

Abstract
Studies were performed to determine the regulation of DMT1 (divalent metal transporter 1) during RA (retinoic acid)-induced differentiation of P19 embryonic carcinoma cells. Protein and mRNA expression for the +/−IRE (iron response element) forms of DMT1, but not the 1A isoform, were down-regulated within the first few hours upon removal of RA, at which time the cells began to differentiate. The turnover of the +/−IRE isoforms of DMT1 protein during this period was found to be dependent on both the proteasomal and lysosomal pathways. Changes in mRNA levels were shown to be regulated by nitric oxide produced by the induction of neuronal nitric oxide synthase after removal of RA. Nitric oxide functions by inhibiting NF-κB (nuclear factor κB) nuclear translocation and the subsequent binding to the putative NF-κB response element (at −19 to −23) within the 1B promoter. Gel-shift analysis and chromatin immunoprecipitation assay indicated that nuclear NF-κB is capable of binding to this response element and that binding decreases during early stages of differentiation. Luciferase reporter gene assay demonstrated that a mutation in this binding domain leads to decreased activity. These results demonstrate that during neuronal differentiation of P19 cells, there is a decrease in specific isoforms of DMT1 via both post-translational and transcriptional mechanisms.
Keywords: divalent metal transporter 1 (DMT1), lysosomal pathway, NF-κB, proteasomal pathway, protein turnover, transcriptional regulation
Abbreviations: ActD, actinomycin D; ChIP, chromatin immunoprecipitation; CHX, cycloheximide; DMT1, divalent metal transporter 1; EMSA, electrophoretic mobility-shift assay; IFN-γ, interferon γ; IκB, inhibitory κB; IRE, iron response element; IRP, iron regulatory protein; L-NMMA, NG-monomethyl-L-arginine; MRE, metal response element; NF-κB, nuclear factor κB; NOS, nitric oxide synthase; iNOS, inducible NOS; nNOS, neuronal NOS; RA, retinoic acid, RT, reverse transcriptase; Sp1, specific protein 1; WT, wild-type
INTRODUCTION
DMT1 (divalent metal transporter 1; DCT1, NRAMP2 or SLC11A2) is a widely expressed putative 12-membrane spanning protein [1] capable of transporting metals such as iron, manganese and cadmium [2]. It has four known isoforms produced by the alternative splicing of a single gene at both the 5′- and/or 3′-ends. The region in between encodes for 543 amino acids, common for all four isoforms. At the 5′-end of the gene, alternative splicing leads to different transcription start sites at either exon 1A or exon 1B, although, in the latter case, protein translation begins at exon 2 since exon 1B is not translated. Exon 1A isoform encodes an additional 30 amino acid peptide segment on the N-terminus in mice, 31 in rats and 29 in humans [3]. Of the two C-terminal spliced variants, one possesses a stem-loop IRE (iron response element) in the 3′-untranslated region of the mRNA (termed +IRE), whereas the other does not (termed −IRE). The presence of the IRE in the mRNA is thought to be responsible for regulating the +IRE species of DMT1 via the binding and stabilization of the mRNA by IRP (iron regulatory protein) [4]. Regulation of gene expression of DMT1, however, is significantly more complex as shown by the fact that the −IRE species of DMT1 can also be regulated by iron and other processes as well [5–7].
Previous studies from our laboratory indicate that the four isoforms of DMT1 are differentially localized within the cell [8–10]. The disparity in the distribution of DMT1 implies distinct regulatory mechanisms controlling the processing of the different isoforms of the transporter. The two transcription start sites (exons 1A and 1B) of the DMT1 gene are thought to be driven by their respective promoters. Lee et al. [4] have indicated that the 5′-regulatory region of DMT1 contains five putative MREs (metal response elements) that are similar to the MREs found in the metallothionein-IIA gene, three potential Sp1 (specific protein 1)-binding sites and a single IFN-γ (interferon γ) regulatory element. A recent study by Wang et al. [11] showed increased expression of both +IRE and −IRE isoforms in bronchial epithelial cells by TNF-α (tumour necrosis factor α), IFN-γ and LPS (lipopolysaccharide) via transcriptional regulation. A recent study from our group [6] has also shown that hypoxia causes an approx. 60-fold increase in mRNAs of the 1A isoform of DMT1 with a smaller increment in both +IRE and −IRE forms of the transporter. Although the paper by Wardrop and Richardson [12] was the first to demonstrate that the IRE in DMT1 mRNA was, indeed, functional, they also demonstrated that the regulation of DMT1 mRNA was not necessarily via the IRE/IRP mechanism in all cells. These investigators showed that in some cell types such as fibroblasts, the −IRE transcript predominates, indicating that other mechanisms besides post-transcriptional regulation is responsible for controlling the expression of DMT1.
DMT1 is present in neuronal cells where it probably functions as the major transporter for carrying divalent metals into cells. Few studies, however, have focused on changes in DMT1 expression during neuronal differentiation. RA (retinoic acid) is a morphogen present in embryonic tissues [13] and is required for proper development of the spinal cord and brain [14,15]. In the embryo, RA acts as a major factor controlling differentiation and maturation of tissues and is necessary to induce neuronal and glial differentiation [16]. Based on this, we decided to examine the effects of RA on DMT1 expression in mouse P19 embryonic carcinoma cells undergoing neuro-ectodermal differentiation. These cells differentiate into both neurons and glia with the neuronal cells forming neurites within 2 days after differentiation [17]. The signalling processes inducing neuronal differentiation of the P19 cells have not been well characterized since there are essentially no studies that have ever examined the cellular events that take place within the first few hours after removal of RA, a procedure which initiates differentiation.
Accordingly, we examined the effects that removal of RA has on DMT1 expression during the initial stages of differentiation. Results reported in this paper reveal, that during the first 24 h after RA removal, both transcriptional and post-translational processes are responsible for the rapid and selective decrease in expression of 1B, +IRE and –IRE isoforms of DMT1.
EXPERIMENTAL
Materials
P19 embryonic carcinoma cells were purchased from the A.T.C.C. Cell culture materials, TRIzol® reagent and the Superscript one-step RT (reverse transcriptase)–PCR system were obtained from Invitrogen (Gibco BRL, Grand Island, NY, U.S.A.). Anti-β-actin antibody and all-trans RA were purchased from Sigma Chemical (St. Louis, MO, U.S.A.) and secondary antibodies and other materials for Western blots were obtained from Pierce (Rockford, IL, U.S.A.). Vectors pGL3-luciferase and pSV-β-galactosidase were purchased from Promega (Madison, WI, U.S.A.) along with the luciferase and galactosidase assay systems. NF-κB (nuclear factor κB) and Sp1 consensus oligonucleotides for gel-shift analysis were also purchased from Promega and Complete™ protease inhibitor cocktail was from Roche Diagnostics (Mannheim, Germany). MG-132 and ubiquitin-luciferase reporter, Ub-FL, were gifts from Dr Dennis Higgins (Department of Pharmacolgy and Toxicology, University at Buffalo); while Bafilomycin A1 was a gift from Dr Michael Garrick (Department of Biochemistry, University at Buffalo). Anti-α-tubulin antibody was a gift from Dr Jian Feng (Department of Physiology and Biophysics, University at Buffalo).
Cell culture
P19 embryonic carcinoma cells were maintained in α-modified minimal essential medium containing 7.5% (v/v) newborn calf serum and 2.5% (v/v) fetal bovine serum in an atmosphere of 5% CO2 at 37 °C. Differentiation to form neuro-ectodermal cells (neurons and glia) was initiated by treating the cells with 500 nM all-trans RA following a previously described method by Jones-Villeneuve et al. [18]. In brief, cells were plated on bacterial grade Petri dishes for 4 days in the presence of 500 nM all-trans RA, after which the cells were treated with trypsin and transferred to tissue culture plates in the absence of RA. At this point, the cells began to differentiate, forming both neurons and glia by the end of the second day after replating.
