

Abstract
RALDH3 (retinal dehydrogenase 3) was characterized by kinetic and binding studies, protein engineering, homology modelling, ligand docking and electrostatic-potential calculations. The major recognition determinant of an RALDH3 substrate was shown to be an eight-carbon chain bonded to the aldehyde group whose kinetic influence (kcat/Km at pH 8.5) decreases when shortened or lengthened. Surprisingly, the β-ionone ring of all-trans-retinal is not a major recognition site. The dissociation constants (Kd) of the complexes of RALDH3 with octanal, NAD+ and NADH were determined by intrinsic tryptophan fluorescence. The similarity of the Kd values for the complexes with NAD+ and with octanal suggests a random kinetic mechanism for RALDH3, in contrast with the ordered sequential mechanism often associated with aldehyde dehydrogenase enzymes. Inhibition of RALDH3 by tri-iodothyronine binding in competition with NAD+, predicted by the modelling, was established kinetically and by immunoprecipitation. Mechanistic implications of the kinetically influential ionizations with macroscopic pKa values of 5.0 and 7.5 revealed by the pH-dependence of kcat are discussed. Analogies with data for non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans, together with the present modelled structure of the thioacyl RALDH3, suggest (a) that kcat characterizes deacylation of this intermediate for specific substrates and (b) the assignment of the pKa of the major ionization (approximating to 7.5) to the perturbed carboxy group of Glu280 whose conjugate base is envisaged as supplying general base catalysis to attack of a water molecule. The macroscopic pKa of the minor ionization (5.0) is considered to approximate to that of the carboxy group of Glu488.
Keywords: enzymology; mouse retinaldehyde dehydrogenase type 3 (RALDH3; ALDH1A3); pH-dependent kinetics; retina; retinoic acid; 3,3′,5-tri-iodothyronine (T3)
Abbreviations: ALDH, aldehyde dehydrogenase; Caps, 3-(cyclohexylamino)propane-1-sulphonic acid; DTT, dithiothreitol; E8, embryonic day 8, etc; GAPN, non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase; IPTG, isopropyl β-D-galactopyranoside; RALDH3, mouse retinaldehyde dehydrogenase type 3 (ALDH1A3); T3, 3,3′,5-tri-iodothyronine
INTRODUCTION
Retinoic acid biosynthesis in the retina involves the generation of retinal from retinol (vitamin A) in reactions catalysed initially by retinol dehydrogenases and subsequently by further oxidation catalysed by RALDHs (retinal dehydrogenases). All are part of the superfamily of ALDHs (aldehyde dehydrogenases). The RALDHs are a group of cytosolic ALDHs that exert high specificity for the NAD+ oxidation of all-trans- or 9-cis-retinal to all-trans- or 9-cis-retinoic acid respectively [1–4]. The recently identified raldh4 gene is mainly expressed in embryonic and adult liver and kidney and has not been identified in the developing retina [4].
Differences in their locations and the domains defined by the expression of each raldh gene and of cyp26, the gene encoding the retinoic acid-catabolizing enzyme CYP26 (cytochrome P450RAI), result in the formation of defined zones containing high concentrations of retinoic acid. During embryogenesis, raldh2 is the first dehydrogenase mRNA expressed in the eye region at E8 (embryonic day 8). It appears transiently in the ventral region and, shortly after, it can be detected in the ectoderm, where raldh3 is also expressed. The dorsal tip of the eye vesicle then expresses cyp26, followed by expression of raldh1 in the dorsal neural retina. By E11.5, distinct spatiotemporal expression patterns are made up of the expression of raldh1, raldh3 and cyp26, but raldh2 is no longer present. CYP26 forms a horizontal boundary between the dorsal RALDH1 region and the ventral RALDH3 region [5,6].
Both RALDH1 and RALDH2 exert substantial substrate specificity for retinals relative to non-physiological aldehydes. Also RALDH2 appears to discriminate markedly in favour of all-trans-retinal, as demonstrated by the reported values of maximal specific activity (incorrectly given the symbol Vmax) divided by Km [7], whereas RALDH1 appears not to so discriminate [1]. These conclusions depend on the assumption that initial rates were measured by the HPLC point assay after 1 h of reaction. The structures of RALDH1 [8] and RALDH2 [9] have been solved by X-ray crystallography and reveal three domains: a novel NAD+-binding domain containing a five-stranded parallel β-sheet, a catalytic domain comprising a six-stranded parallel β-sheet, and a three-stranded anti-parallel β-sheet oligomerization domain. The substrate-binding tunnel of ovine RALDH1 [8] is the largest of any of the ALDH structures solved so far. The entrance is wide and contains amino acid residues whose side chains adopt conformations that maintain the tunnel open and allow access for the retinal substrates [8]. The active site of RALDH1 contains the conserved cysteine residue (Cys302) of the ALDHs, which is transiently acylated during catalysis [10]. The kinetic mechanism [11] involves the sequential binding of NAD+, followed by the aldehyde, which reacts with Cys302 to form a thiohemiacetal intermediate. Hydride transfer to the nicotinamide ring of NAD+ then results in formation of a thiol ester intermediate. This undergoes base-catalysed hydrolysis by Glu268 [12] and, finally, the carboxylic acid product and NADH are released sequentially. The structure of rat RALDH2 in the presence of NAD+ [9] suggests mobility in the catalytic machinery. The substrate-access tunnel is considerably smaller than that in RALDH1 and slightly larger than that in the class 2 ethanal-metabolizing enzyme ALDH2. There is a lack of inter-monomer contacts and a disordered loop that might contribute to the mobility of the catalytic domain. Also, two residues involved in the aromatic cluster in ALDH2, namely Phe453 and the neighbouring Gly454, are replaced by Leu477 and Asn478 in RALDH2, causing movement leading to disorder in this region. Lamb and Newcomber [9] suggested that binding of retinal restores an appropriate conformation in the binding tunnel and catalytic site through an induced-fit mechanism. A recent paper on mouse RALDH2 [13] suggests, on the basis of a combination of kinetic, spectroscopic and crystallographic evidence, that a disorder-to-order transition is linked to catalytic activity.
