

Abstract
The introduction of apo-ferritin or the iron chelator DFO (desferrioxamine) conjugated to starch into the lysosomal compartment protects cells against oxidative stress, lysosomal rupture and ensuing apoptosis/necrosis by binding intralysosomal redox-active iron, thus preventing Fenton-type reactions and ensuing peroxidation of lysosomal membranes. Because up-regulation of MTs (metallothioneins) also generates enhanced cellular resistance to oxidative stress, including X-irradiation, and MTs were found to be capable of iron binding in an acidic and reducing lysosomal-like environment, we propose that these proteins might similarly stabilize lysosomes following autophagocytotic delivery to the lysosomal compartment. Here, we report that Zn-mediated MT up-regulation, assayed by Western blotting and immunocytochemistry, results in lysosomal stabilization and decreased apoptosis following oxidative stress, similar to the protection afforded by fluid-phase endocytosis of apo-ferritin or DFO. In contrast, the endocytotic uptake of an iron phosphate complex destabilized lysosomes against oxidative stress, but this was suppressed in cells with up-regulated MT. It is suggested that the resistance against oxidative stress, known to occur in MT-rich cells, may be a consequence of autophagic turnover of MT, resulting in reduced iron-catalysed intralysosomal peroxidative reactions.
Keywords: apoptosis, autophagocytosis, lysosome, metallothionein, oxidative stress, redox-active iron
Abbreviations: AO, Acridine Orange; DCF, 2′,7′-dichlorofluorescein; DFO, desferrioxamine; DMEM, Dulbecco's modified Eagle's medium; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); FBS, fetal bovine serum; HBSS, Hanks balanced salt solution; H2DCF, non-fluorescent 2′,7′-dichlorodihydrofluorescein; H2DCF-DA, non-fluorescent 2′,7′-dichlorodihydrofluorescein diacetate; HMM-DFO, high-molecular-mass DFO; HRP, horseradish peroxidase; LAP, α-lipoic acid plus; MT, metallothionein; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; PI, propidium iodide
INTRODUCTION
Oxidative stress influences cells in a variety of ways, dose dependently and usually with a delayed onset of effects. A limited amount of oxidative stress often stimulates cell replication, while a little more results in growth arrest, DNA damage and reparative autophagocytosis. Moderate or advanced oxidative stress, finally, results in apoptosis or necrosis respectively [1,2]. The up-regulation of many phase II proteins, including ferritin, heat-shock proteins, MTs (metallothioneins) and thioredoxin, protects against oxidative stress-induced cellular injury [3,4]. In spite of substantial research efforts concerning the underlying molecular mechanisms, it is, however, not yet generally understood, either how oxidative stress induces injury or how these impairments are counteracted by the above phase II proteins.
During the last 5–10 years, a growing number of reports have been presented in favour of the hypothesis that oxidative stress-induced cellular damage to a large extent is a function of lysosomal rupture, with release to the cytosol of potent cathepsins, other lysosomal hydrolases and low mass redox-active iron [2,5–9]. Major reasons to believe that this is a correct view are findings that the quantity of damage is paralleled by the degree of lysosomal rupture, that lysosomal rupture induced by different mediators gives the same result and that lysosomal stabilizers eliminate the harmful effect of agonists that normally cause both lysosomal rupture and cell damage [2,8,10].
Exposure of cells in culture to DFO (desferrioxamine), both of standard type and conjugated to starch [HMM-DFO (high-molecular-mass DFO)], or apo-ferritin, which are all taken up by fluid-phase endocytosis, effectively protects cells and lysosomes against oxidative stress, including X-irradiation, suggesting that lysosomal rupture and its consequences following such stress are effects of iron-catalysed peroxidation of lysosomal membranes. This hypothesis is strengthened by findings that minute concentrations of a lysosomotropic iron chelator, LAP (α-lipoic acid plus), is highly effective in the protection of cells and lysosomes against oxidative stress [2,5,11,12]. Further support for the idea that lysosomal membranes are peroxidized over time following an oxidative attack, which by itself may be of short duration, is provided by the finding that the chain-breaking antioxidant vitamin E delays and partially protects against lysosomal destabilization [13].