Western blots
The cell homogenates were prepared in lysis buffer containing 50 mM Tris/HCl (pH 8.0), 2 mM EDTA, 100 mM NaCl, 1% Triton-X 100 and Complete™ protease inhibitor cocktail. Western blots were performed using affinity-purified antibodies prepared against rat peptide sequences recognizing the +IRE, −IRE and 1A epitopes of DMT1 as described previously [9]. These antibodies were shown to cross-react with the appropriate mouse sequences, as pretreatment with the respective peptide was able to inhibit the bands formed on the Western blots (see the Results section; Figure 1B). For these experiments, cell lysates (∼15 μg of protein/lane) were electrophoresed on 12% (w/v) polyacrylamide gels and subsequently transferred on to nitrocellulose membranes which were incubated with affinity-purified primary antibody (+IRE, −IRE or 1A) overnight. Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was used followed by incubation in chemiluminescent substrate. Anti-β-actin antibody was employed as a gel loading control and the intensities of the bands were evaluated by scanning the blots in a densitometer and then quantified using Quantity One software (Bio-Rad). For each experiment, Western blots were repeated at least three times.
Figure 1. Down-regulation of specific isoforms of DMT1 during neural differentiation.

(A) Western-blot shows down-regulation of +IRE and −IRE isoforms of DMT1 on day 5, a day after removal of RA. The expression of +IRE and −IRE DMT1 isoforms is recovered and is similar to the UD (undifferentiated cells) level by day 11. Western-blot using anti-1A antibody shows no significant change during differentiation. Anti-β-actin antibody was used as gel loading control. Protein loading was 15 μg/lane and electrophoresis was on 12% polyacrylamide gel. (B) Western-blot showing preincubation of antibody with the respective blocking peptide results in inhibition of all the bands on the blot, indicating specificity of the bands. (C) RT-PCR of cells during differentiation shows decrease in +IRE, −IRE and 1B DMT1 mRNA on day 5 after removal of RA. The mRNA expression recovered by the seventh day (day 11) after differentiation was initiated by removal of RA. RT–PCR for the 1A isoform shows no change during differentiation. RT-PCR for β-actin was done as the RNA control. Total RNA (500 ng) was used for each RT-PCR assay. PCR was performed for 30 cycles in an Eppendorf Mastercycler and the products were electrophoresed on a 2% (w/v) agarose gel.
RT–PCR
Total RNA from undifferentiated and differentiating cells was collected using TRIzol® reagent following the manufacturer's method (Invitrogen manual; Invitrogen). RT–PCR was performed using the Superscript one-step RT–PCR system. The primers used for +IRE, −IRE and 1B mRNAs were previously described by Hubert and Hentze [3]. For the 1A isoform, the primers used were as follows: TCCGATGGGGAAGAAGCAGCC (forward) and CCCAGAAGCACCATCGTCTG (reverse). For β-actin, the primers were: CACCACAGCTGAGAGGGAAATCGTGCGTGA (forward) and ATTTGCGGTGCACGATGGAGGGGCCGGACT (reverse). For nNOS (neuronal nitric oxide synthase), the primers were GACTGATGGCAAGCATGACTTC (forward) and GCCCAAGGTAGAGCCATCTG (reverse).
Protein/mRNA turnover studies
To estimate protein turnover, RA-treated cells (4 days) were replated into media containing CHX (cycloheximide; 20 μg/ml) in the absence of RA. The cell lysates were collected at various time points after addition of CHX (0, 2, 4 and 8 h) and Western blots were performed using the DMT1 isoform-specific antibodies. As a control, undifferentiated cells were treated with CHX and cell lysates were collected at the same time points. The intensities of the bands were assessed by scanning the blots in a densitometer and then quantified using Quantity One software (Bio-Rad). For mRNA turnover, cells were incubated with ActD (actinomycin D; 2 μg/ml) at various time intervals after removal of RA and total RNA was collected for RT–PCR analysis using TRIzol®. The gels, stained with ethidium bromide, were scanned and the intensity of the bands was quantified as described above. The band density was plotted against time to determine mRNA turnover rate. The band intensities were shown as means±S.D. in graphical form.
Lysosomal/proteasomal inhibitor
After treatment of cells with RA for 4 days, the cells were replated in the presence of either the proteasomal inhibitor MG-132 (10 μM) and/or the lysosomal inhibitor bafilomycin A1 (500 nM) for 10 h in the absence of RA. For control, the cells were treated with the solvent DMSO. The cells were collected after 6 h and Western blots were performed using the specific anti-DMT1 antibodies described above.
Reporter construct and mutagenesis
The 960-bp 5′-flanking region of 1B exon of DMT1 was isolated by PCR from a bacterial clone (BAC RP23-226N20) purchased from BAC-PAC Resources (Oakland, CA, U.S.A.). The PCR was carried out in a reaction containing high-fidelity platinum Taq polymerase (Invitrogen) using the following primers: AATGGTACCGCATTTAAGAAGCCTTTCTCCA (forward) and ATACTCGAGCGACCCAGTCAACCTCCTGCCC (reverse). The underlined bases indicate restriction sites for KpnI and XhoI respectively. To obtain the 3′-half of the promoter region (+15 to −504), the above-mentioned reverse primer was used along with the following forward primer: AATGGTACCATGAGTCTTGGGAGTAGGACGA. The PCR product was gel purified using a Gel Extraction kit (Qiagen) and then ligated to KpnI- and XhoI-treated pGL3 luciferase vector. The resulting plasmid was amplified in DH5-α cells overnight and then isolated using a Miniprep kit (Qiagen). Mutations were introduced at specific sites using Quik Change site-directed mutagenesis kit (Stratagene) using the manufacturer's method (Stratagene manual; Stratagene, La Jolla, CA, U.S.A.). In the case of NF-κB site mutation, bases GG (−20) were changed to TT, and in the case of Sp1 site mutation, bases CCG (−9) were changed to TTT. For all the above-mentioned plasmids, DNA sequencing was performed to confirm the orientation and/or mutations in the sequences.
Luciferase assay
Undifferentiated P19 cells were treated for 2 days with RA and then transfected, using Lipofectamine™ 2000, with pGL3 and pSV-β-gal plasmids for two more days along with RA. Cell lysates were collected at the end of 4 days of RA treatment or 1 day after removal of RA and luciferase assays were performed on the lysates using the manufacturer's method (Promega manual; Promega). For this, a Berthold luminometer (Lumat LB9507) was used with 3 s measuring time. β-Galactosidase enzyme assay was also performed as a transfection control on the lysate using the manufacturer's method (Promega manual; Promega).
EMSA (electrophoretic mobility-shift assay)/gel-shift
Nuclear extracts from undifferentiated and differentiating P19 cells were collected using a modification of the Dignam et al. method [19]. For this, cells were first lysed with hypo-osmotic buffer (20 mM Hepes, pH 7, 10 mM KCl, 1 mM MgCl2, 0.5 mM dithiothreitol, 0.1% Triton X-100, 20% (v/v) glycerol and protease inhibitors) and then centrifuged to collect the pelleted nuclear fraction which was subsequently resuspended in nuclear extraction buffer (hypo-osmotic buffer plus 420 mM NaCl) for 20 min at 4 °C. After centrifugation at 15000 g for 10 min, the supernatant solutions were collected and used as the nuclear extract. The nuclear extract was incubated with specific DNA probes for 20 min at room temperature (25 °C) and resolved on a non-denaturing gel (6% acrylamide). For NF-κB and Sp1 probes (Promega), the gels were stained with SYBR Green EMSA nucleic acid stain (Molecular Probes) and visualized using 300 nm UV transilluminator. For the DMT1 promoter region, biotin-labelled 30 bp region upstream of 1B exon (sense and anti-sense) was obtained from Invitrogen as single-stranded DNAs (from GenBank®; GI: 63681430). After annealing, the product was gel-purified and used as a probe for gel-shift assay using the LightShift chemiluminescent EMSA kit (Pierce). The reaction was resolved on 6% polyacrylamide gel; the manufacturer's method (Pierce manual; Pierce) was followed to observe the gel-shift.
ChIP (chromatin immunoprecipitation) assay
Proteins were cross-linked with chromatin in undifferentiated P19 cells using 1% formaldehyde. The cells were subsequently sonicated in lysis buffer [20], and an aliquot of the lysate was used in PCR reaction as input. The remaining lysate was cleared with salmon sperm DNA/Protein G–agarose slurry. One-half of the cleared lysate was incubated with p50 or p65 antibody, while the other half was used as a negative control, without the antibody. After reversing the cross-linking, the immunocomplex was digested with proteinase K, and the DNA was purified by phenol extraction. DNA was precipitated for detection by PCR with primers specific to the DMT1-1B promoter region.