RALDH3 was first identified by linking it with the production of large amounts of retinoic acid in the developing retina [6]. The protein sequence of mouse RALDH3 (GenBank® accession number AF28044) has 70% identity with that of RALDH1, 71% identity with that of mouse RALDH2 and 94% identity with human RALDH3 [14] (originally designated ALDH6 [15]). More recently, chicken RALDH3 (ALDH6) was identified and cloned [3]. The catalytic characteristics of RALDH3 have been little studied, being limited to showing that the Km values for the oxidation of all-trans-retinal catalysed by the chicken and mouse enzymes are similar (≈0.3 μM) and the Km for the oxidation of the short-chain aldehyde, ethanal, by the chicken enzyme is much larger (≈3 mM). Also, by contrast with RALDH1 and RALDH2, the three-dimensional structure of RALDH3 has not been reported. Differential regulation of RALDHs in the developing retina results in the formation of a steep gradient of retinoic acid, which defines the dorsal and ventral domains. Differences in the kinetic characteristics of the various RALDHs would be expected to contribute to the mechanisms available for the control of retinoic acid synthesis. In view of the lack of structural and kinetic information available for RALDH3, the work reported in the present paper was undertaken to remedy this deficit as a contribution to the eventual understanding of retina development. We here report a modelled three-dimensional structure for RALDH3 and key kinetic and binding characteristics relevant to specificity and catalytic mechanism, including the involvement of ionizing groups. In addition, a study of the possible involvement of the thyroid hormone T3 (3,3′,5-tri-iodothyronine) as a physiological regulator of RALDH3 was initiated.
MATERIALS AND METHODS
Chemicals and reagents
Most chemicals were purchased from Sigma, Poole, Dorset, U.K. Other materials were provided by the following suppliers: restriction enzymes and modification enzymes were from Promega, Chilworth, Southampton, U.K.; Chelating Sepharose Fast Flow resin and gel-filtration columns were from Amersham Biosciences, Little Chalfont, Bucks., U.K.; pET14b was from Novagen, Madison, WI, U.S.A.; tryptone and yeast extract were from Oxoid, Basingstoke, U.K.; sodium dithionite was from Roche, Poole, U.K.; and primers were from Invitrogen, Paisley, Renfrewshire, Scotland, U.K.
Cloning and site-directed mutagenesis
Mouse Raldh3 was amplified from Raldh3-pcDNA (kindly donated by Dr P. McCaffery, Eunice Kennedy Shriver Center at the University of Massachusetts Medical School, Waltham, MA, U.S.A.) with primers specific for the 5′ and 3′ of each cDNA incorporating NdeI or BamHI restriction sites respectively. Following PCR, the product was ligated into the cloning vector pGEM-T Easy (Promega) and subcloned into the expression vector pET14b (Novagen). Sequence analysis confirmed the correct clone of mouse Raldh3-pET14b. Site-directed mutagenesis of mouse Raldh3-pET14b was performed using the GeneEditor™ in vitro Site-Directed Mutagenesis System (Promega). The mutagenic oligonucleotide (TACAATGCACTTTATGCAC) was used in conjunction with the sense-strand selection oligonucleotide, Top Select Oligo, following the manufacturer's instructions. To confirm the T1411C mutation, raldh3F471L-pET14b was digested with NsiI and NdeI, and the absence of a 1.4 kb fragment indicated the incorporation of the base change at position 1411. The positive clone was sequenced using the DNA sequencing service at Qiagen (Hilden, Germany).
Recombinant-protein production and purification
Raldh3-pET14b was transformed into BL21(DE3) pLysS cells and grown to an attenuance (D600) of 0.6, and protein production was induced with 0.4 mM IPTG (isopropyl β-D-galactopyranoside) at 16 °C overnight. Cells were collected by centrifugation (8000 g, 4 °C, 10 min) and resuspended in lysis buffer (0.5 M NaCl/20 mM Tris/HCl, pH 7.9) containing 5 mM imidazole and frozen at −20 °C. Cells were defrosted and lysed by sonication in an ice bath in the presence of 1 mM PMSF. The cell suspension was centrifuged (27000 g, 25 min, 4 °C) and the cell cytosol loaded on to a pre-charged nickel-chelating Sepharose column. The column was washed with 10 vol. of lysis buffer containing 5 mM imidazole, followed by 10 vol. containing 80 mM imidazole. The protein was eluted with 5 vol. of lysis buffer containing 400 mM imidazole. The eluted protein was separated on a S300 HR Sephacryl gel-filtration column (26/60; Amersham Biosciences), equilibrated in 25 mM Tris/HCl pH 8.0/50 mM NaCl/5 mM DTT (dithiothreitol). Proteins were analysed by both denaturing and non-denaturing PAGE and concentrated in Vivaspin 10K 20 ml concentrators (Vivascience AG, Göttingen, Germany). Protein concentration was calculated by measuring the A280 under denaturing conditions on a Hitachi Europe Ltd (Maidenhead, Berks., U.K.) 2010 UV–visible spectrophotometer. An ϵ value of 46380 M−1·cm−1 was used.
Immunoprecipitation
The binding of T3 to human and mouse RALDH3 was assessed by immunoprecipitating proteins bound to T3 with an anti-T3 antibody, αT3 (Fitzgerald Industries International, Concord, MA, U.S.A.). In 500 μl of RALDH3 reaction buffer, 50 μg of cell extract supernatant was incubated in the presence or absence of 100 μM T3 for 30 min on ice. T3-bound protein was immunoprecipitated with anti-αT3 (1:100000) for 1 h on ice and bound to Protein G beads (20 μl of a 50% slurry) by incubating on a rotating wheel at 4 °C for 1 h. The beads were pelleted by centrifugation (14000 g, 4 °C, 2 min) and washed in 1 ml of IP (immunoprecipitation) buffer with six cycles of 10 min rotation and centrifugation (14000 g, 4 °C, 2 min). The beads were then resuspended in SDS sample buffer, boiled for 5 min, centrifuged briefly and the supernatant was separated by denaturing 10%-(w/v)-PAGE. Proteins on the gel were transferred to a nitrocellulose membrane and the Western blot probed with an antibody to the histidine tag (αHis). This approach specifically identified histidine-rich proteins to which T3 is bound.