If we do assume that oxidative stress initiates most of its toxic effects by causing intralysosomal, iron-catalysed peroxidation with ensuing lysosomal rupture, obviously the concentration of lysosomal redox-active iron, and how it is regulated, must be considered of fundamental importance, even if other aspects of antioxidative defence, such as capacity to degrade H2O2, would be important as well. Low-mass iron occurs in the lysosomal compartment as a result of degradation of iron-containing metalloproteins, while its redox-active capacity is promoted by the acidic pH and reducing milieu of lysosomes [2,5,12,14–16]. Autophagocytosis is a normal cellular activity by which organelles and most long-lived proteins are turned over. Exactly how autophagocytosis is regulated is unknown, but it may be partly a random process [17,18]. In resting cells without much iron uptake from outside, a major part of the cytosolic labile iron pool may be derived from autophagocytotic turnover of iron-containing macromolecules with release of low-mass iron from autophagolysosomes to the cytosol [5,12].
In cells with up-regulated ferritin, the autophagocytotic process would bring this iron-binding protein into the lysosomal compartment and make it less sensitive to oxidative stress by preventing iron from participating in Fenton-type reactions. Probably, autophagocytosed ferritin retains its iron-binding capacity for a while intralysosomally. As long as ferritin is not fully Fe-saturated and is continuously autophagocytosed, it would work as a permanent intralysosomal iron chelator. The amount of cytosolic non-saturated ferritin would be reflected in the degree of lysosomal stabilization during oxidative stress [19].
Perhaps some phase II proteins other than ferritin, up-regulated following oxidative stress, may also have iron-binding capacity. In the present study, we focused on MTs, a family of small (∼6–7 kDa), heat-resistant proteins containing 25–30% cysteine residues that are evolutionarily highly conserved in a broad range of species from yeast to mammals. MTs are up-regulated by glucocorticoids, oxidative stress and a variety of heavy metals, such as copper, cadmium, mercury and zinc [20]. Isoforms range from MT-I to MT-IV and have slightly different amino acid composition. MTs bind metals and protect against their toxicity, as was first demonstrated in aquatic species, such as fish, arthropods and molluscs from contaminated waters [20–22]. Apart from binding heavy metals, MTs are considered to act as antioxidants, although by undetermined mechanisms. Thus MTs have been found to protect against apoptosis/necrosis induced by oxidative stress, etoposide, cisplatin, doxorubicin and X-irradiation [23–25]. Apo-MTs are effectively degraded by lysosomal cathepsins, while metal-conjugated MTs are more stable [26,27].
Here, we show that exposure of J774 cells in culture to 100 μM Zn for 12–24 h results in a substantial MT up-regulation and increased resistance to oxidative stress-induced apoptosis/necrosis in parallel with enhanced lysosomal stability. Since MTs were found to be able to chelate low mass iron in a non-redoxactive form under acidic and reducing lysosomal-like conditions, we suggest that the decreased sensitivity to oxidative stress is a function of MT autophagocytosis, resulting in reduced concentration of lysosomal redox-active iron, less intralysosomal peroxidation and, consequently, diminished release of lysosomal apoptogenic factors to the cytosol.
EXPERIMENTAL
Chemicals
DMEM (Dulbecco's modified Eagle's medium), HBSS (Hank's balanced salt solution), FBS (fetal bovine serum), penicillin and streptomycin were obtained from Gibco (Paisley, U.K.). AO (Acridine Orange) base was from Gurr (Poole, Dorset, U.K.), while silver lactate was from Fluka AG (Buchs, Switzerland). Glutaraldehyde and SDS were from Bio-Rad (Cambridge, MA, U.S.A.), and ammonium sulphide, hydrochloric acid and quinol from BDH Ltd (Poole, Dorset, U.K.). DFO was from Ciba-Geigy (Basel, Switzerland) and DFO conjugated to starch (HMM-DFO) was a gift from Professor John W. Eaton (James Graham Brown Cancer Center, University of Louisville, Louisville, KY, U.S.A.). CaspACE™ FITC-VAD-FMK (where VAD-FMK is Valylalanyl-DL-aspartylfluoromethane) In Situ Marker was from Promega (Madison, WI, U.S.A.), while HRP (horseradish peroxidase) was from Roche Diagnostics (Indianapolis, IN, U.S.A.). Monoclonal mouse anti-MT antibodies (clone E9) were from Zymed (San Francisco, CA, U.S.A.), anti-(Pan)Actin (Ab-5) antibodies were from Neomarkers (Fremont, CA, U.S.A.), polyclonal goat anti-mouse immunoglobulins (HRP-conjugated) were from Dako (Glostrup, Denmark) and polyclonal rabbit anti-mouse immunoglobulins (FITC-conjugated) from Calbiochem (Darmstadt, Germany). H2DCF-DA (non-fluorescent 2′,7′-dichlorodihydrofluorescein diacetate) was from Molecular Probes (Eugene, OR, U.S.A.). MT from rabbit liver (a mixture of form I and II) and all other chemicals were from Sigma (St. Louis, MO, U.S.A.).
