part of the nucleotide as is known for other G proteins (39). As expected, the xanthosine mant derivative (mXMPPNP) was recognized only by the D123N mutant (B).
The G Protein Is Required for Fe(II) Uptake.
The results obtained for the D123N mutation also suggested that the introduction of this mutation into a full-length protein should allow testing whether the G protein is required per se for Fe(II) uptake. To this end, a histidine-tagged full-length FeoBD123N mutant was constructed and both FeoB and the mutant were examined for their ability to rescue the Fe(II) uptake defect of a feoB-mutant host strain. As shown in Fig. 3, only FeoB was able to rescue Fe(II) uptake in the feoB-deficient host, whereas the FeoBD123N mutant and the vector-only control were indistinguishable from the feoB-mutant host strain alone. Notably, the Fe(II)-uptake-deficient phenotype remained stable even in the presence of very high concentrations of extracellular iron (tested up to 400 μM), suggesting that any leakage through FeoB-independent uptake systems was negligible. Because the G protein appeared essential for Fe(II) uptake, investigating its properties may provide clues about the molecular mechanism through which the G protein controls Fe(II) uptake.
FeoB's N Terminus Catalyzes Fast Nucleotide Binding and Release.
To establish how FeoB's G protein compares with other G proteins, we determined the nucleotide-binding kinetics for mant-GMPPNP and mant-GDP. Fig. 4A shows an example, following mant-GMPPNP binding at increasing concentrations of NFeoBh6. Plotting of the observed rate constants (kobs) revealed a linear increase as a function of the protein concentration. The plot provided an off-rate constant (koff) of 1.4 s−1 for mant-GMPPNP binding and an on-rate constant (kon) of 0.36 μM−1⋅s−1 was obtained from the slope of the linear relationship (Table 1). The measured mant-GMPPNP binding rate constants corresponded to an equilibrium dissociation constant of KD ≈4 μM (at 20°C), similar to those observed for other bacterial G proteins such as Era, which is involved in cell cycle control (20). Nevertheless, NFeoB's affinity for GTP is about three orders of magnitude lower than the binding of GTP by small regulatory G proteins such as p21-ras (21) or the α-subunit of heterotrimeric G proteins (22).
Fig 4.

Determination of kinetic parameters for mant-GMPPNP and mant-GDP binding. (A) Hexahistidine-tagged NFeoB (NFeoBh6) was rapidly mixed at various protein concentrations in the presence of mant-labeled nucleotides. (Inset) Traces of mant-GMPPNP binding at the indicated final protein concentrations. Each trace represents the average of ≈10 individual measurements. The averages were fitted with a single-exponential function to obtain the observed rate constants (kobs). When plotted as a function of the protein concentration, the observed rate constants followed a linear relationship, which allowed determination of the on- and off-rate constants as well as calculation of the equilibrium binding constant Kd = koff/kon. (B) Displacement of GDP from NFeoBh6 occurred instantaneously. From these measurements, the off-rate and equilibrium binding constants were obtained as described in Materials and Methods.
Table 1.
Rate constants for the association (kon) and dissociation (koff) of mant-nucleotides with NFeoBh6, NFeoBh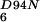 , and NFeoBh
, and NFeoBh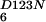
| Protein | Constant (20°C) | mant-GMPPNP | mant-XMPPNP |
|---|---|---|---|
| NFeoBh6 (WT) | kon, μM−1⋅s−1 | 0.36 | No binding |
| koff, s−1 | 1.42 | ||
| Kd, μM | 3.94 | ||
NFeoBh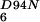
|
kon, μM−1⋅s−1 | 0.50 | No binding |
| koff, s−1 | 2.48 | ||
| Kd, μM | 4.98 | ||
NFeoBh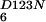
|
kon, μM−1⋅s−1 | No binding | 0.05 |
| koff, s−1 | 1.83 | ||
| Kd, μM | 36.6 |
Highest protein concentration used: 20 μM.
A hallmark of regulatory G proteins is their slow release of GDP (21, 23). Unexpectedly, the binding and release of GDP by NFeoB were too fast to be measured by stopped-flow approaches at temperatures >10°C. In part, this problem was overcome by directly measuring the off-rate constant for GDP through a direct competition experiment (koff-direct, Table 2 for NFeoB and Fig. 4B for NFeoBh6). The GDP-binding experiments showed that both association and dissociation of GDP are faster than for GTP. As discussed in more detail below, such a binding behavior suggests that FeoB's G protein domain is subject to regulation by additional factors such as the intracellular loops, the C terminus, or other, as yet unidentified, accessory proteins.