RESULTS
Initial studies were performed to determine expression of the specific isoforms of DMT1 at various times before and after initiating P19 cell differentiation by removal of RA. Western blots shown in Figure 1(A) reveal that on day 5, one day after removal of RA, expression of the +IRE form of DMT1 was down-regulated when compared with the undifferentiated cells. Expression subsequently increased during the first few days of differentiation and was completely recovered by day 11 in the fully differentiated cells. Similar results were seen using the −IRE-specific antibody, though down-regulation was not as prominent as that observed with the +IRE antibody. Surprisingly, Western blots performed using the 1A isoform-specific antibody revealed minimal change in expression at any time point monitored before and during differentiation. It should be noted that the presence of multiple bands in Western blot is consistent with previous reports using different antibodies for DMT1 [21,22]. The reason for multiple bands is not clear but may suggest the presence of unidentified isoforms of DMT1 due to mRNA splicing or protein modifications. To determine the specificity of the antibodies, we preincubated them along with the respective peptides used for their preparation. Results of these experiments, indicated in Figure 1(B), reveal that the peptides inhibited antibody binding, indicating that they recognized the expected epitopes on the different isoforms of DMT1.
It was also of interest to determine whether the down-regulation of DMT1 observed was a direct response of exposure to RA for 4 days prior to its removal. Accordingly, cells were treated with RA for an additional fifth day at which time RA was removed. Although not shown, Western blots performed on cell lysates collected on day 5 and 24 h after RA removal revealed a similar pattern as that seen in Figure 1(A) using a 4 day exposure regimen to RA, i.e. there was no change in DMT1 expression at day 5, yet there was a significant decrease on day 6. These results indicate that the down-regulation of DMT1 is a result of biochemical changes occurring after removal of RA and not as a direct consequence of exposure to the retinoid.
The rapid decrease in expression of DMT1 observed within the first few hours after removal of RA could be caused by either a change in transcription or a change in protein turnover or both. To examine whether changes in the rate of gene transcription could account for the observed alteration in protein levels, RT–PCR experiments were performed to determine the levels of mRNA in samples collected during the initial stages of differentiation after removal of RA. Results of these experiments to a large extent parallel the findings presented above for the Western blots. As illustrated in Figure 1(C), mRNA levels for the −IRE and +IRE isoforms of DMT1 exhibited a down-regulation on day 5, a day after removal of RA, which subsequently recovered during the following days of differentiation. A similar sequence of events also occurred with primers that specifically recognize the 1B mRNA. Consistent with our Western-blot findings, there was no change in mRNA levels using primers for the 1A sequence at any of the time points chosen.
The above results possibly suggest that protein levels may be regulated by expression of mRNA during the initial stages of differentiation. To ascertain whether this is the case, protein levels for the +IRE and −IRE isoforms of DMT1 were assessed during the first 24 h after removal of RA. Western blots, shown in Figure 2(A), reveal that the +IRE and −IRE proteins rapidly decreased within the first 4 h, while the 1A isoform, as expected, displayed no change in protein content. A parallel study to determine changes in mRNA levels during this time frame was also performed and results, shown in Figure 2(B), reveal that mRNA levels for the −IRE and +IRE isoforms of DMT1 do not decrease until approx. 10 h after RA removal. As previously noted, there was no change in the 1A-containing mRNA during this period. These results indicate that the levels of the +IRE and −IRE proteins decrease more rapidly than the corresponding mRNA and suggest that mRNA levels are not the primary factor regulating the rapid down-regulation of these isoforms of DMT1 after RA removal.
Figure 2. DMT1 protein down-regulation is earlier than mRNA down-regulation.

(A) Western-blots for DMT1 isoforms using cell lysates during the first 24 h after removal of RA. Cells were collected at 0, 2, 4, 10 and 24 h after replating without RA and Western-blot showed that +IRE and −IRE isoforms were decreased after 4 h, while the 1A isoform did not show any significant change. (B) RT-PCR for DMT1 isoforms during the first 24 h after removal of RA. Total RNA was collected at 0, 4, 10 and 24 h after replating without RA, and RT-PCR was performed as described. The results showed that+and−isoforms were decreased after 10 h, while there was no change in the 1A mRNA isoform. UD, undifferentiated cells.
Based on these observations, we considered the possibility that DMT1 expression was regulated post-translationally by increased protein turnover. Accordingly, studies were performed to determine the half-lives of the different isoforms of DMT1 by treating cells with CHX, an inhibitor of translation, before and after removal of RA. Results of these experiments, illustrated in Figure 3(A), reveal that protein turnover increased significantly for both the +IRE and −IRE isoforms of DMT1 after removal of RA when compared with that for undifferentiated cultures. To quantify protein expression, blots were scanned and the density of the bands with molecular mass between 60 and 65 kDa were determined. These bands were used since they represent the approximate calculated molecular mass of the protein. The half-life of the +IRE isoform of DMT1 decreased from approx. 15 h for the undifferentiated cells to approx. 2.5 h for the RA-treated cells. Similarly, the half-life of the −IRE species changed from 12 h in undifferentiated cells to approx. 3 h in the RA-exposed cells (Figure 3B). Consistent with our previous findings noted above, the half-life for the 1A species of DMT1 did not change during RA-induced differentiation of P19 cells. To ascertain whether the increase in protein turnover is the direct consequence of RA exposure, protein half-life was also assessed on day 4 during RA treatment. Results of these experiments, shown in Figure 3(C), indicate that there was no change in the half-life of any of the DMT1 isoforms at this time point, further demonstrating that the increased turnover observed was caused by the removal of RA.
Figure 3. Increased turnover of specific DMT1 isoforms after RA removal.

(A) Representative Western-blots showing the turnover of DMT1 isoforms after RA removal as compared with undifferentiated cells (UD). See the Experimental section for details. The left panel shows turnover of protein in undifferentiated cells, while the right panel shows that of cells treated with RA for 4 days. The decrease in +IRE and −IRE isoforms was earlier in cells first treated with RA as compared with undifferentiated cells, indicating faster turnover. (B) Graphical representation of band densitometry (band around 61 kDa) from Western-blots for protein turnover. The solid line represents undifferentiated cells, while the dotted line represents cells treated with RA. The graph shows that half-life of +IRE and −IRE DMT1 was decreased in cells after RA treatment as compared with that of undifferentiated cells. There is no significant change in half-life of 1A isoforms of DMT1. Results are presented as means±S.D. for three Western-blots. (C) Western-blot for turnover of DMT1 +IRE and −IRE isoforms during RA treatment. CHX (20 μg/ml) was added to the cells during RA treatment on day 4 and lysates were collected at 0, 2, 4 and 8 h. There was no change in the turnover of proteins as compared with the undifferentiated cells as shown in (A).
The above results suggest that changes in protein degradation are probably responsible for the observed decrease in expression of DMT1 after removal of RA. Since DMT1 is a membrane protein, the expected candidate for its degradation is the lysosomal pathway, although the proteasomal pathway has recently been suggested to play a role in the proteolytic degradation of this transporter [23]. This becomes particularly relevant since RA is known to increase proteasomal activity within cells. Thus, to distinguish which of these pathways is responsible for increased DMT1 turnover after RA treatment, studies were performed in cells treated with the lysosomal inhibitor bafilomycin A1 and/or the proteasomal inhibitor MG-132 after removal of RA. After scanning the blots, the bands around molecular mass 63 kDa were quantified using Quantity One software (Bio-Rad) and statistical analysis was performed using paired t test. As illustrated in Figure 4, +IRE isoform was rescued by MG-132 but not by bafilomycin A1, suggesting that the proteasomal pathway is responsible for the down-regulation of this isoform of DMT1. Interestingly, the −IRE isoform was rescued partially by both MG-132 as well as by bafilomycin A1, suggesting that both proteasomal and lysosomal pathways are responsible for the down-regulation of −IRE isoform during the initial stages of P19 cell differentiation. This is further supported by the observation that the combined treatment of bafilomycin and MG-132 completely prevented the degradation of the −IRE isoform of DMT1 produced by the removal of RA.
Figure 4. DMT1 degradation occurs via both lysosomal and proteasomal pathways after RA removal.