Dehydrogenase assay
In a total reaction volume of 1 ml, 2–200 nM enzyme was preincubated with 500 μM NAD+ in reaction buffer (50 mM Hepes/50 mM MgCl2/5 mM DTT) for 10 min at 37 °C. The reaction was initiated by the addition of 0–100 μM substrate and the rate of NADH production at 37 °C was measured as a rate of ΔA340/s using a Hitachi 3010 UV–visible spectrophotometer. For inhibitor assays, RALDH3 was pre-incubated with a range of inhibitor concentrations at 37 °C for 10 min and the reaction was initiated by the addition of both substrate and cofactor. For substrates with sub-micromolar Km values, the dehydrogenase assay was performed using an Applied Photophysics (Leatherhead, Surrey, U.K.) SX•18MV-R stopped-flow reaction analyser. The reactants were allowed to equilibrate to 37 °C in the chamber for 10 min before firing. Each reaction was measured for 20–50 s as ΔA340/s. The reaction was repeated four times for each data point and each experiment repeated three times. Kinetic data were analysed using DeltaGraph 4.5 to provide values of Km and Vmax.
Determination of dissociation constants of enzyme–ligand complexes
These were determined from changes in intrinsic tryptophan fluorescence. RALDH3 has six tryptophan residues, and at least one residue (Trp189) is part of the substrate-binding tunnel. The fluorescence of 0.4 μM protein in a total volume of 3 ml in a 10 mm quartz cuvette was measured by excitation of 280 nm and emission at 340 nm at 20 °C in a Hitachi 2010 spectrofluorimeter. The change in fluorescence (ΔF) was measured after sequential addition of substrate, with account taken of the change in concentration. The dissociation constant (Kd) was calculated using eqn (1) and DeltaGraph 4.5:
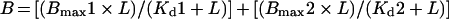 |
(1) |
where B is ΔF, Bmax is the maximal ΔF, L is the ligand concentration and Kd is the dissociation constant. The first term of the right-hand side of the equation represents specific binding (specific ΔF) and the second incorporates non-specific binding (non-specific ΔF). These terms are plotted separately as a function of substrate concentration.
pH-dependence studies
These were carried out over the pH range 6.25–10.5 at pH intervals of 0.25 using a substrate concentration (100 μM) that was 100 times the Km values to provide values of vi that approximate closely to Vmax values. Values of kcat were calculated by dividing these Vmax values by [RALDH]. Buffers used were Mes (pH 6.0– 7.25), Hepes (pH 7.0–8.75) and Caps [3-(cyclohexylamino)-propane-l-sulphonic acid; pH 9.75–10.25].
Computer evaluation of pH-dependent kinetic data
These were evaluated using the basis described in [16]. pH-dependent kinetic studies, sometimes involving a multiplicity of reactivity states, require a rapid method of evaluating a series of kinetic models differing in the number and reactivity of the reactive states (see [17]). This is the situation that obtains in the present work. A useful method is provided by the multitasking application program SKETCHER [18,19]. SKETCHER permits rapid estimation of characterizing parameters in the general set of equations for the various models by means of interactive manipulation of calculated curves. This approach allows different models to be readily distinguished and the parameters thus obtained provide provisional estimates for analysis by weighted non-linear regression. The regression values are generally close to the SKETCHER values, particularly in the case of the complex models, when one or more of the parameters is known from an independent experiment, which obviates problems deriving from multiple local minima.
Structural modelling of RALDH3
The RALDH3 protein structure was modelled using MODELLER 4 [20] and RALDH1 (Protein Databank code 1bxs; [8]) as the template. The structure of retinal was extracted from the Cambridge Structural Database [21]. Retinal binding to RALDH3 and NAD+ in the in conformation was modelled using the graphics program O [22]. Electrostatic potentials were calculated using DELPHI [23], and Figure 1 (below) was produced using MOLSCRIPT [24].
Figure 1. Graphic representations of the model of mouse RALDH3.

(A) Overview of the subunit structure of RALDH3 looking down into the substrate-binding tunnel with Cys314 (nucleophile) and NAD+ shown at the bottom of the tunnel. (B) A model of retinal binding to RALDH3 showing NAD+ and Cys314. In this model the NAD+, shown in ball-and-stick representation, is in the out position and Glu280 in the in position. Also shown are hydrophobic amino acids that line the tunnel, namely Phe182, Trp189, Phe308, Phe471, Tyr472, Phe477 and Val131. The β-ionone ring sits above the binding tunnel and may make few or no interactions with the enzyme. (C) Modelled tetrahedral intermediate state showing the proposed tetrahedral species with the oxyanion stabilized by hydrogen bonding to Asn181 and the amide of Cys314. The water molecule is activated by Glu280 in the in position in this model. Glu280 is adjacent to Glu488.
RESULTS AND DISCUSSION
The RALDH variants
The crystal structures of RALDH1 and RALDH2 and some kinetic characterization of these enzymes have been reported (see the Introduction). By contrast, neither the structure of RALDH3 nor substantial kinetic characterization has been published. In the present work the three-dimensional structures of RALDH3 and the key tetrahedral intermediate in the deacylation process were modelled using computational methods to provide a basis for the design and interpretation of the experiments also reported here.
Sequence alignment of mouse RALDH3 and sheep RALDH1
There is considerable conservation in sequence across a range of ALDHs. A BLASTp alignment showed sheep ALDH1A1 (RALDH1) to be the most appropriate for comparison with mouse RALDH3 (results not shown). The sequence identity is distributed throughout the protein sequence, except for the first 17 residues of RALDH3. There are no insertions or deletions in the RALDH3 sequence, which strongly suggests that the three-dimensional structure will be closely similar to that of RALDH1, including the conformations of loops connecting the secondary-structure elements. The structure of RALDH1 has been solved by X-ray crystallography, and its high sequence identity with that of RALDH3 made possible its use as a template for modelling.