Table 2.
Rate constants for the association (kon) and dissociation (koff) of mant-nucleotides with untagged NFeoB
| Substrate
|
Constant
|
Temperature, °C | ||
|---|---|---|---|---|
| 10 | 20 | 35 | ||
| mant-GMPPNP | kon1, μM−1⋅s−1 | ND | 0.54 | 0.76 |
| koff1, s−1 | ND | 1.83 | 9.5 | |
| koff1-direct, s−1 | ND | 2.23 | 11.6 | |
| Kd, μM | 4.1 | 15.3 | ||
| mant-GDP | kon2, μM−1⋅s−1 | 7.07 | ||
| koff2, s−1 | 32.1 | |||
| koff2 direct, s−1 | 37.7 | 144 | ||
| Kd, μM | 5.3 | |||
ND, not determined.
At temperatures >10°C, apparent rate constants kobs >1000 s−1 were obtained for some/most of the protein concentrations used, indicating that GDP binding and dissociation occurred on a time scale faster than what can be reliably measured by stopped-flow approaches.
The N Terminus Is a GTPase.
Because of the fast on- and off-rate constants for GTP and GDP, the NTPase activity of NFeoB could be determined by steady-state methods. In line with the properties of regulatory G proteins, NFeoB catalyzed the slow hydrolysis of GTP to GDP with a kcat of ≈0.0015 s−1 (37°C, data not shown). In agreement with the nucleotide-binding data, no significant hydrolysis was observed for ATP (Fig. 5), which suggested that the N-terminal nucleotide-binding domain of FeoB is not responsible for the reported ATP dependence of Fe(II) uptake (2). Furthermore, the addition of orthovanadate, a potent ATPase inhibitor reported to abolish FeoB-dependent Fe(II) uptake in vivo (2), did not influence the rate of GTP hydrolysis by NFeoB, nor did the GTPase activity of the full-length FeoB differ from the isolated NFeoB alone (data not shown). These findings suggested that FeoB itself could serve as a transporter for Fe(II) only if assisted by other factors.
Fig 5.

The N-terminal domain of FeoB is a GTPase. No significant ATPase activity was detected even at elevated temperatures, fully consistent with the nucleotide-binding specificity shown in Fig. 1. The NTPase activity was measured as described in Materials and Methods.
Discussion
G protein coupling of membrane processes is traditionally thought of as being unique for higher organisms. In this study, we show that the polytopic bacterial membrane protein FeoB contains a G protein with unusual properties, and that the G protein is required for Fe(II) uptake.
A G Protein Coupled to a Polytopic Membrane Protein.
Our results establish that the N-terminal ≈160 residues of FeoB constitute a G protein. Although rare, the covalent linkage between a G protein and a membrane protein is not unprecedented, as exemplified by the β-subunit of the signal recognition particle receptor (24, 25) or Fzo1p, a membrane protein involved in mitochondrial fusion (26). However, these proteins have only one or two transmembrane α-helices, which primarily function as membrane anchors for the GTPase domain. In contrast, the membrane-embedded core of FeoB is likely to have (at least) seven transmembrane α-helices, whose overall conservation suggests that they play more than just a membrane-anchoring role. Indeed, it has been proposed that FeoB is an Fe(II)-transporting ATPase (1–3). However, the amino acid sequence of FeoB does not support this model. In particular, FeoB does not seem to be a heavy metal-pumping ATPase because it lacks the signature motifs GXXCXXC close to the N terminus and CPC/H in its sixth (or any other) transmembrane α-helix (4). FeoB also does not seem to be a P-type transport ATPase because it lacks the phosphate-acceptor motif (DKTGTXT) and may have fewer than 10 transmembrane α-helices, which constitute a structural hallmark of P-type ATPases (4, 27–30). Moreover, FeoB does not resemble ABC-type transporters. These transporters share a characteristic “four-core-domain organization” (5, 6, 31) consisting of two sets of six membrane-spanning α-helices and two ATPase domains that can either be part of the same polypeptide or be provided in trans through binding to independent ATPase modules. To date, there is no evidence that FeoB forms higher-order oligomers, nor has it been shown that FeoB interacts with separate, soluble ATPase modules, raising the question of how the apparent ATP-dependence of Fe(II) uptake relates to FeoB function. Moreover, FeoB does not share sequence homology with other transporters for Fe(II) (7–11), thus raising the question of whether FeoB itself does indeed transport Fe(II) or fulfills a different function in the uptake of Fe(II). Notably, the membrane part of FeoB lacks homology with any other class of membrane protein identified to date, thus classifying FeoB as, to our knowledge, unique and precluding the possibility of inferring its function from its sequence. Our study shows that FeoB is a G protein-coupled membrane protein and that the function of the G protein is critical for Fe(II) uptake in prokaryotes.