(A) Representative Western-blot showing down-regulation of +IRE and −IRE isoforms after removal of RA (C/day5, lane 2), as shown before. Incubation of the same cells with MG-132 (10 μM) and/or bafilomycin A1 (500 nM) for 10 h prevents the decrease in specific isoforms. Western-blot for 1A does not show any significant change with any treatment. (B) Graphical representation of band densities (band around 63 kDa) from three Western-blot experiments as above. The relative density of bands from each cell treatment is indicated for specific DMT1 isoforms. Results are presented as means±S.D. for three Western blots. Two-tailed t test was performed. *Significant difference from only RA-treated cells with P<0.001; **Significance with P<0.05. (C) Western-blot showing that the down-regulation of α-tubulin after removal of RA (day 5) as compared with undifferentiated cells (UD). The expression is partially restored by day 6. (D) Western-blot showing that the down-regulation of α-tubulin after removal of RA (day 5) was reversed when the cells were incubated with MG-132 (10 μM) for 10 h.
These results suggest that the proteosomal pathway may be up-regulated during differentiation immediately after removal of RA. To further validate these findings, we also examined expression of α-tubulin, a protein known to be degraded via the proteosomal pathway [24]. As illustrated in Figure 4(C), expression of α-tubulin was reduced immediately after RA removal. To verify that this decrease was indeed caused by proteolytic digestion via the proteasomal pathway, we also examined the effect of the proteasomal inhibitor MG-132 (10 μM) on this process. As shown in Figure 4(D), this inhibitor reversed the loss of α-tubulin expression, suggesting that the proteosomal pathway may be up-regulated in the early stages of P19 cell differentiation. Although not shown, we also examined the proteasomal activity using the ubiquitin-luciferase reporter, Ub-FL [25], and results of these experiments duplicate the results with α-tubulin, further confirming increased proteasomal activity after RA removal.
Since there was also down-regulation of +IRE, −IRE and 1B mRNA after removal of RA, studies were performed to determine whether this decrease is due to changes in mRNA turnover. To accomplish this, mRNA levels for the different species of DMT1 in undifferentiated and differentiating cells (first 24 h) were determined in the presence and absence of the transcriptional inhibitor, ActD. As illustrated in Figure 5(A), no difference was observed between the turnover rates for the +IRE, −IRE or 1B mRNA isoform of DMT1 between undifferentiated and differentiating cells. The band densitometry showed that the half-life for these isoforms of DMT1 mRNA was approx. 5, 3 and 4 h respectively. Interestingly, stability of the 1A isoform mRNA remained constant and was considerably greater (>12 h) compared with that for the other species of DMT1 mRNA (Figure 5B).
Figure 5. No change in turnover of DMT1 isoform mRNA after RA removal.

(A) A representative RT-PCR showing the turnover of DMT1 mRNAs after RA removal as compared with undifferentiated cells. See the Experimental section for details. The left panel shows turnover of mRNA in undifferentiated cells, while the right panel shows that of cells treated with RA for 4 days. There was no significant difference in the turnover of mRNA between the two conditions. (B) Graphical representation of band densitometry from RT-PCR for mRNA turnover. The solid line represents undifferentiated cells, while the dotted line represents cells treated with RA. The graph shows that there was no change in half-lives of DMT1 isoforms in cells after RA treatment as compared with that of undifferentiated cells. Results are presented as means±S.D. for three RT-PCR gels.
Studies were then performed to further characterize the mechanism responsible for the down-regulation of the mRNA for the 1B isoforms of DMT1 in P19 cells undergoing differentiation. Prior studies have reported that nitric oxide repressed c-myc gene transcription in a protein synthesis-independent manner in P19 cells by inactivation of NF-κB [26]. Since RA is known to induce production of nitric oxide, we also tested whether the decrease observed in DMT1 mRNA during differentiation is due to nitric oxide. For these studies, RA-treated cells were exposed to the NOS inhibitor, L-NMMA (NG-monomethyl-L-arginine; 500 μM), for 18 h after RA removal. Total RNA from the cells was collected and RT–PCR was performed to detect changes in either the 1A or 1B isoform of the transporter. The results shown in Figure 6(A) indicate that the decrease in 1B isoform of DMT1 that occurred after removal of RA was prevented by incubation with L-NMMA. Consistent with our prior observation, there was essentially no change in expression of the 1A isoform in the presence or absence of the NOS inhibitor. These results imply that the decrease in the 1B mRNA seen after RA removal may be caused by production of nitric oxide. To test whether NOS is being induced during P19 cell differentiation, we performed RT–PCR for inducible and neuronal forms of nitric oxide [iNOS (inducible NOS) and nNOS]. The results, using two different pairs of primers for iNOS, demonstrated lack of expression of iNOS during differentiation (results not shown). In contrast, results illustrated in Figure 6(B) demonstrate induction of nNOS expression within 10 h after removal of RA and are consistent with increased nitric oxide generation being responsible for inhibition of the 1B species of DMT1 during P19 cell differentiation.
Figure 6. Nitric oxide production is responsible for the down-regulation of 1B DMT1.
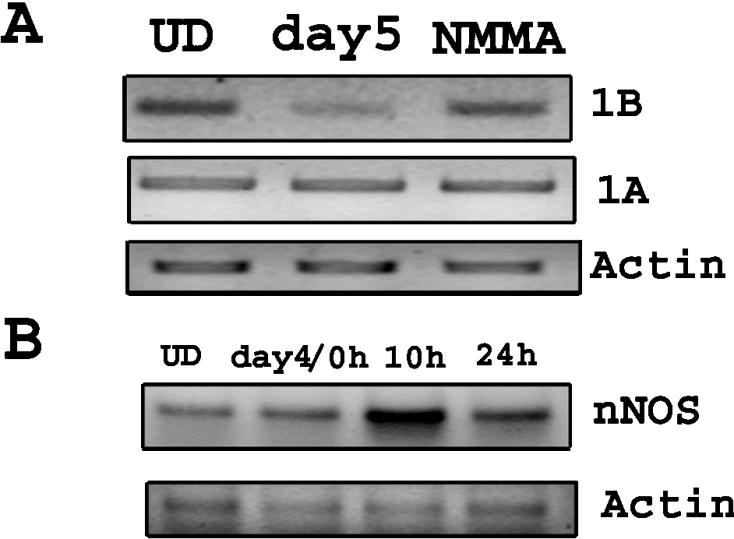
(A) RT-PCR was done using total RNA from undifferentiated cells (UD), cells 18 h after removal of RA (day5) and cells after removal of RA treated with 500 μM L-NMMA (NMMA) for 18 h. The results showed that the expression of the 1B isoform was rescued in cells treated with the NOS inhibitor L-NMMA. No change was seen in 1A isoform mRNA. (B) RT-PCR was done using total RNA from cells during differentiation. The results show increased expression of neuronal NOS 10 h after removal of RA.
As noted above, Park and Wei [26] reported that exposure to nitric oxide caused a decrease in NF-κB nuclear binding activity. Liu et al. [27] reported that the NF-κB site in the NR1 promoter region in P19 cells can interact not only with NF-κB but also with Sp factors. Since both nuclear NF-κB and Sp1 have been reported to be constitutively expressed in undifferentiated P19 cells [26,27], it was necessary to distinguish which of the transcription factors is responsible for the nitric oxide response observed. To establish a link between nNOS induction and decrease in NF-κB DNA binding activity, EMSAs were performed using nuclear extracts from undifferentiated P19 cells using commercially available NF-κB and Sp1 probes (Promega). The results reported in Figures 7(A) and 7(B) confirm previous findings that undifferentiated P19 cells do, in fact, constitutively express nuclear NF-κB and Sp1. To ascertain whether NF-κB is, in fact, responsible for regulation of DMT1, EMSAs were also performed using nuclear extracts from P19 cells at various times after initiating differentiation by the removal of RA. As shown in Figure 7(A), binding of NF-κB to the probe decreased after removal of RA within 4 h, indicative of the fact that nuclear NF-κB binding was attenuated during differentiation. In contrast, as shown in Figure 7(B), there was essentially no change in Sp1 binding after RA removal.
Figure 7. Decrease in NF-κB DNA binding activity with no change in Sp1 binding activity after removal of RA.