Cell culture and exposure to ZnSO4, DFO, iron and oxidative stress
Murine macrophage-like J774 cells (A.T.C.C., Manassas, VA, U.S.A.) were grown in DMEM supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 i.u./ml penicillin and 100 μg/ml streptomycin, at 37 °C in humidified air with 5% CO2. The cells were subcultivated twice a week, plated at a concentration of 1×106 cells per 35 mm dish, with or without coverslips, and typically subjected to oxidative stress (or not) within the following 24 h.
H2O2 and ZnSO4 concentrations and exposure times (in relation to cell density) were established in preliminary experiments. In the final experiments, cells were exposed before oxidative stress to fresh complete medium with or without 100 μM ZnSO4 for 1, 6, 12 or 24 h. After Zn exposure, cells were rinsed in PBS. In some experiments, cells were returned to standard culture conditions for 1 h after completed exposure to Zn. Control and Zn pretreated cells were then oxidatively stressed (or not) for 30 min by exposure to a bolus dose of 100 μM H2O2 in 2 ml of PBS at 37 °C. Note that under these conditions the H2O2 concentration declines quickly (t1/2∼15 min) to <20 μM after 30 min (see below). Cells were then returned to standard culture conditions and further analysed after 6–8 h. In some experiments, cells with/without Zn pretreatment were incubated for 3 h in complete medium with 1 mM DFO or HMM-DFO or 30 μM FeCl3 (resulting in the formation of an iron phosphate complex that is endocytosed and transported into the lysosomal compartment) followed by 1 h under standard conditions before exposure to oxidative stress.
Western blots
Control and Zn-treated cells were collected, washed in PBS, pelleted, resuspended in lysis buffer [62.5 mM Tris/HCl (pH 6.8), 10% (v/v) glycerol and 2% (w/v) SDS] and heated for 7 min at 95 °C. To some control cell suspensions, 1 μg of MT was added as an internal standard. Western blots for MTs were performed by the method of Mizzen et al. [28]. Briefly, equal amounts of protein (∼40 μg) were loaded on to an SDS/15% polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad) with the addition of 2 mM CaCl2 to the transfer buffer (25 mM Tris, pH 8.2). Membranes were then fixed in 2.5% (v/v) glutaraldehyde for 1 h, washed in TBS (50 mM Tris/HCl and 150 mM NaCl; pH 7.5) (3×5 min) and incubated with mouse IgG anti-MT antibodies (1 μg/ml, 4 °C, overnight) in 0.1% non-fat milk in TBS with 0.05% Tween 20 after being blocked with 5% (w/v) non-fat milk in TBS with 0.1% Tween 20. Finally, membranes were incubated with HRP-conjugated secondary goat anti-mouse antibodies (1:1500; 20 °C; 60 min), and assayed with an ECL®+ (enhanced chemiluminescence plus) Western detection kit (Amersham Biosciences, U.K.). The antibodies were then stripped off the membrane by incubating for 1 h at 60 °C with 2% SDS, 100 mM 2-mercaptoethanol and 62.5 mM Tris/HCl (pH 6.8) and membranes were reprobed with an anti-(Pan)Actin antibody to demonstrate equal loading of cell lysate.