Modelled structure of RALDH3
Mouse RALDH3 has 70 and 71% identity with sheep RALDH1 and rat RALDH2 respectively. The RALDH1 structure, however, is refined at higher resolution (2.35 Å; 1 Å=0.1. nm) than that of RALDH2 (2.7 Å), and the RALDH1 structure has a defined position for the nicotinamide group of NAD+ which is not defined in the co-ordinates of RALDH2 [PDB (Protein Databank) code 1bi9] [9]. Additionally 20 residues of RALDH2 which form one side of the substrate access tunnel are disordered in the binary complex, whereas the corresponding residues in RALDH1 are well defined. The lack of insertions and deletions, together with the high sequence identity, increase confidence in the overall features of the model which is necessarily very closely similar to RALDH1 (Figure 1A shows the subunit structure). Because the RALDH1 structure is a binary complex with NAD+, the binary complex of RALDH3 and NAD+ can also be readily modelled. This we call the out conformation. The in conformation of the coenzyme can be modelled using the phosphate positions defined in the RALDH2 binary complex and predicting the position of the nicotinamide moiety. The catalytic machinery of RALDH3 is closely similar to that of RALDH1, comprising the nucleophile Cys314, the predicted general base Glu280, and Gln181, which may contribute to transition-state stabilization. The authors of the RALDH1 structure [8] suggest that the disorder present in the position of the residue equivalent to Glu280 and the observation that NAD+ binds in two distinct modes indicates that flexibility is a key facet of the reaction mechanism. In our RALDH1-based structure of RALDH3, the C-4 atom of the nicotinamide is too far away (7.1 Å) from Cys314 for direct hydride transfer from a thiohemiacetal intermediate to occur during the dehydrogenase reaction; this is the NAD+ out position. In the NAD+ in conformation modelled on the RALDH2–NAD+ phosphate positions the distance is closer at 3.8 Å. These observations are in agreement with a reaction mechanism in which the nicotinamide can move in and out of the active-site pocket, anchored to the protein by the adenine moiety, with the nicotinamide phosphate and ribose acting as a flexible linker. Glu280 can also be in a proximal or distal position with respect to Cys314. In the Glu in position the thiohemiacetal would be dehydrogenated to form the thiol ester intermediate, and in the Glu out position the enzyme–thiol ester would be hydrolysed. The nicotinamide moiety of NAD+ proximal to, and Glu280 distal from, Cys314 would be expected to increase the availability of the cysteine thiolate and also allow hydride transfer from Cys314 thiohemiacetal to the C-4 atom of the nicotinamide of the NAD+, leading to formation of the thiol ester intermediate. Electrostatics calculations support the argument for the enhanced availability of Cys314 when NAD+ is in and Glu280 is out. The nicotinamide may then move away from, and Glu280 towards, Cys314, allowing Glu280 to fulfil the role of general base catalyst by orientating and activating a water molecule to hydrolyse the thiol ester bond. The size and shape of the RALDH3 substrate-access tunnel is similar to that of RALDH1, with aromatic amino acid residues lining the tunnel. As shown in Figure 1(B), the eight-carbon chain binds in the tunnel and the β-ionone ring is relatively exposed on the surface of the enzyme. This may explain the similar affinities that RALDH3 has for octanal and retinal. One clear difference between the access tunnels is the substitution of Phe471 in RALDH3 for Leu458 in RALDH1. The NAD+ cofactor and Cys314 nucleophile are at the bottom of the substrate-access tunnel (Figure 1B).
The tetrahedral intermediate state in deacylation
On the basis of the model of retinal binding to RALDH3 (Figure 1B), a model of a tetrahedral intermediate can readily be constructed. C-15 of the thiol ester intermediate is attacked by a base-activated water molecule, and the electrons move from carbonyl–oxygen double bond of the intermediate to form the oxyanion of the quasi-tetrahedral transition state (Figure 1C). Glu280 acts as the base activating and orientating the water molecule, and the model suggests that the oxyanion may be stabilized by interactions with Asn181 and by the amide group of Cys314.
Establishment of reaction conditions for RALDH3 catalysis assay
Reaction conditions for a general kinetic assay of the oxidation of a number of substrates catalysed by RALDH3 were established using octanal as substrate (see the subsection on substrate specificity). Increase in the concentration of MgCl2 over the range 0–50 mM produced an increase in activity that became essentially constant above 10 mM. MgCl2 had been shown to stimulate catalysis by mouse RALDH2, but to inhibit catalysis by RALDH1 [1,7]. Assay conditions at 37 °C for particular concentrations of NAD+ that produced optimal activity (50 mM Hepes buffer, pH 8.5, containing 20 mM MgCl2, 2 mM DTT, 150 mM KCl and 1 mM EDTA) were adopted as a standard assay for the study of substrate specificity. Because sequence analysis suggested a possible ATP/GTP-binding site in RALDH3, the influence of these potential cofactors was investigated. Neither affected the catalytic activity towards octanal.
Kinetic and binding studies of substrate and cofactor specificity of RALDH3 and the effect of the F471L mutation
The naturally occurring retinoids, all-trans-, 13-cis-, 11-cis- and 9-cis-retinals are highly sensitive to both light and oxygen. In the present work the kinetic and binding characteristics were determined initially using the eight-carbon-chain aldehyde octanal as substrate and wild-type RALDH3. These studies were then extended to obtain analogous characteristics for shorter- and longer-chain non-physiological aldehydes, all-trans-retinal, NAD+ and NADH, with both wild-type enzyme and the F471L mutant. Examples of kinetic saturation curves and binding curves are shown in Figure 2, and parameter values for all of the 11 data sets are listed in Table 2. The kinetic studies were carried out at pH 8.5 as discussed above under Michaelis–Menten conditions, i.e. using [S]0≫[E]T, where [S]0 is initial substrate concentration and [E]T is total enzyme concentration, and measuring initial rates (v). When the concentration of the pathway substrate was varied, the concentration of NAD+ was fixed at close-to-saturating concentration and vice versa when the concentration of NAD+ or NADH was varied. Hyperbolic saturation curves such as those in Figures 2(A) and 2(B) were obtained and the data were used to obtain the values of Km, kcat, kcat/Km and Kd shown in Table 2.
Figure 2. Examples of (A) kinetic saturation curves at 37 °C and pH 8.5 and (B) binding curves.

(A) Saturation of RALDH3 (10 nM) by NAD+ with an octanal concentration of 100 μM. (B) Specific binding of octanal by the RALDH3 F471L mutant.
Table 2. Kinetic and binding parameters at pH 8.5 and 37 °C for wild-type RALDH3 and the RALDH3 F471L mutant.