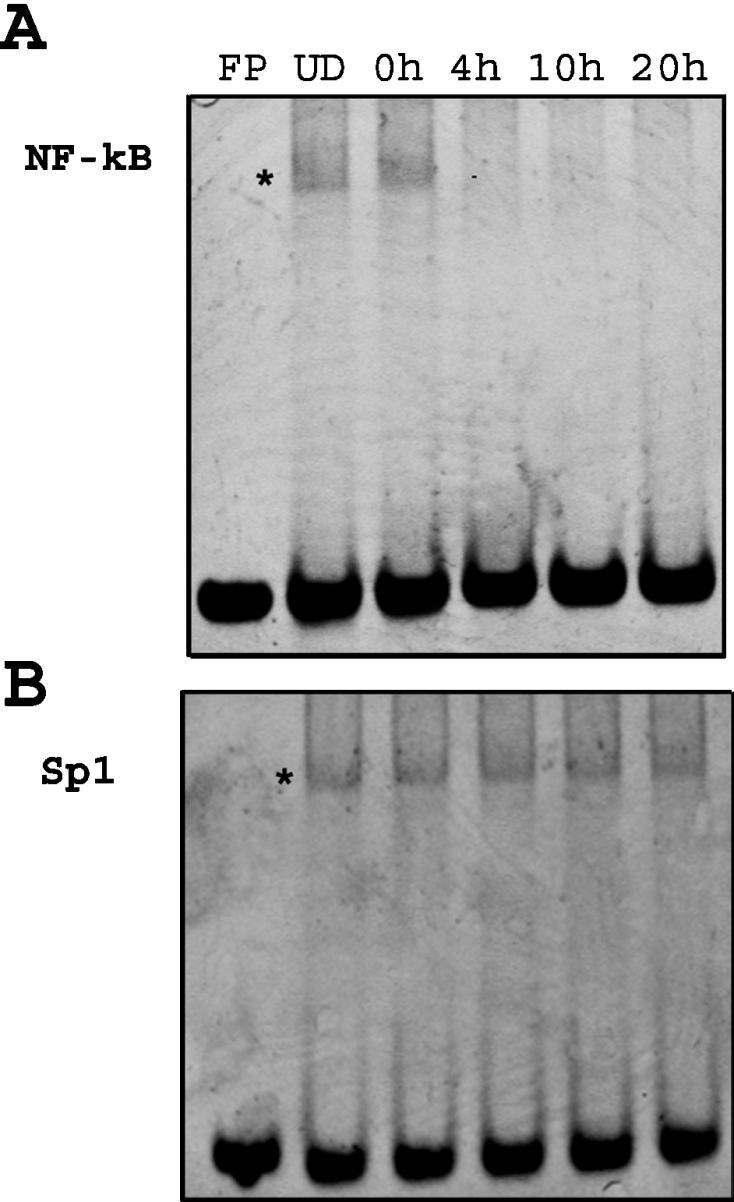
(A) EMSA was performed on nuclear extracts from cells at various times after removal of RA using consensus NF-κB probe. The results showed constitutive binding activity of NF-κB in P19 undifferentiated cells (UD). There was a time-dependent decrease within 4 h after removal of RA. FP stands for free probe without nuclear extract. The asterisk represents the shifted band. (B) EMSA was performed on nuclear extracts from cells at various times after removal of RA using consensus Sp1 probe. The results showed constitutive binding activity of Sp1 in P19 undifferentiated cells (UD). There was no change in DNA binding activity of Sp1 after removal of RA. The asterisk represents the shifted band.
To establish whether the decrease observed in mRNA is due to NF-κB transcriptional regulation of the DMT1 isoforms starting at exon 1B, we needed to first establish the presence of this response element within the promoter region of the 1B species. Accordingly, the 5′-flanking region of the transcription start site of exon 1B was analysed using MatInspector software (Genomatrix) [28] for probable transcription binding sites. We failed to find any RA receptor (RAR-RXR, where RAR is retinoic acid receptor and RXR is retinoid X receptor) binding domains within the promoter, supporting our observation that the responses to DMT1 expression could not be due to the direct effect of RA via its receptor. The analysis revealed the presence of two NF-κB and three Sp1 (GC box) sites, one of which overlaps with one of the NF-κB sites. Since it is considered that important regulatory regions are mostly conserved among species, we also performed phylogenetic footprinting using FootPrinter 2.0 [29]. The comparison of mouse promoter region with rat and human sequences yielded 21 conserved regions. By comparing these results with those obtained with MatInspector revealed two Sp1 sites and one NF-κB site, which are common between all three 1B promoter sequences (Figure 8A).
Figure 8. Luciferase reporter assay determined the site on the 1B promoter region responsible for regulation of the isoform after RA removal.

(A) Diagrammatic representation of 5′-isoforms of DMT1. The Figure shows two alternative start sites of transcription (1A and 1B) with their respective 5′-flanking regions (indicated ‘Pr’). The analysis of that region showed probable binding sites for NF-κB and Sp1. (B) Luciferase reporter assay showing decrease in relative luciferase activity a day after removal of RA when WT promoter (WT-day 4 versus WT-day 5) and 3′-end of the promoter [Prom (−504)-day 4 versus Prom (−504)-day 5] was used. The results are shown as relative luciferase activity (RLU) normalized to β-galactosidase (b-gal) activity. Results are means±S.D. for three separate experiments. A graphical representation of the transfected plasmid is shown. (C) Luciferase reporter assay showing lack of RA removal response when promoter with NF-κB mutation (NFm-day 4 versus NFm-day 5) was used. The response was preserved when promoter with Sp1 mutation (Sp1m-day 4 versus Sp1m-day 5) was used. As a control, luciferase activity was measured in cells transfected with pGL3-control plasmid, which showed no change after RA removal (pGL3c-day 4 versus pGL3-day 5). The results are shown as relative luciferase activity (RLU) normalized to β-galactosidase (b-gal) activity. Results are means±S.D. for three separate experiments. A graphical representation of the transfected plasmid is shown.
Initial studies were performed to determine whether the decrease in 1B isoform expression was caused by changes in NF-κB activation. For these experiments, P19 cells were transiently transfected with reporter plasmids containing the 1B promoter region upstream of the firefly luciferase gene (see the Experimental section). As a transfection control, cells were also transfected with pSV-β-galactosidase plasmid. Luciferase activity was measured in cell lysates collected before (day 4) and 24 h after removal of RA (day 5) and subsequently adjusted for β-galactosidase activity. Results reported in Figure 8(B) reveal that removal of RA caused an approx. 70% reduction in luciferase activity with the WT (wild-type) construct of the 1B promoter (compare WT-day 4 with WT-day 5). When the promoter region equivalent to approximately half of the full-length promoter (+16 to −504) was placed upstream of reporter, a similar response (∼65% decrease) was observed, although the total luciferase activity for each condition was approx. 2.5 times greater when the whole WT promoter was used. These results indicate that the region responsible for down-regulation of the 1B isoform of DMT1 lies between the transcription start site and −504 of the 1B promoter. Within this region, there are two Sp1 sites and one NF-κB site.
It should be noted that several reports indicate that nitric oxide can by itself decrease luciferase activity. To exclude this as a possibility, we included a control where cells were transfected with pGL3-control vector alone. As shown in Figure 8(C), we did not see any effect on luciferase activity after removal of RA using this vector.
In order to determine which of these sites is responsible for regulating DMT1 expression, studies were performed in which the promoter regions were mutated at the overlapping Sp1 and NF-κB sites. Two mutations were made, one in the Sp1 site and one in the NF-κB response element, as described in the Experimental section, and luciferase assays were performed on cell lysates obtained after RA treatment. The results shown in Figure 8(C) demonstrate that mutation of the NF-κB site essentially total reversed the effects that RA removal had on luciferase activity, while mutation of Sp1 binding site had little effect on the decrease in luciferase activity. These results strongly imply that reduced NF-κB binding to the response element between −23 and −19 is most likely responsible for the decrease in the 1B isoforms of DMT1 seen during P19 neural differentiation.