NFeoB Is a G Protein with Unusual Properties.
Lacking sequence homology otherwise, the spacing between NFeoB's G1–G4 motifs almost matched those found in the human oncogene p21-ras. Based on this observation, we first expected NFeoB's biochemical properties to be similar to those of p21-ras-like G proteins. Indeed, NFeoB possessed the slow intrinsic GTPase turnover rate typical for small regulatory G proteins involved in signal transduction cascades. However, NFeoB bound guanine nucleotides with only micromolar affinities. Weak nucleotide binding has previously been reported for other bacterial GTPases such as Era (20), the CgtA protein from Caulobacter crescentus (32), the signal recognition particle receptor FtsY in E. coli (33), as well as for the human guanylate-binding protein hGBP1 (13), and the antiviral GTPase MxA (34) (see also Table 3). The latter two catalyze GTP hydrolysis at a high rate, which was not the case for NFeoB or the purified full-length FeoB. Therefore, FeoB's G protein seemed more closely related to the Era family of bacterial GTPases (35), which biochemically are characterized by a slow rate of GTP hydrolysis coupled with a fast and spontaneous release of GDP. Although NFeoB readily exchanged GDP, its ability to do so at a rate exceeding that of any other G protein by at least one order of magnitude came as a surprise. In fact, the fast dissociation of GDP from the nucleotide-binding site resulted in a reversal of the relative nucleotide-binding affinities, a property shared with hGBP1 and the antiviral MxA protein (13, 34). This particular combination of extremely fast GDP release and slow GTP hydrolysis seemed odd, because coupling of the G protein to solute transport would require a higher rate of GTP hydrolysis, whereas a function as a signaling molecule would require tight GDP binding to stabilize the “off-state” of the G protein. This apparent contradiction could be explained if, like its eukaryotic counterparts, FeoB's G protein is a target for other regulatory proteins such as GTPase-activating factors or guanine-dissociation inhibitors (36).
Table 3.
Comparison of binding-affinity ranges and hydrolytic activities of various types of G proteins
| Type | Overall affinity for nucleotides | Kd(GTP) < Kd(GDP) | GTPase | Refs. |
|---|---|---|---|---|
| Ras-like | pM-nM | No | Slow | 21, 23 |
| Era, CgtA, FtsY | μM | No | Slow | 20, 32, 33, 35 |
| hGBP, MxA | μM | Yes | Fast | 13, 34 |
| NFeoB | μM | Yes | Slow | This work |
Intrinsic hydrolytic activity without a GTPase-activating protein.
The Functional Role of FeoB's G Protein.
Having shown that the nucleotide-binding domain of FeoB acts as a G protein, how can this be reconciled with previous reports suggesting that FeoB functions as a transport ATPase? One solution is to postulate that FeoB is a G protein-coupled receptor directly or indirectly activating a downstream Fe(II) transport-ATPase (see Fig. 7, which is published as supporting information on the PNAS web site). For this model to be correct, one would expect a slow GTPase activity coupled with slow GDP release to keep the G protein in its “off-state” until the membrane-spanning part senses the presence of Fe(II) in the periplasm similar to the Salmonella PmrA/PmrB signal-transduction system (37). In the case of FeoB, the observed fast GDP release by NFeoB combined with its slow rate of GTP hydrolysis would result in a constitutive activated state. This behavior does not make much sense unless as-yet-unidentified factors slow down GDP release by locking the nucleotide into the binding site. On the other hand, fast GDP release would be the prerequisite were FeoB to function as a transport GTPase for Fe(II). However, the slow turnover rate of the GTPase, which could also be measured using detergent-solubilized and purified FeoB (data not shown), argues against this mode of action because the rate of Fe(II) uptake would be too slow to provide sufficient iron (≈105 iron ions per cell; ref. 38) for cell growth and division. The opposite would be true were FeoB to function as a G protein-coupled Fe(II) channel. In this case, the combination of slow GTP turnover and fast GDP release would likely cause a constitutive open state of the channel, thus resulting in Fe(II) overload. Taken together, these arguments emphasize that the established working model of FeoB as an Fe(II) transport ATPase is too simplistic. As a first step in reevaluating the function of FeoB, our study has established that FeoB represents a membrane protein in which a G protein is covalently tethered to a polytopic hydrophobic domain. Furthermore, the function of the G protein is essential for rescue of Fe(II)-uptake defects in feoB-deficient strains, yet the target for the G protein remains unknown. FeoB's unusual molecular design raises the possibility that FeoB represents a “missing link” in the evolution of G protein-coupled membrane processes in higher organisms.