S.E.M. values for the the kinetic parameters were less than 10% of the values given. Abbreviation: ND, not determined.
| Substrate | Enzyme | Km (μM) | kcat (s−1) | kcat/Km (M−1·s−1) | Kd (μM) |
|---|---|---|---|---|---|
| all-trans-Retinal | Wild-type | 0.2 | 3.9 | 1.95× 107 | ND |
| Mutant | 0.2 | 13.0 | 5.2×107 | ND | |
| Octanal | Wild-type | 0.7 | 4.8 | 6.9×106 | 0.25 |
| Mutant | 0.9 | 5.4 | 6.0×106 | 0.16 | |
| Hexanal | Wild-type | 22.1 | 0.3 | 1.4×104 | ND |
| Decanal | Wild-type | 6.5 | 0.4 | 6.15×104 | ND |
| Benzaldehyde | Wild-type | 103.0 | 0.2 | 1.9×103 | ND |
| NAD+ | Wild-type | 52.8 | 3.3 | 1.0×105 | 0.16±0.03 |
| Mutant | 43.1 | 3.1 | 1.0×105 | 0.13±0.08 | |
| NADH | Wild-type | ND | ND | ND | 39.74±2.22 |
| Mutant | ND | ND | ND | 25.60±3.76 |
Studies on wild-type RALDH3
The catalytic effectiveness (kcat/Km) of wild-type RALDH3 towards non-physiological aldehydes varies markedly with chain length of the substrate. Of the aldehydes investigated, the value of kcat/Km is smallest for benzaldehyde (1.9×103 M−1·s−1), increases 7-fold to 1.36×104 M−1·s−1 for hexanal and by a further 500-fold to 6.86×106 M−1·s−1 for octanal. An additional increase in chain length to decanal results in a decrease in kcat/Km to 6.15×104 M−1·s−1. The variation in kcat/Km for most of these substrates derives mainly from the variation in Km (6.5–103 μM), variation in kcat being smaller (0.2–0.4 s−1). For the most effective of these substrates (octanal), however, the large increase in kcat/Km derives from a combination of a much smaller Km (0.7 μM) and a much larger kcat (4.8 s−1). Insight into the major recognition determinant of RALDH3 is obtained by comparing the values of the Michaelis–Menten parameters for octanal with those for the naturally occurring retinal, namely all-trans-retinal. Km for the retinal is only about one-third of that for octanal, with kcat similar (3.9 s−1) to that for octanal, and thus kcat/Km (1.95×107 M−1·s−1) is only about three times greater than that for octanal (6.86×106 M−1·s−1). Both substrates have an eight-carbon chain, although whereas that in octanal is fully saturated and hence conformationally flexible, that in the retinal is rigid and planar, as a result of the four double bonds in the conjugated system, and possesses two methyl substituents. The surprising result is that the β-ionone ring does not appear to be a significant recognition site for RALDH3. Rather, the major requirement appears to be an eight-carbon chain whose kinetic effectiveness through binding decreases both when shortened to six carbon atoms and lengthened to 10 carbon atoms. This result is in accord with the model of Figure 1(B), which shows that the eight-carbon chain binds in the access tunnel, whereas the β-ionone ring is relatively exposed to the solvent. Comparison of the sequence of RALDH3 with those of other class 1 ALDHs led to the classification of this enzyme as an NAD-dependent dehydrogenase. In the present work the saturation of RALDH3 by NAD+ was established (Figure 2A) and kcat/Km for NAD+ was shown to be about 200 times smaller than for all-trans-retinal and about 70 times smaller than for octanal (Table 2). Preliminary work showed that NAD+ is at least 100 times more effective in the catalysed oxidation of octanal than NADP+.
To complement the kinetic studies, the dissociation constant (Kd) for the complexes of RALDH3 with octanal, NAD+ and NADH were determined by using the intrinsic tryptophan fluorescence of the enzyme as described in the Materials and methods section. Although ALDH from sheep liver has been reported to adhere to an ordered sequential mechanism [11], and this seems often to be assumed for other ALDHs, the similarity of the Kd values for NAD+ (0.16 μM) and octanal (0.25 μM) in the RALDH3 catalysis demonstrates a substantial degree of randomness in this mechanism. The NADH product of the forward reaction binds much less tightly (approx. 40 μM) than NAD+, and the differences between the Km and Kd values for the forward reaction, particularly for those of NAD+ (Kd=0.16 μM; Km=52.8 μM) demonstrate that Km is not purely a binding constant, but is perturbed by one or more additional rate constants, e.g. k+2 as in (k−1+k+2)/k+1.
Effect of mutation of Phe471 to leucine
The model of RALDH3 (Figure 1A) discussed above highlights the differences between the structures of RALDH3 and RALDH1, in particular the nature of residue 471 in RALDH3 (458 in RALDH1). This residue in the model of RALDH3 is phenylalanine, whereas in RALDH1 it is leucine, which provides a more spherical and less rigid steric bulk than phenylalanine. The possibility that access to the catalytic-site cysteine residue (Cys314) might be differentially affected in RALDHs 3 and 1 by residue 471 (458) was investigated in the present work by mutation of Phe471 to leucine in RALDH3. The kinetic and binding parameters for the RALDH3 F471L mutant are compared with those for the wild-type enzyme in Table 2. The largest change resulting from the change of phenylalanine to leucine at position 471 is that the enzyme discriminates somewhat more effectively in favour of all-trans-retinal relative to octanal [kcat/Km (retinal)/kcat/Km (octanal) 5.2×107/6.0×106=8.7 for the mutant and 1.95×107/6.9×106=2.8 for the wild-type enzyme]. The values of Km and kcat for octanal are closely similar for wild-type and mutant enzymes, and kcat/Km for the retinal is approximately three times larger for the mutant than for the wild-type. The Kd values for octanal, NAD+ and NADH change only slightly as a result of the mutation. It is concluded, therefore, that decreasing the steric bulk of the side chain of residue 471 results in only a small increase in kinetic specificity in favour of the larger substrate (retinal) and emphasizes that the major recognition determinant in RALDH3 is the eight-carbon chain.
Inhibition of RALDH3 by T3
Residues 91–107 in RALDH1 have been identified as a thyroid- hormone-binding site [10]. This region is within the NAD+-binding domain which accounts for the competitive nature of the inhibition by T3 of NAD+ binding to RALDH1 [10,14]. In the present work, sequence alignment of RALDH1 and RALDH3 showed that RALDH3 contains a region with 80% sequence identity with the T3-binding site of RALDH1. Kinetic analysis using a fixed concentration of substrate (octanal) and different concentrations of NAD+ at several fixed concentrations of T3 identified this hormone as a competitive inhibitor (see Figure 3). Kmapp is the observed value of Km in the presence of a given concentration of inhibitor, I. The value of Ki for the T3 inhibition of RALDH3, calculated to be 37.7 μM by using eqn (2) and the parameters given in the legend of Figure 3, is somewhat larger than the values reported for the analogous inhibition of RALDH1 (7.4 μM) and ALDH2 (12 μM) [10,14]:
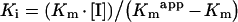 |
(2) |
Direct evidence that T3 binds to RALDH3 was obtained by immunoprecipitation using an antibody to T3 coupled to Protein G beads, followed by an antibody to the histidine-rich region of RALDH3. Proteins with a histidine-rich region and immunoprecipitated by αT3 were detected on the Western blot with the αHis. Hence T3 specifically binds to RALDH3, in accord with the observation of the competitive inhibition described above. Inhibition of RALDH1 activity by T3 has been reported previously (Ki=7.4 μM) [10] and is five times more effective than that obtained for RALDH3 in the present study (Ki=37 μM). It seems possible that this is one mechanism that might be used to maintain precise control of retinoic acid levels in the retina.