EMSA was also performed using biotin-labelled 30 bp region of the DMT1 promoter, containing one NF-κB and one Sp1 site to determine whether there is a nuclear component within P19 cells capable of binding to this region and whether the magnitude changes after removal of RA. Results shown in Figure 9(A) reveal that there is a component within the nuclear fraction that is capable of binding to this base sequence and further demonstrate decreased binding within the first 12 h upon initiating cell differentiation. Although not shown, the specificity of the observed band-shift was further verified using excess of unlabelled probe. Results of these experiments demonstrate that in the presence of excess probe, the band disappeared using WT construct of the 1B promoter, whereas the same experiment using unlabelled probe containing a mutation in the NF-κB site displayed no change in the band-shift. To further confirm that NF-κB proteins actually bind to 1B promoter region of DMT1, ChIP was performed using anti-p50 and anti-65 antibodies. The results as shown in Figure 9(B) indicate that both p50 and p65 bind to the NF-κB-binding domain on DMT1-1B promoter. Also, there was a selective decrease in signal intensity in the immunoprecipitate obtained with anti-p65 antibody by 10 h after removal of RA. These results indicate that a decrease in p65 binding activity and not that of p50 is responsible for the decreased NF-κB response observed. To confirm the specificity of the immunoprecipitate, PCR was also performed on the same samples using specific primers for +IRE to precipitate the complex and results showed the absence of an amplified signal.
Figure 9. NF-κB binding to the promoter region of the 1B isoform of DMT1 is decreased after removal of RA.

(A) EMSA was performed using nuclear extracts from cells (undifferentiated and 0, 4, 8, 12 and 20 h after removal of RA). Biotin-labelled probe representing promoter region of 1B isoform was incubated with nuclear extracts, and using LightShift chemiluminescent EMSA kit (Pierce), gel-shift was observed. There was constitutive binding of nuclear factor (denoted by *) in nuclear extracts from undifferentiated cells (UD). There was a decrease in binding (less gel-shift band intensity) after removal of RA, also seen by an increase in free probe at the bottom (FP). (B) ChIP assay using primers specific for 1B promoter region shows constitutive binding of p50 and p65 in undifferentiated cells. There was no change in the binding of p50, 10 h after RA removal (RA), but there was a decrease in binding of p65. The first lane (In, input) indicates total chromatin DNA used in PCR reaction, while as a control, no antibody control was used in immunoprecipitation reaction (No Ab). As a PCR control, same samples were used for PCR using +IRE primers. Bands are seen only in the first lane and the no antibody lane.
DISCUSSION
DMT1 is generally considered to be the major transport protein required for cellular uptake of a variety of divalent transition metals including iron, manganese, cobalt, copper and cadmium [21]. As noted above, it exists in at least four distinct isoforms comprised of two N-terminal forms which may be attached to either the +IRE or −IRE carboxy peptide. Transcription of the different isoforms is regulated by two discrete promoters, one controlling expression of the exon 1A species and the other controlling transcription of exon 1B isoforms [3]. Although all of these isoforms are capable of transporting divalent metals, the role of each in maintaining divalent metal homoeostasis in the adult as well as during development has not been clearly defined.
As explained in the Introduction section, P19 embryonic carcinoma cells used in the present study represent a good model for studying changes in the different isoforms of DMT1 during neuronal differentiation. It is reasonable to speculate that during the differentiation process, requirements for iron may be altered to meet the demands associated with the morphological and biochemical changes required for this process. Since DMT1 is the major transporter of iron, we chose to examine whether expression of this transporter would adjust to the needs of the cells during RA-induced differentiation. The results presented in this paper demonstrate that all four forms of DMT1 are present in the undifferentiated cells as well as at all stages of neuro-ectodermal differentiation of the P19 cells. The change in expression was selective for the isoforms of DMT1 in which the start codon begins at exon 2 since there was no apparent change in expression of the 1A species of the transporter. At this point, we cannot speculate as to why these forms of DMT1 are selectively down-regulated during differentiation or whether this change actually contributes to the differentiation process, but the unique selectivity of this raises the important question as to functional role that each of the different species of DMT1 have regarding the maintenance of iron homoeostasis during P19 differentiation.
We initially considered the possibility that the decrease in protein levels observed were caused by changes in mRNA levels and, therefore, studies were performed to examine mRNA content for the different species of DMT1 during the differentiation process. Our findings reveal that mRNA levels for the +IRE and −IRE isoforms of DMT1 as well as the 1B-containing species decreased upon removal of RA, although the changes were delayed relative to the decrease seen in protein content. The half-lives of mRNA for all forms of DMT1 including the 1A form were unaffected during or after treatment with RA, indicating that the decrease in mRNA levels observed must have been caused by changes in transcription of the gene. Since the changes in mRNA levels were considerably slower than those observed for protein during the first few hours after RA removal, we conclude that the loss of mRNA is not responsible for the initial rapid decrease in protein content of the transporter. The decreased transcription of the gene, however, may be responsible for the delayed recovery of DMT1 during the extended differentiation process.
We also needed to exclude the possibility that the process of trypsin treatment and replating may be responsible for the apparent reduction in protein expression of the +IRE- and −IRE-specific DMT1 isoforms. This scenario, however, is unlikely as both isoforms of DMT1 were present immediately after RA removal (see Figure 2A, 0 h). This is further demonstrated by studies indicating that there was no change in expression of these isoforms of DMT1 in trypisnized undifferentiated cells maintained in the absence of RA for 5 days (results not shown). Thus these results establish that the decrease in expression of DMT1 was not produced as a response to the harvesting or physical treatment of the cells but confirm that the selective alteration in +IRE and −IRE forms of DMT1 during the initial stages of P19 cell differentiation are caused by post-translational changes in protein turnover. This is validated by the results demonstrating a 3–6-fold increase in protein turnover respectively within 6 h after removal of RA.
Tabuchi et al. [22] demonstrated that DMT1 is present in the late endosomes and lysosomes in HEp-2 cells, suggesting that DMT1 may be degraded via the lysosomal pathway. Our results, however, indicate that the proteolytic enzymes within this latter compartment are not alone in influencing DMT1 turnover, at least under the conditions employed in our studies. Our findings further demonstrate that the proteasomal pathway may also play a key role in degradation of these membrane-bound proteins. Although membrane-bound proteins are normally not degraded via this pathway, there are several reports indicating that other membrane proteins such as growth hormone receptor [30] and LDLR [LDL (low-density lipoprotein) receptor-related protein] [31] are degraded by the ubiquitin-proteasomal pathway. Other proteins such as the gap junction protein, connexin43, are, in fact, degraded by both the proteasomal and lysosomal pathways [32]. We cannot, however, exclude the possibility that an ancillary protein, which itself is regulated by the proteasomal system, may function as a modulator for DMT1 turnover, similar to growth hormone receptor [33]. Interestingly, DMT1 is not the only protein involved in iron transport to be regulated by the ubiquitin-proteasomal pathway. For example, both IRP2 [34] and the transferrin receptor [35] are also degraded by this pathway and this suggests that this may be a common regulatory process monitoring iron homoeostasis in the body.
Since RA has been previously shown to increase proteasomal activity in cells [36], it was necessary to determine whether the increase in DMT1 turnover occurs as a direct consequence of RA or actually occurs after its removal. When cells were exposed for an additional fifth day with RA, expression of DMT1 did not change, indicating that the increased turnover only occurs after removal of RA. The elevated turnover of DMT1 suggests that there was either a very rapid increment in activity of the degradative enzyme associated with the lysosomal and proteasomal pathways or that removal of RA results in DMT1 becoming more accessible thus allowing facile digestion by these degradative enzymes. The former is most likely since an increase in activity of the proteasomal pathway was evidenced by the observation that α-tubulin, a protein known to be degraded via the proteasomal pathway [24], was also rapidly down-regulated after removal of RA.
It was also noted that the levels of mRNA for the 1B, +IRE and −IRE isoforms of DMT1 in P19 cells were attenuated during the differentiation process although at a slower rate than that observed for protein content. Turnover of mRNA was not altered during differentiation, indicating that mRNA levels were, most likely regulated by gene transcription. Previous studies have reported that transcription of c-myc was down-regulated by nitric oxide via inhibition of NF-κB in P19 cell undergoing differentiation [26]. Based on this prior observation, we considered the possibility that DMT1 was similarly regulated by nitric oxide inhibition of NF-κB interaction with the 1B promoter of DMT1. We, however, could not exclude the possibility that the responses observed to DMT1 expression were not caused by changes in Sp1 activity, since prior studies have shown that Sp1 was also capable of interacting with the NF-κB binding sequences in the promoter region of NR-1 in P19 cells [27]. As noted previously, using MatInspector software and phylogenetic footprinting analysis, we determined that this region contained two Sp1 sites and one NF-κB binding sites, both of which were regarded as possible candidates for the regulation of expression of the 1B isoform of DMT1.