Supplementary Material
Acknowledgments
We thank Dr. Klaus Hantke for providing the FeoB gene and the feoB-mutant strain H1771. We also thank Thomas Yae for excellent technical assistance. We acknowledge Dr. Werner Kuhlbrandt for supporting this project during its initial phase at the Max Planck Institute for Biophysics (Frankfurt/Main, Germany). This research was supported through a Long Term Fellowship (ALTF 596-1998) from the European Molecular Biology Organization, and from the Max Kade Foundation, New York (to T.C.M.). Further support was furnished by the Austrian National Bank (to T.C.M.), the Robert Leet and Clara Guthrie Patterson Trust (to V.M.U.), and a National Institutes of Health grant (GM66145 to V.M.U.). Oligonucleotides for mutagenesis were produced by the Howard Hughes Medical Institute Keck Facility, Yale University.
Abbreviations
mant, N-methylanthraniloyl
GMPPNP, AMPPNP, and XMPPNP, guanosine, adenosine, and xanthosine 5′-[β,γ-imido]triphosphate, respectively
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kammler M., Schon, C. & Hantke, K. (1993) J. Bacteriol. 175, 6212-6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velayudhan J., Hughes, N. J., McColm, A. A., Bagshaw, J., Clayton, C. L., Andrews, S. C. & Kelly, D. J. (2000) Mol. Microbiol. 37, 274-286. [DOI] [PubMed] [Google Scholar]
- 3.Katoh H., Hagino, N., Grossman, A. R. & Ogawa, T. (2001) J. Bacteriol. 183, 2779-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nucifora G., Chu, L., Misra, T. K. & Silver, S. (1989) Proc. Natl. Acad. Sci. USA 86, 3544-3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higgins C. F. (1992) Annu. Rev. Cell Biol. 8, 67-113. [DOI] [PubMed] [Google Scholar]
- 6.Higgins C. F. (1995) Cell 82, 693-696. [DOI] [PubMed] [Google Scholar]
- 7.Dix D. R., Bridgham, J. T., Broderius, M. A., Byersdorfer, C. A. & Eide, D. J. (1994) J. Biol. Chem. 269, 26092-26099. [PubMed] [Google Scholar]
- 8.Askwith C. & Kaplan, J. (1997) J. Biol. Chem. 272, 401-405. [DOI] [PubMed] [Google Scholar]
- 9.Eide D., Broderius, M., Fett, J. & Guerinot, M. L. (1996) Proc. Natl. Acad. Sci. USA 93, 5624-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunshin H., Mackenzie, B., Berger, U. V., Gunshin, Y., Romero, M. F., Boron, W. F., Nussberger, S., Gollan, J. L. & Hediger, M. A. (1997) Nature 388, 482-488. [DOI] [PubMed] [Google Scholar]
- 11.Stearman R., Yuan, D. S., Yamaguchi-Iwai, Y., Klausner, R. D. & Dancis, A. (1996) Science 271, 1552-1557. [DOI] [PubMed] [Google Scholar]
- 12.Lenzen C., Cool, R. H. & Wittinghofer, A. (1995) Methods Enzymol. 255, 95-109. [DOI] [PubMed] [Google Scholar]
- 13.Praefcke G. J., Geyer, M., Schwemmle, M., Robert Kalbitzer, H. & Herrmann, C. (1999) J. Mol. Biol. 292, 321-332. [DOI] [PubMed] [Google Scholar]
- 14.Hantke K. (1987) FEMS Microbiol. Lett. 44, 53-57. [Google Scholar]
- 15.Leipe D. D., Wolf, Y. I., Koonin, E. V. & Aravind, L. (2002) J. Mol. Biol. 317, 41-72. [DOI] [PubMed] [Google Scholar]
- 16.Sprang S. R. (1997) Annu. Rev. Biochem. 66, 639-678. [DOI] [PubMed] [Google Scholar]