MT immunocytochemistry
MT in cells pre-exposed to 100 μM ZnSO4 for 12 h was detected immunocytochemically using mouse anti-MT antibodies. In brief, cells were fixed for 10 min at 4 °C in 4% (w/v) paraformaldehyde in PBS. The fixed cells were washed with PBS and permeabilized with 0.1% (w/v) saponin in PBS for 20 min. Cells were then incubated with 1 μg/ml mouse IgG anti-MT antibodies for 60 min in the dark, washed with 0.1% saponin in PBS, exposed to the secondary antibody (FITC-conjugated anti-mouse IgG) and again washed in saponin buffer. Fluorescence was examined under a Nikon microphot-SA fluorescence microscope (Nikon, Tokyo, Japan) equipped with a Hamamatsu C4742-95 (Hamamatsu, Bridgewater, NJ, U.S.A.) digital camera. Cells were photographed using phase and transmitted light modes and results were analysed using Adobe Photoshop software.
Degradation of H2O2
To ensure that the observed Zn-induced resistance to oxidative stress was not an effect of enhanced H2O2 catabolism, the rate of H2O2 clearance was determined. Control cells and cells pretreated with Zn (in concentrations described above) were exposed to a bolus dose of 100 μM H2O2 in 2 ml of PBS at 37 °C. During a 60 min period, aliquots (50 μl) were sampled for H2O2 analysis by the HRP-mediated H2O2-dependent p-hydroxy-phenylacetic acid oxidation technique [29]. Fluorescence intensity was read (λex=315 and λem=410 nm) using an RF-540 spectrofluorimeter (Shimadzu, Kyoto, Japan) connected to a DR-3 data recorder.
Measurement of total thiols
Total cellular reduced thiols (including GSH and protein thiols) were measured by their reaction with DTNB [5,5′-dithiobis-(2-nitrobenzoic acid)], based on the method described by Boyne and Ellman [30]. Cells were washed three times in cold PBS 6 h after oxidative stress, and scraped and sonicated for 2 min. SDS (final concentration 5%) and DTNB (final concentration 30 μM) were then added, and the samples were vortex-mixed and incubated while stirring at room temperature (22 °C) for 30 min. The absorbance was measured spectrophotometrically at 412 nm.
Lysosomal membrane stability assay
At 6 h after the oxidative-stress period (see above), cells were stained with 5 μg/ml AO in complete medium at 37 °C for 15 min, detached by scraping and collected for flow cytofluorimetric assessment. AO is a metachromatic fluorophore and a lysosomotropic base (pKa=10.3), which becomes charged (AOH+) and retained by proton trapping within acidic compartments, mainly secondary lysosomes (pH 4.5–5.5). When normal cells are excited by blue light, highly concentrated lysosomal AO emits an intense red fluorescence, while nuclei and cytosol show weak diffuse green fluorescence. Red fluorescence was measured (FL3 channel) using a Becton Dickinson FACScan (Becton Dickinson, Mountain View, CA, U.S.A.) equipped with a 488 nm argon laser. Cells with a reduced number of intact, AO-accumulating lysosomes (here termed ‘pale’ cells), were detected as described earlier [5,6,10–12]. CellQuest software was used for acquisition and analyses.
Assessment of cell viability
Cell viability was assessed by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay [31]. Briefly, appropriate controls and cells exposed to oxidative stress, with or without pretreatment with Zn, were washed and incubated with 0.5 mg/ml MTT in HBSS for 4 h. The crystals were dissolved in 10 mM HCl with 10% SDS, and the absorbance was measured at 570 nm.
Apoptosis assays
DNA fragmentation assay
The Nicoletti DNA fragmentation assay is based on PI (propidium iodide) staining of nuclear DNA and was performed essentially as described in [32]. Cell pellets from individual dishes were resuspended in 1.5 ml of a hypo-osmotic and membrane-disrupting solution of PI (50 μg/ml in 0.1% sodium citrate with 0.1% Triton X-100), kept in the dark overnight at 4 °C and the PI-induced red fluorescence of suspended individual nuclei was measured by flow cytofluorimetry using the FL3 channel. Nuclei with partially degraded DNA were counted and their frequency was expressed as a percentage of the total number of analysed nuclei (10000).