Figure 3. Demonstration of competitive inhibition by T3 of the NAD+ oxidation of octanal catalysed by RALDH3.

The concentration of octanal was maintained at 100 μM; the plotted v–[NAD+] data pairs are means of replicates with S.D. values less than 10% of the v values.
Kinetically influential ionizations of RALDH3
The importance of the protonation state of specific side chains in the enzyme mechanism make pH-dependent kinetic studies a valuable contributor to the characterization of enzyme function. The principal objective of such studies is the determination of pKa values approximating to those of individual ionizing groups and rate constants characteristic of reactions of specific ionic forms of the free enzyme molecule and its intermediates on the catalytic pathway. The former can be obtained by the study of the pH-dependence of kcat/Km and the latter by analogous study of kcat. In the present work, using octanal as substrate, it was possible to use the condition [S]0≫Km over an appropriate pH range, but not [S]0≪Km. As a consequence the study was restricted to the pH-dependence of kcat. It was established that, at pH values 7.0, 8.5 and 10.5, the initial rates were closely similar, with [S]0=100 μM and [S]0=1 mM, and thus approximated closely to Vmax values. Division of these by [E]T provided values of kcat over the approximate pH range 6–10. A typical data set is shown in Figure 4. In the approximate pH range 6.0–9.0 the data appear to adhere to a sigmoidal ionization curve. By contrast, at pH values above approx. 9.4, the values of kcat decrease steeply with increase in pH in a manner characteristic of a co-operative denaturation process (results not shown). Closer examination of the data over the pH range 6.0–9.0 using SKETCHER (see the Materials and methods section) demonstrates that the pH-dependence of kcat does not adhere to a single ionization curve, but rather corresponds to the kinetic influence of two ionizations with macroscopic pKa values of 5.0 and 7.5 respectively (eqn 3). The good fit of the data to the equation for two kinetically influential ionizations (equation 3) is shown in Figure 4.
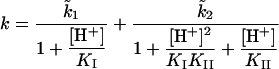 |
(3) |
Figure 4. pH-dependence of kcat for the NAD+ oxidation of octanal at 37 °C catalysed by RALDH3: demonstration of a major and a minor kinetically influential ionization.

The data points are means for three replicates with S.D. values less than 10% of the kcat values. The plotted data points are means for three replicates with S.D. values less than 10% of the kcat values, and appear to conform to a single ionization curve over the pH range ≈6–9. These data fit well to the continuous line theoretical for a pH-dependent rate equation for two reactive protonic states (eqn 3 of the text) with pKI=5.0,  cat(1)=0.16 s−1 (the minor ionization) and pKII=7.5, cat(2)=2.14 s−1.
cat(1)=0.16 s−1 (the minor ionization) and pKII=7.5, cat(2)=2.14 s−1.
Mechanistic implications
Evidence from studies on several ALDHs using kinetics, fluorescence spectroscopy, affinity labelling and site-directed mutagenesis (see [25]) has demonstrated the formation of a covalent intermediate via nucleophilic attack of the conserved cysteinyl thiolate anion on the aldehyde carbon atom of the substrate. The ternary enzyme–cofactor–substrate complex leads to a thioacyl intermediate and reduced cofactor. Chemical logic requires the redox step to involve hydride transfer from a tetrahedral species preceding the thioacyl-enzyme. Detailed kinetic studies on GAPN (non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase; [26,27]) have provided a useful mechanistic model for the investigation of aldehyde dehydrogenases. Major features of the GAPN mechanism are summarized below and related to the results obtained for RALDH3 in the present work.
For GAPN, cofactor binding induces a conformational change within the active centre that leads to increased reactivity of the catalytic-site cysteinyl thiol group and a shift in its pKa from 8.5 to 6.1, detected by the use of thiol-specific time-dependent inhibitors. The kinetically influential ionizations observed in catalysis by GAPN have pKa values of 6.2 and 7.5 in acylation and 6.1 and 7.4 in deacylation. The apparently analogous ionizations observed in deacylation (kcat) for RALDH3 have pKa values of 5.0 for the minor ionization [ cat(1)=0.16 s−1] and 7.5 for the major ionization [cat(2)=2.14 s−1]. For GAPN, the ionization with pKa 7.5 in acylation and pKa 7.4 in deacylation was assigned to the carboxy group of the invariant glutamic acid residue reported as Glu268 [25,26], but now known to be Glu250 [28,29]. The catalytic base and the catalytic-site thiol group in GAPN were designated Glu280 and Cys203 respectively [26,27] by using the numbers of the conserved residues in ALDHs (class I and class II). These residues are Glu250 and Cys284 in the crystal structure of GAPN [28,29]. The evidence for this assignment is strong: mutation of Glu268 (i.e. Glu250) to alanine transforms the double-sigmoidal pH-dependence of the rate constant for acylation (with pKa values 6.2 and 7.5) to a single sigmoidal curve with pKa 6.2. This mutation did not significantly affect the efficiency of the acylation process, but resulted in a large decrease (by at least three orders of magnitude) in the rate of deacylation. The low residual rate did not permit observation of any ionization phenomena.
cat(1)=0.16 s−1] and 7.5 for the major ionization [cat(2)=2.14 s−1]. For GAPN, the ionization with pKa 7.5 in acylation and pKa 7.4 in deacylation was assigned to the carboxy group of the invariant glutamic acid residue reported as Glu268 [25,26], but now known to be Glu250 [28,29]. The catalytic base and the catalytic-site thiol group in GAPN were designated Glu280 and Cys203 respectively [26,27] by using the numbers of the conserved residues in ALDHs (class I and class II). These residues are Glu250 and Cys284 in the crystal structure of GAPN [28,29]. The evidence for this assignment is strong: mutation of Glu268 (i.e. Glu250) to alanine transforms the double-sigmoidal pH-dependence of the rate constant for acylation (with pKa values 6.2 and 7.5) to a single sigmoidal curve with pKa 6.2. This mutation did not significantly affect the efficiency of the acylation process, but resulted in a large decrease (by at least three orders of magnitude) in the rate of deacylation. The low residual rate did not permit observation of any ionization phenomena.