- 17.Kang C., Sun, N., Honzatko, R. B. & Fromm, H. J. (1994) J. Biol. Chem. 269, 24046-24049. [PubMed] [Google Scholar]
- 18.Schmidt G., Lenzen, C., Simon, I., Deuter, R., Cool, R. H., Goody, R. S. & Wittinghofer, A. (1996) Oncogene 12, 87-96. [PubMed] [Google Scholar]
- 19.Hwang Y. W. & Miller, D. L. (1987) J. Biol. Chem. 262, 13081-13085. [PubMed] [Google Scholar]
- 20.Sullivan S. M., Mishra, R., Neubig, R. R. & Maddock, J. R. (2000) J. Bacteriol. 182, 3460-3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.John J., Sohmen, R., Feuerstein, J., Linke, R., Wittinghofer, A. & Goody, R. S. (1990) Biochemistry 29, 6058-6065. [DOI] [PubMed] [Google Scholar]
- 22.Bokoch G. M., Katada, T., Northup, J. K., Ui, M. & Gilman, A. G. (1984) J. Biol. Chem. 259, 3560-3567. [PubMed] [Google Scholar]
- 23.Bourne H. R., Sanders, D. A. & McCormick, F. (1991) Nature 349, 117-127. [DOI] [PubMed] [Google Scholar]
- 24.Keenan R. J., Freymann, D. M., Stroud, R. M. & Walter, P. (2001) Annu. Rev. Biochem. 70, 755-775. [DOI] [PubMed] [Google Scholar]
- 25.Tajima S., Lauffer, L., Rath, V. L. & Walter, P. (1986) J. Cell Biol. 103, 1167-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermann G. J., Thatcher, J. W., Mills, J. P., Hales, K. G., Fuller, M. T., Nunnari, J. & Shaw, J. M. (1998) J. Cell Biol. 143, 359-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auer M., Scarborough, G. A. & Kuhlbrandt, W. (1998) Nature 392, 840-843. [DOI] [PubMed] [Google Scholar]
- 28.Stokes D. L., Auer, M., Zhang, P. & Kuhlbrandt, W. (1999) Curr. Biol. 9, 672-679. [DOI] [PubMed] [Google Scholar]
- 29.Toyoshima C., Nakasako, M., Nomura, H. & Ogawa, H. (2000) Nature 405, 647-655. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P., Toyoshima, C., Yonekura, K., Green, N. M. & Stokes, D. L. (1998) Nature 392, 835-839. [DOI] [PubMed] [Google Scholar]
- 31.Locher K. P., Lee, A. T. & Rees, D. C. (2002) Science 296, 1091-1098. [DOI] [PubMed] [Google Scholar]
- 32.Lin B., Covalle, K. L. & Maddock, J. R. (1999) J. Bacteriol. 181, 5825-5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moser C., Mol, O., Goody, R. S. & Sinning, I. (1997) Proc. Natl. Acad. Sci. USA 94, 11339-11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter M. F., Schwemmle, M., Herrmann, C., Wittinghofer, A. & Staeheli, P. (1995) J. Biol. Chem. 270, 13512-13517. [PubMed] [Google Scholar]
- 35.Caldon C. E., Yoong, P. & March, P. E. (2001) Mol. Microbiol. 41, 289-297. [DOI] [PubMed] [Google Scholar]
- 36.Fukumoto Y., Kaibuchi, K., Hori, Y., Fujioka, H., Araki, S., Ueda, T., Kikuchi, A. & Takai, Y. (1990) Oncogene 5, 1321-1328. [PubMed] [Google Scholar]
- 37.Wosten M. M., Kox, L. F., Chamnongpol, S., Soncini, F. C. & Groisman, E. A. (2000) Cell 103, 113-125. [DOI] [PubMed] [Google Scholar]
- 38.Braun V., Hantke, K. & Koster, W. (1998) Met. Ions Biol. Syst. 35, 67-145. [PubMed] [Google Scholar]
- 39.Brunger A. T., Milburn, M. V., Tong, L., deVos, A. M., Jancarik, J., Yamaizumi, Z., Nishimura, S., Ohtsuka, E. & Kim, S. H. (1990) Proc. Natl. Acad. Sci. USA 87, 4849-4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}