Assessment of caspase activation
Caspase activation associated with apoptosis was evaluated by microscopic analysis of living cells, according to the manufacturer's instructions, using an FITC-conjugated broad spectrum inhibitor of caspases, CaspACE™ FITC-VAD-FMK In Situ Marker, that irreversibly binds to activated caspases. Briefly, 7 h after oxidative stress, the marker was added to the medium at a final concentration of 10 μM, and cells were incubated in the dark for 20 min, rinsed three times in PBS (pH 7.4; 5 min in total), and observed, counted and photographed using the Nikon fluorescence microscope.
Assay of Fenton-type reactions under lysosomal conditions
In order to assay Fenton-type chemistry under lysosomal conditions (a reducing and acidic milieu, pH ∼5.0), and to find out whether MT might chelate iron under these circumstances, experiments were done in vitro using a modification of a technique described by Myhre et al. [33]. Briefly, ferric iron (10 μM) was partially reduced to its ferrous form by cysteine (100 μM) in 150 mM acetate buffer (pH 5.0). H2O2 (100 μM) was added to initiate the production of hydroxyl radicals (HO•). The latter oxidize H2DCF (non-fluorescent 2′,7′-dichlorodihydrofluorescein; 5 μM) to fluorescent DCF (2′,7′-dichlorofluorescein) [H2DCF was obtained by hydrolysing its acetate ester (H2DCF-DA)]. DMSO (10%) and DFO (10 μM) were used to demonstrate the formation of HO• and the involvement of iron respectively. Finally, MT at various concentrations was assayed for its iron-chelating capacity. Fluorescence was measured in an FL600 Microplate Fluorescence reader (Bio-Tek, Winooski, VT, U.S.A.) at λex 485 nm and λem 530 nm.
Cytochemical assay of lysosomal reactive iron
For evaluation of cellular low-mass iron, we used the ‘autometal-lographic’ sulphide-silver method as previously described [34]. Cells, grown on coverslips, were rinsed briefly in PBS (22 °C) prior to fixation with 2% glutaraldehyde in 0.1 M NaOH/cacodylic acid buffer with 0.1 M sucrose (pH 7.2) for 2 h at 22 °C. The fixation was followed by short rinses (5×) in glass-distilled water at 22 °C. Cells were then sulphidated at pH ∼9 with 1% (w/v) ammonium sulphide in 70% (v/v) ethanol for 15 min. Following careful rinsing in glass-distilled water for 10 min at 22 °C, development was performed using a physical, colloid-protected developer containing silver lactate. The reaction was performed in the dark at 26 °C for various periods of time (20–60 min). Following dehydration in a graded series of ethanol solutions and mounting in Canada balsam, the cells were examined and photographed, using transmitted light, under the Nikon microscope.
Statistical analysis
Results are given as means±S.D. Statistical comparisons were made using ANOVA. P<0.05 (*), P<0.01 (**) and P<0.001 (***).
RESULTS
As expected, and in agreement with the findings of many others, cells exposed to Zn showed MT up-regulation over time, as demonstrated by Western blots (Figure 1). Immunocytochemistry was also used to show increased amounts of cellular MT following 12 h exposure to Zn (Figure 2).
Figure 1. Demonstration of MT up-regulation by Western blotting.

Immunoblots showed increased expression of MT I and II in cells exposed to 100 μM ZnSO4 for 6–24 h. Each lane was loaded with 40 μg of total cell lysate. Demonstration of actin was used as a loading control. As an internal standard, 1 μg of MT was added to some control cell suspensions.
Figure 2. Immunocytochemical demonstration of MT up-regulation.

Compared with control cells (A, B), cells exposed for 12 h to 100 μM Zn under otherwise standard conditions (C, D) showed elevated levels of MT I and II. Immunofluorescence (A, C) and phase contrast (B, D) images are shown. Micrographs were taken with fixed settings. The lighter background in (C) compared with (A) is an effect of a stronger light scattering from the fluorescence-emitting cells with up-regulated MT.
In order to exclude the possibility that the effects of MT up-regulation were due to non-specific Zn-induced enhancement of H2O2 degrading capacity (e.g. increase of catalase and/or glutathione peroxidase), control and Zn-exposed cells were subjected to H2O2 degradation studies. However, as shown in Figure 3(A), both groups of cells degraded H2O2 in a similar manner.