In the case of RALDH3 the values of kcat for the various substrates (Table 2) support the view that deacylation may be rate-determining for the specific substrates, all-trans-retinal and octanal, whereas for the non-specific substrates with much smaller kcat values, a step prior to deacylation may be rate-determining. The modelled structure of the quasi-transition state for deacylation (Figure 1C) suggests that the orientation and activation of the water molecule (base catalysis) is brought about by Glu280 in the in conformation where it is adjacent to Glu488. This result provides the simplest interpretation for the double sigmoidal pH-dependence of kcat, the rate constant for deacylation. The Glu280/Glu488 couple constitutes a two-site acid, protonic dissociation from which would occur in two stages (Scheme 1). The kinetically determined pKa values are macroscopic (molecular) constants pKI and pKII, which are related to the microscopic (group) constants pKA, pKB, pK′A, pK′B of Scheme 1 by the well-known expressions eqns 4 and 5 (see, e.g., [27]).
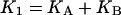 |
(4) |
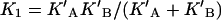 |
(5) |
If the upper route of Scheme 1 predominates, CO2H(a) represents Glu488 and only a small fraction of Glu280 becomes the catalytic base [CO2−(b)] as the pH is increased across pKI (5.0). Most of Glu280 becomes Glu280-CO2− as the pH is increased across pKII (7.5), which will approximate to the intrinsic pKa of this group in the presence of Glu488-CO2−, i.e. pK′B. An analogous interpretation is not available for the minor ionization (pKa 6.1) in GAPN because the structure [28,29] does not contain another carboxy side chain in the vicinity of Glu250. The fact that both pKa values are relatively high for carboxy groups, even after the perturbation downwards in the case of Glu488-CO2H by its proximity to Glu280, may be explained by the hydrophobic nature of the environment of the carboxy couple in RALDH3 provided principally by the side chains of Trp189, Leu439, Phe477 and Tyr497. Examples of carboxy groups with high pKa values resulting from hydrophobic environments include those in lysozyme (pKa 6.5), α-lactoglobulin (pKa 7.5) and lysozyme–glycochitin complex (pKa approx. 8.2) [30].
Scheme 1. Protonic dissociation from a two-site acid.

The relationships between the microscopic (group) acid dissociation constants (KA, KB, K′A, K′B) and the macroscopic (molecular) constants (KI and KII) characteristic of the two stages of ionization of the dicarboxy couple (Glu280 and Glu488) XH2 to XH− to X2− are given by eqns (4) and (5) in the text.
Concluding comment
The combination of modelling and kinetic and binding studies produced a number of advances in understanding RALDH3 and its relationship to some other ALDHs. Surprisingly, the major recognition determinant of RALDH3 is the eight-carbon chain and not the β-ionone ring of all-trans-retinal, which modelling suggests protrudes into solvent. This explains why this enzyme, like RALDH1, but unlike RALDH2, does not discriminate markedly in favour of the physiological substrate relative to non-physiological aldehydes. Also, surprisingly, RALDH3 appears to follow a random kinetic mechanism rather than the ordered sequential mechanism often associated with ALDH enzymes. Inhibition of RALDH3 by T3 in competition with NAD+, predicted by the modelled structure and characterized kinetically, might contribute to control of the concentration of retinoic acid in the retina, in view of the marked difference between RALDH1 and RALDH3 in binding T3. The modelled structure of RALDH3 underscores the notion that the double-sigmoidal pH-dependence of kcat, suggested to be the rate constant for the rate-determining deacylation of specific thioacylenzyme intermediates, is due to successive protonic dissociations from the Glu280/Glu488 couple with Glu280-CO2− acting as the catalytic base.
Table 1. Aromatic residues lining the substrate tunnels of some RALDHs and ALDHs.
Sequences were compiled from PubMed databases and the mouse sequence for each ALDH is shown, except for ALDH6, which is the human RALDH3. The amino acid one-letter code is used for the sake of brevity.
| Enzyme | Aromatic residues | ||||||
|---|---|---|---|---|---|---|---|
| RALDH3 | F182 | W189 | F307 | F308 | F471 | Y472 | F477 |
| RALDH1 | F171 | W178 | F296 | Y297 | V460 | S461 | F466 |
| RALDH2 | F193 | W200 | F313 | F314 | L477 | N478 | F483 |
| ALDH2 | F164 | W171 | F289 | F290 | F453 | G454 | F459 |
| ALDH3 | Y116 | Q123 | F235 | M236 | T396 | L397 | F402 |
| ALDH5 | F187 | W194 | F312 | F313 | V476 | T477 | F482 |
| ALDH6 | F182 | W189 | F307 | F308 | L471 | Y472 | F477 |
| ALDH7 | Y116 | V123 | Y236 | F237 | T406 | L407 | F412 |
| ALDH12 | L156 | W163 | F280 | A281 | R451 | E452 | F457 |
Acknowledgments
We are grateful to Dr Peter McCaffery for providing Raldh3-pcDNA and to Dr David Cobessi for clarifying aspects of the structure of GAPN. C. E. G. was supported by a Wellcome Trust Prize Studentship. Financial support from the Wellcome Trust, the BBSRC (Biotechnology and Biological Sciences Research Council) and the European Union Viteomics Consortium (EU-FPV RTN HPRN-CT-2002-00244) is acknowledged.