Unpublished studies in our laboratory have revealed that nitric oxide is capable of generating isoform-specific changes in DMT1 expression in a fashion almost identical with that seen upon RA removal in differentiating P19 cells [36a]. These results imply that nitric oxide may be responsible for the decreased expression of DMT1 during the initial phases of P19 cell differentiation. The results reported in the present paper are consistent with this hypothesis in that inhibition of nitric oxide production during P19 cell differentiation prevents the decrease in DMT1 expression. Nitric oxide is considered as an atypical neurotransmitter, produced in vivo by NOS, and has been shown to cause cell-cycle arrest as well as to be required in neural differentiation. There are three known isoforms of NOS; iNOS, endothelial NOS and nNOS. In vitro studies in PC12 cells [37] have shown that nitric oxide produced by nNOS is necessary but not sufficient to produce neuronal differentiation by nerve growth factor. It has also been proposed that nNOS serves as a growth arrest gene, initiating the switch to cytostasis during differentiation [38]. Our results indicating a temporary induction of nNOS after removal of RA in P19 cells gives further credence to the role of nitric oxide as being the initial event ultimately responsible for the down-regulation of DMT1. This increase in nNOS is analogous to the results obtained by Bredt and Snyder [39] showing that there is transient induction of nNOS in cerebral cortical plate during E15–E19 (where E15 is embryonic day 15), since this time period corresponds to the differentiation stage of P19 cells.
If NF-κB is indeed one of the down-stream targets of nitric oxide produced during RA-induced differentiation of P19 cells, we would anticipate that inhibitors of nitric oxide would prevent the decrease in expression of DMT1. As demonstrated in the present paper, inhibition of nitric oxide not only prevents the loss of DMT1 seen after removal of RA (see Figure 6A) but also correlates with a decrease in nuclear DNA binding of NF-κB upon removal of RA. We also did not observe any change in Sp1 binding during differentiation, suggesting that it does not play a role in regulating DMT1 expression. These findings strongly suggest that NF-κB may be the principal nuclear factor regulating transcription of the 1B isoforms of DMT1.
To further verify that the NF-κB response element in the 1B promoter is responsible for regulating expression, luciferase reporter assays revealed that the region within the first 500 bp of 5′-flanking region of the promoter (+15 to −504) is most likely responsible for regulating synthesis of the 1B isoforms of DMT1. Within this region, there exists one putative NF-κB site and two Sp1 binding sites. The luciferase reporter studies using mutant constructs of NF-κB and SP1 verify that the specific NF-κB site which lies between −23 and −19 is responsible for regulating the expression of the 1B isoforms of DMT1. There are multiple proteins in the NF-κB family forming homo- or hetero-dimers, including p50, p65 (RelA), c-Rel, RelB and p52, usually present in the cytosol bound to IκB (inhibitory κB). Certain stimuli can cause phosphorylation of IκB leading to its degradation by the proteasomal pathway and nuclear localization of the NF-κB complex, where it can bind to the promoter region of genes. It has been shown earlier that undifferentiated P19 cells show constitutive nuclear expression of p50–p65 heterodimer and that nitric oxide exposure leads to inactivation of the NF-κB complex [26]. Our results confirmed these findings and further reveal that nuclear binding of the p65 component of NF-κB is specifically decreased during the initial stages of differentiation. These results reveal, for the first time, a mechanism other than post-translational by IRE–IRP interactions that is responsible for regulating DMT1 expression in vivo.
It is interesting to note that the overall luciferase activity was attenuated almost 60% in cell transfected with the entire promoter as compared with those transfected with approximately the first 500 bases (compare WT-day 4 versus Prom(−504)-day 4 in Figure 8B). Since this decrease occurs in P19 cell cultures before and after removal of RA (both day 4 and 5), we suggest that there may be another response element on the 5′-end of the promoter which is responsible for attenuating luciferase activity. Studies are currently underway to determine the exact region responsible for the observed regulation of DMT1.
One of the most surprising findings in the present study was the observation that the 1A-containing isoforms of DMT1 appear to be considerably more resistant to the effects of RA removal when compared with either the −IRE or +IRE species. The reason for this is not clear, though it may be possible that the extra 30 amino acids, encoded by exon 1A, may block ubiquitin-binding sites on the protein, preventing its degradation. It is also feasible, as revealed in prior studies from our laboratory [9], that the different isoforms of DMT1 are present in separate and distinct endosomal vesicles which can be sorted and modified independently within the cell. Thus some vesicles may be transported to and processed within the lysosomes, whereas others may terminate at other sites within the cells. It is also difficult to rationalize why there is no change in expression of the 1A protein or mRNA after RA removal, given the fact that it is presumably coupled with the +IRE and/or −IRE carboxy tail. We cannot rule out the possibility that the 1A isoform exists in the P19 cell without the +IRE or −IRE C-termini. The fact that 1A and 1B forms of DMT1 are under control of the different promoters can explain the observation that there was a change in the 1B form of DMT1 mRNA without a concomitant change in 1A form.
The functional significance of the decrease in specific isoforms of DMT1 mRNA is not known at this time. Since the decrease of DMT1 occurs at the beginning of the differentiation process, it may indicate low requirement of cells for metals. It is not known whether this decrease in DMT1 is necessary or just a by-product for the differentiation process. It has been shown that chelation of iron by desferrioxamine leads to cell-cycle arrest in G1 phase [40]. Since cell-cycle arrest is one of the first steps of differentiation, the decrease in DMT1 may indeed be necessary for the differentiation.
In summary, our results suggest that the molecular events which occur during the first 24 h after removal of RA in P19 cells lead to a rapid increase in proteasomal/lysosomal degradation of +/−IRE forms of DMT1 along with induction of nNOS, which subsequently regulates DMT1 transcription by decreasing NF-κB translocation to the nucleus and decreasing binding to the 1B promoter region of DMT1.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences grant no. R01 ES1112.