Figure 3. Assessment of antioxidative defence.

(A) Degradation of H2O2. Cells pre-exposed for 12 h to 100 μM Zn (or not) showed the same H2O2 degradation kinetics. A representative experiment is shown. (B) Measurement of total reduced thiols. Compared with control cells, the amount of total reduced thiols did not change following exposure of cells to 100 μM Zn for 12 h. H2O2-induced oxidative stress (a single bolus dose of initially 100 μM for 30 min) decreased reduced thiols by approx. 50%. This decrease was slightly (but not significantly) less in cells pre-exposed to 100 μM Zn for 12 h. Results are expressed as means±S.D. for four experiments. Comparisons were made against untreated controls.
The total level of reduced thiols in Zn-exposed cells did not change compared with controls. When normal cells were subjected to H2O2, the thiol level decreased approx. 50%, while pre-exposure of cells to Zn resulted in a slightly smaller decrease in thiols following oxidative stress, perhaps a result of reduced lysosomal break (Figure 3B).
To demonstrate that Fenton-type reactions would occur intralysosomally if H2O2 diffused into this acidic and reducing compartment and came into contact with redox-active iron, and that MT would suppress such reactions, we assayed the production of hydroxyl radicals in vitro under lysosomal conditions. To allow the reduction of ferric iron to its ferrous form, we used the reducing amino acid cysteine, which occurs at high concentrations in lysosomes [16], and the conversion of H2DCF into DCF as an indicator of hydroxyl radical production [33].
By the linear oxidation of H2DCF to DCF [before the reaction levelled off soon after 10 min when all cysteine was consumed (results not shown)], it was demonstrated that production of hydroxyl radicals took place (Figure 4). This was further substantiated by the decrease in the level of hydroxyl radicals in 10% DMSO, which is a well-known hydroxyl radical scavenger when used at high concentrations. The importance of iron in redox-active form for Fenton-type reactions was demonstrated by the use of DFO in a concentration equimolar to iron. DFO is a chelator that binds all six co-ordinates of iron. In the final tests, it was found that MT prevents the production of hydroxyl radicals and that one molecule of MT seems to bind two atoms of iron under the test conditions (Figure 4).
Figure 4. Iron binding by MT under lysosomal conditions.

Production of hydroxyl radicals was demonstrated by oxidation of the non-fluorescent probe H2DCF to fluorescent DCF in an acidic and reducing environment of the lysosomal type. To induce production of HO•, 100 μM H2O2 was added to 150 mM acetate buffer (pH 5.0) containing 10 μM FeCl3 and 100 μM cysteine. Suppression of fluorescence was observed by a high concentration of the well-known hydroxyl radical scavenger DMSO (10%). The strong iron chelator DFO bound iron in equimolar concentration (higher concentrations did not add further protection; results not shown), which is in agreement with its capacity to bind all six co-ordinates of iron. MT was found to give maximum protection at a ratio to iron of 1:2 (higher concentrations did not add further protection; results not shown), suggesting that one MT molecule binds two atoms of iron. A representative experiment is shown.
Since we have previously found that oxidative stress causes apoptosis/necrosis and DNA damage by inducing lysosomal rupture, with ensuing relocation of lytic enzymes and low-mass iron, we next wanted to see if the Zn exposure had any influence on lysosomal stability. Using the AO uptake method, increased lysosomal resistance to oxidative stress was indeed found to parallel the MT up-regulation. The number of cells with a reduced number of intact lysosomes (‘pale’ cells) following oxidative stress was significantly diminished in the 6–24 h Zn-exposed cells (Figure 5). The results were the same if cells were exposed to oxidative stress directly after the end of Zn exposure, or if they were rinsed and returned to standard conditions for 1 h before oxidative stress, excluding a direct Zn-mediated stabilizing effect on lysosomal membranes (results not shown).
Figure 5. Lysosomal stabilization following exposure to Zn.