References
- 1.Gagnon I., Duester G., Bhat P. V. Enzymatic characterization of recombinant mouse retinal dehydrogenase type. Biochem. Pharmacol. 2003;65:1685–1690. doi: 10.1016/s0006-2952(03)00150-3. [DOI] [PubMed] [Google Scholar]
- 2.Wang X., Penzes P., Napoli J. L. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. J. Biol. Chem. 1996;271:16288–16293. doi: 10.1074/jbc.271.27.16288. [DOI] [PubMed] [Google Scholar]
- 3.Grün F., Hirose Y., Kawauchi S., Ogura T., Umesono K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J. Biol. Chem. 2000;275:41210–41218. doi: 10.1074/jbc.M007376200. [DOI] [PubMed] [Google Scholar]
- 4.Lin M., Zhang M., Abraham M., Smith S. M., Napoli J. L. Mouse RALDH4: molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. J. Biol. Chem. 2003;278:9856–9861. doi: 10.1074/jbc.M211417200. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Wagner E., McCaffery P., Smith D., Andreadis A., Dräger U. C. A retinoic acid synthesizing enzyme in ventral retina and telencephalon of the embryonic mouse. Mech. Dev. 2000;95:283–289. doi: 10.1016/s0925-4773(00)00352-x. [DOI] [PubMed] [Google Scholar]
- 6.Mic F. A., Molotkov A., Fan X., Cuenca A. E., Duester G. RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, optic vesicle and olfactory pit during mouse development. Mech. Dev. 2000;97:227–230. doi: 10.1016/s0925-4773(00)00434-2. [DOI] [PubMed] [Google Scholar]
- 7.Gagnon I., Duester G., Bhat P. V. Kinetic analysis of mouse retinal dehydrogenase type-2 (RALDH2) for retinal substrates. Biochim. Biophys. Acta. 2002;1596:156–162. doi: 10.1016/s0167-4838(02)00213-3. [DOI] [PubMed] [Google Scholar]
- 8.Moore S. A., Baker H. M., Blythe T. J., Kitson K. E., Kitson T. M., Baker E. N. Sheep liver cytosolic aldehyde dehydrogenase: The structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure. 1998;6:1541–1551. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 9.Lamb A. L., Newcomber M. E. The structure of retinal dehydrogenase type II at 2.7 Å resolution: Implications for retinal specificity. Biochemistry. 1999;38:6003–6011. doi: 10.1021/bi9900471. [DOI] [PubMed] [Google Scholar]
- 10.Yamauchi K., Nakajima J., Hayashi H., Horiuchi R., Tata J. R. Xenopus cytosolic thyroid hormone-binding protein (xCTBP) is aldehyde dehydrogenase catalyzing the formation of retinoic acid. J. Biol. Chem. 1999;274:8460–8469. doi: 10.1074/jbc.274.13.8460. [DOI] [PubMed] [Google Scholar]
- 11.MacGibbon A. K., Blackwell L. F., Buckley P. D. Kinetics of sheep-liver cytoplasmic aldehyde dehydrogenase. Eur. J. Biochem. 1977;77:93–100. doi: 10.1111/j.1432-1033.1977.tb11645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X., Weiner H. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry. 1995;34:237–243. doi: 10.1021/bi00001a028. [DOI] [PubMed] [Google Scholar]
- 13.Bordelon T., Montegudo S. K., Pakhomova S., Oldham M. L., Newcomer M. E. A disorder to order transition accompanies catalysis in retinaldehyde dehydrogenase type II. J. Biol. Chem. 2004;279:43085–43091. doi: 10.1074/jbc.M406139200. [DOI] [PubMed] [Google Scholar]
- 14.Yamauchi K., Tata J. R. Characterisation of Xenopus cytosolic thyroid hormone-binding protein (xCTBP) with aldehyde dehydrogenase activity. Chem. Biol. Interact. 2001;130–132:309–321. doi: 10.1016/s0009-2797(00)00274-x. [DOI] [PubMed] [Google Scholar]
- 15.Napoli J. L. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim. Biophys. Acta. 1999;1440:139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 16.Brocklehurst K. A sound basis for pH-dependent kinetic studies on enzymes. Protein Eng. 1994;7:291–299. doi: 10.1093/protein/7.3.291. [DOI] [PubMed] [Google Scholar]
- 17.Pinitglang S., Watts A. B., Patel M., Reid J. D., Noble M. A., Gul S., Bokth A., Naeem A., Patel H., Thomas E. W., et al. A classical enzyme active center motif lacks catalytic competence until modulated electrostatically. Biochemistry. 1997;36:9968–9982. doi: 10.1021/bi9705974. [DOI] [PubMed] [Google Scholar]
- 18.Brocklehurst S. M., Topham C. M., Brocklehurst K. A general kinetic equation for multihydronic state reactions and rapid procedures for parameter evaluation. Biochem. Soc. Trans. 1990;18:598–600. doi: 10.1042/bst0180598. [DOI] [PubMed] [Google Scholar]
- 19.Topham C. M., Salih E., Frazao C., Kowlessur D., Overington J. P., Thomas M., Brocklehurst S. M., Patel M., Thomas E. W., Brocklehurst K. Structure–function relationships in the cysteine proteinases actinidin, papain and papaya proteinase omega. Three-dimensional structure of papaya proteinase omega deduced by knowledge-based modelling and active-centre characteristics determined by two-hydronic-state reactivity probe kinetics and kinetics of catalysis. Biochem. J. 1991;280:79–92. doi: 10.1042/bj2800079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sali A., Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 21.Allen F. H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002;58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 22.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A Found. Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Roccia W., Alexov E., Honig B. Extending the applicability of the non-linear Poisson–Boltzmann equation: Multiple dielectric constants and multivalent ions. J. Phys. Chem. 2001;105:6507–6514. [Google Scholar]
- 24.Kraulis P. J. MOLSCRIPT; a program to produce both detailed and schematic plots of proteins. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 25.Marchal S., Branlant G. Evidence for the chemical activation of essential Cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 1999;38:12950–12958. doi: 10.1021/bi990453k. [DOI] [PubMed] [Google Scholar]
- 26.Marchal S., Rahuel-Clermont S., Branlant G. Role of glutamate-268 in the catalytic mechanism of nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 2000;39:3327–3335. doi: 10.1021/bi9914208. [DOI] [PubMed] [Google Scholar]
- 27.Brocklehurst K. pH-dependent enzyme kinetics in Enzymology Labfax. In: Engel P. C., editor. San Diego: Bios Scientific Publishers Ltd and Academic Press; 1996. pp. 175–198. [Google Scholar]
- 28.Cobessi D., Tete-Favier F., Marchal S., Azza S., Branlant G., Aubry A. Apo and holo crystal structures of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 1999;290:161–173. doi: 10.1006/jmbi.1999.2853. [DOI] [PubMed] [Google Scholar]
- 29.Cobessi D., Tete-Favier F., Marchal S., Branlant G., Aubry A. Structural and biochemical investigations of the catalytic mechanism of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J. Mol. Biol. 2000;300:141–152. doi: 10.1006/jmbi.2000.3824. [DOI] [PubMed] [Google Scholar]
- 30.Fersht A. The pH dependence of enzyme catalysis. In: Fersht A., editor. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: W. H. Freeman and Company; 1999. pp. 169–190. [Google Scholar]