References
- 1.Cellier M., Prive G., Belouchi A., Kwan T., Rodrigues V., Chia W., Gros P. Nramp defines a family of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1995;92:10089–10093. [Google Scholar]
- 2.Gunshin H., Mackenzie B., Berger U. V., Gunshin Y., Romero M. F., Boron W. F., Nussberger S., Gollan J. L., Hediger M. A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature (London) 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 3.Hubert N., Hentze M. W. Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12345–12350. doi: 10.1073/pnas.192423399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee P. L., Gelbart T., West C., Halloran C., Beutler E. The human Nramp2 gene: characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol. Dis. 1998;24:199–215. doi: 10.1006/bcmd.1998.0186. [DOI] [PubMed] [Google Scholar]
- 5.Ghio A. J., Wang X., Silbajoris R., Garrick M. D., Piantadosi C. A., Yang F. DMT1 expression is increased in the lungs of hypotransferrinemic mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;284:L938–L944. [Google Scholar]
- 6.Lis A., Paradkar P. N., Singleton S., Kuo H. C., Garrick M. D., Roth J. A. Hypoxia induces changes in expression of isoforms of the divalent metal transporter (DMT1) in rat pheochromocytoma (PC12) cells. Biochem. Pharmacol. 2005;69:1647–1655. doi: 10.1016/j.bcp.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 7.Roth J. A., Garrick M. D. Iron interactions and other biological reactions mediating the physiological and toxic actions of manganese. Biochem. Pharmacol. 2003;66:1–13. doi: 10.1016/s0006-2952(03)00145-x. [DOI] [PubMed] [Google Scholar]
- 8.Kuo H. C., Smith J. J., Lis A., Zhao L., Gonsiorek E. A., Zhou X., Higgins D. M., Roth J. A., Garrick M. D., Garrick L. M. Computer-identified nuclear localization signal in exon 1A of the transporter DMT1 is essentially ineffective in nuclear targeting. J. Neurosci. Res. 2004;76:497–511. doi: 10.1002/jnr.20112. [DOI] [PubMed] [Google Scholar]
- 9.Lis A., Barone T. A., Paradkar P. N., Plunkett R. J., Roth J. A. Expression and localization of different forms of DMT1 in normal and tumor astroglial cells. Mol. Brain Res. 2004;122:62–70. doi: 10.1016/j.molbrainres.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 10.Roth J. A., Horbinski C., Feng L., Dolan K. G., Higgins D., Garrick M. D. Differential localization of divalent metal transporter 1 with and without iron response element in rat PC12 and sympathetic neuronal cells. J. Neurosci. 2000;20:7595–7601. doi: 10.1523/JNEUROSCI.20-20-07595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., Garrick M. D., Yang F., Dailey L. A., Piantadosi C. A., Ghio A. J. TNF, IFN-gamma, and endotoxin increase expression of DMT1 in bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:L24–L33. doi: 10.1152/ajplung.00428.2003. [DOI] [PubMed] [Google Scholar]
- 12.Wardrop S. L., Richardson D. R. The effect of intracellular iron concentration and nitrogen monoxide on Nramp2 expression and non-transferrin-bound iron uptake. Eur. J. Biochem. 1999;263:41–49. doi: 10.1046/j.1432-1327.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 13.Horton C., Maden M. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev. Dyn. 1995;202:312–323. doi: 10.1002/aja.1002020310. [DOI] [PubMed] [Google Scholar]
- 14.Corcoran J., So P. L., Barber R. D., Vincent K. J., Mazarakis N. D., Mitrophanous K. A., Kingsman S. M., Maden M. Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J. Cell Sci. 2002;115:3779–3786. doi: 10.1242/jcs.00046. [DOI] [PubMed] [Google Scholar]
- 15.Maden M., Gale E., Kostetskii I., Zile M. Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 1996;6:417–426. doi: 10.1016/s0960-9822(02)00509-2. [DOI] [PubMed] [Google Scholar]
- 16.Maden M. Role and distribution of retinoic acid during CNS development. Int. Rev. Cytol. 2001;209:1–77. doi: 10.1016/s0074-7696(01)09010-6. [DOI] [PubMed] [Google Scholar]
- 17.Jones-Villeneuve E. M., McBurney M. W., Rogers K. A., Kalnins V. I. Retinoic acid induces embryonal carcinoma cells to differentiate into neurons and glial cells. J. Cell Biol. 1982;94:253–262. doi: 10.1083/jcb.94.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones-Villeneuve E. M., Rudnicki M. A., Harris J. F., McBurney M. W. Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol. Cell. Biol. 1983;3:2271–2279. doi: 10.1128/mcb.3.12.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dignam J. D., Martin P. L., Shastry B. S., Roeder R. G. Eukaryotic gene transcription with purified components. Methods Enzymol. 1983;101:582–598. doi: 10.1016/0076-6879(83)01039-3. [DOI] [PubMed] [Google Scholar]
- 20.Hu X., Bi J., Loh H. H., Wei L.-N. An intronic Ikaros-binding element mediates retinoic acid suppression of the kappa opioid receptor gene, accompanied by histone deacetylation on the promoters. J. Biol. Chem. 2001;276:4597–4603. doi: 10.1074/jbc.M005477200. [DOI] [PubMed] [Google Scholar]
- 21.Burdo J. R., Menzies S. L., Simpson I. A., Garrick L. M., Garrick M. D., Dolan K. G., Haile D. J., Beard J. L., Connor J. R. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J. Neurosci. Res. 2001;66:1198–1207. doi: 10.1002/jnr.1256. [DOI] [PubMed] [Google Scholar]
- 22.Tabuchi M., Yoshimori T., Yamaguchi K., Yoshida T., Kishi F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 2000;275:22220–22228. doi: 10.1074/jbc.M001478200. [DOI] [PubMed] [Google Scholar]
- 23.Touret N., Martin-Orozco N., Paroutis P., Furuya W., Lam-Yuk-Tseung S., Forbes J., Gros P., Grinstein S. Molecular and cellular mechanisms underlying iron transport deficiency in microcytic anemia. Blood. 2004;104:1526–1533. doi: 10.1182/blood-2004-02-0731. [DOI] [PubMed] [Google Scholar]
- 24.Hyun D., Lee M., Halliwell B., Jenner P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem. 2003;86:363–373. doi: 10.1046/j.1471-4159.2003.01841.x. [DOI] [PubMed] [Google Scholar]
- 25.Luker G. D., Pica C. M., Song J., Luker K. E., Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nat. Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 26.Park S. W., Wei L. N. Regulation of c-myc gene by nitric oxide via inactivating NF-kappa B complex in P19 mouse embryonal carcinoma cells. J. Biol. Chem. 2003;278:29776–29782. doi: 10.1074/jbc.M303306200. [DOI] [PubMed] [Google Scholar]
- 27.Liu A., Hoffman P. W., Lu W., Bai G. NF-kappaB site interacts with Sp factors and up-regulates the NR1 promoter during neuronal differentiation. J. Biol. Chem. 2004;279:17449–17458. doi: 10.1074/jbc.M311267200. [DOI] [PubMed] [Google Scholar]
- 28.Quandt K., Frech K., Karas H., Wingender E., Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchette M., Tompa M. FootPrinter: a program designed for phylogenetic footprinting. Nucleic Acids Res. 2003;31:3840–3842. doi: 10.1093/nar/gkg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strous G. J., Vankerkhof P., Govers R., Ciechanover A., Schwartz A. L. The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. EMBO J. 1996;15:3806–3812. [PMC free article] [PubMed] [Google Scholar]
- 31.Melman L., Geuze H. J., Li Y., McCormick L. M., Van Kerkhof P., Strous G. J., Schwartz A. L., Bu G. Proteasome regulates the delivery of LDL receptor-related protein into the degradation pathway. Mol. Biol. Cell. 2002;13:3325–3335. doi: 10.1091/mbc.E02-03-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laing J. G., Tadros P. N., Westphale E. M., Beyer E. C. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- 33.Govers R., ten Broeke T., van Kerkhof P., Schwartz A. L., Strous G. J. Identification of a novel ubiquitin conjugation motif, required for ligand-induced internalization of the growth hormone receptor. EMBO J. 1999;18:28–36. doi: 10.1093/emboj/18.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamanaka K., Ishikawa H., Megumi Y., Tokunaga F., Kanie M., Rouault T. A., Morishima I., Minato N., Ishimori K., Iwai K. Identification of the ubiquitin-protein ligase that recognizes oxidized IRP2. Nat. Cell Biol. 2003;5:336–340. doi: 10.1038/ncb952. [DOI] [PubMed] [Google Scholar]
- 35.Kotamraju S., Tampo Y., Keszler A., Chitambar C. R., Joseph J., Haas A. L., Kalyanaraman B. Nitric oxide inhibits H2O2-induced transferrin receptor-dependent apoptosis in endothelial cells: role of ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10653–10658. doi: 10.1073/pnas.1933581100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi T., Shinozaki A., Momoi T., Arahata K., Tsukahara T. Identification of an interleukin-1 beta converting enzyme-like activity that increases upon treatment of P19 cells with retinoic acid as the proteasome. J. Biochem. (Tokyo) 1996;120:699–704. doi: 10.1093/oxfordjournals.jbchem.a021467. [DOI] [PubMed] [Google Scholar]
- 36a.Paradkar P. N., Roth J. A. Nitric oxide transcriptionally down-regulates specific isoforms of divalent metal transporter (DMT1) via NF-κB. J. Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.03702.x. in the press. [DOI] [PubMed] [Google Scholar]
- 37.Phung Y. T., Bekker J. M., Hallmark O. G., Black S. M. Both neuronal NO synthase and nitric oxide are required for PC12 cell differentiation: a cGMP independent pathway. Brain Res. Mol. Brain Res. 1999;64:165–178. doi: 10.1016/s0169-328x(98)00315-5. [DOI] [PubMed] [Google Scholar]
- 38.Peunova N., Enikolopov G. Nitric oxide triggers a switch to growth arrest during differentiation of neuronal cells. Nature (London) 1995;375:68–73. doi: 10.1038/375068a0. [DOI] [PubMed] [Google Scholar]
- 39.Bredt D. S., Snyder S. H. Transient nitric oxide synthase neurons in embryonic cerebral cortical plate, sensory ganglia, and olfactory epithelium. Neuron. 1994;13:301–313. doi: 10.1016/0896-6273(94)90348-4. [DOI] [PubMed] [Google Scholar]
- 40.Chenoufi N., Drenou B., Loreal O., Pigeon C., Brissot P., Lescoat G. Antiproliferative effect of deferiprone on the Hep G2 cell line. Biochem. Pharmacol. 1998;56:431–437. doi: 10.1016/s0006-2952(98)00071-9. [DOI] [PubMed] [Google Scholar]