After incubation with 100 μM Zn for 1, 6, 12 or 24 h, cells were exposed to an initial bolus dose of 100 μM H2O2 for 30 min, followed by 6 h under standard culture conditions. AO staining and analysis by flow cytofluorimetry showed time-dependently increased lysosomal stability in cells incubated with Zn for 6 h or more. Results (percentage of cells with reduced number of intact lysosomes) are expressed as mean±S.D. and represent a minimum of four experiments. Comparisons were made with cells exposed to H2O2 (third bar). Examples of flow cytofluorimetry results are shown.
We then confirmed that the Zn-induced stabilization of lysosomes resulted in reduced cell death via apoptosis or necrosis, using three assays testing different parts of the cell death pathway. The Nicoletti assay showed that oxidative stress-induced DNA fragmentation increased over time after H2O2 exposure, while Zn incubation was able to decrease this damage (Figure 6A). A fluorescent pan-caspase inhibitor was used to show that Zn incubation decreased caspase activation found after H2O2 exposure by approx. 60% (Figure 6B). Finally, the MTT assay, measuring an endpoint of cell death, showed that Zn incubation increased cell viability 8 h after H2O2 exposure by 36% (Figure 6C).
Figure 6. Assessment of apoptosis and cell viability.

(A) The Nicoletti method for assaying apoptosis. After incubation with 100 μM Zn for 12 h, cells were exposed to a bolus dose of 100 μM H2O2 for 30 min, followed by 6, 7 or 8 h under standard culture conditions. Cells were then scraped, centrifuged and resuspended in membrane-disrupting solution with PI. Following incubation overnight, the percentage of subdiploid nuclei was analysed by flow cytofluorimetry. Increased apoptosis was found in cells exposed to H2O2. Zn preincubation reduced this level. Results are expressed as means±S.D. and represent a minimum of four experiments. Comparisons were made with control cells exposed to H2O2 and assayed after another 6 h under standard conditions (third bar). (B) The CaspACE inhibitor method for assaying apoptosis. After incubation with 100 μM Zn for 12 h, cells were exposed to an initial bolus dose of 100 μM H2O2 for 30 min, followed by 7 h under standard conditions. The cells were then exposed to 10 μM CaspACE inhibitor for 20 min at 37 °C in the dark. The percentage of strongly fluorescent cells was determined in a fluorescent microscope at ×600. A minimum of 500 cells were counted in each sample. Incubation with Zn reduced the activation of caspases. Results are expressed as means±S.D. and represent a minimum of four experiments. Comparisons were made with cells exposed to H2O2. (C) Assessment of cell viability by MTT metabolism. After incubation with 100 μM Zn for 12 h, cells were exposed to an initial bolus dose of 100 μM H2O2 for 30 min, followed by 8 h under standard conditions. Cells were then washed and incubated with 0.5 mg/ml MTT in HBSS for 4 h. The crystals formed were dissolved in SDS/HCl and the absorbance was measured at 570 nm. Incubation with Zn reduced loss of cellular viability. Results are expressed as means±S.D. and represent a minimum of four experiments. Comparisons were made with cells exposed to H2O2.
To demonstrate the normal occurrence of lysosomal iron and the endocytotic uptake of a hydrated iron phosphate complex (obtained by adding 30 or 100 μM FeCl3 to the medium), we used a modified (high pH; high S2−) Timm [34a] sulphide-silver method, an extremely sensitive cytochemical technique used to confirm the presence of free heavy metals. In most normal cells, the only free metal at any detectable concentration is iron. As shown (Figure 7), the vast majority of free iron was detected as a strong perinuclear lysosomal-type pattern. Some cytosolic ‘staining’ also occurred. Following exposure to the iron phosphate complex, the lysosomal pattern became much more exaggerated.
Figure 7. Cytochemical demonstration of iron by the sulphide-silver method (autometallography).
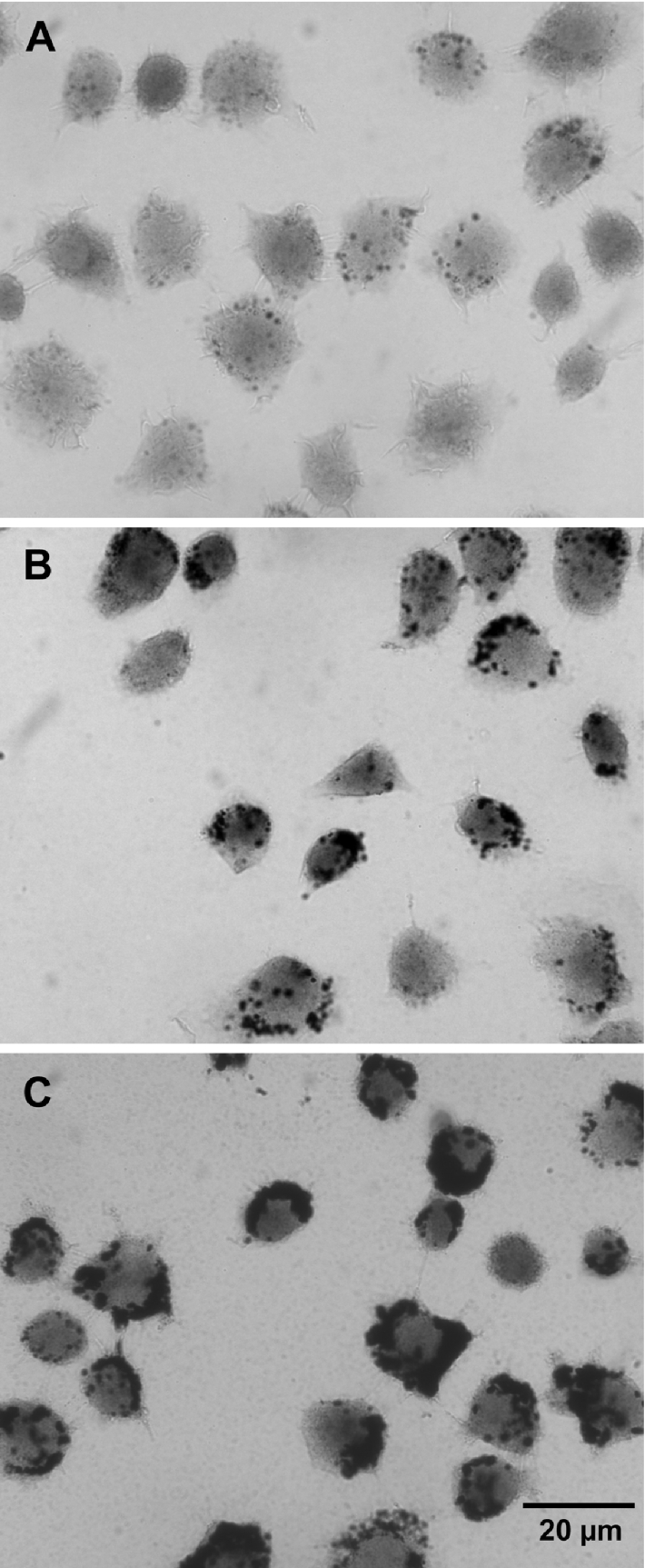
Cells showed a distinct lysosomal pattern of black, granular silver precipitates, indicating the presence of concentrated lysosomal low-molecular-mass iron. In control cells (A), only a few granules were found, reflecting a limited number of autophagolysosomes with Fe-containing macromolecules under degradation, while cells exposed for 3 h to a hydrated iron phosphate complex [obtained by adding FeCl3 to the culture medium to final concentrations of 30 μM (B) or 100 μM (C)] showed considerably more granules, reflecting fluid-phase endocytosis into late endosomes/lysosomes of the Fe complex. There was also a weak (A) or more substantial (B, C) yellowish diffuse colorization, reflecting the occurrence of cytosolic low mass iron (development time=50 min).
Since lysosomal stability against oxidative stress would be a function of the magnitude of oxidative stress and the lysosomal content of redox-active iron, we then exposed cells to either the potent iron chelator DFO, conjugated to starch or not, or the above hydrated iron phosphate complex, allowing the endocytotic uptake and transport to the lysosomal compartment of these three compounds with antagonistic effects of oxidative stress on lysosomal stability [6]. The result was significantly greater lysosomal stabilization or destabilization respectively following oxidative stress. There was no difference between DFO and HMM-DFO, suggesting that both are taken up by fluid phase endocytosis. Zn-induced MT up-regulation was still able to significantly reduce the enhanced sensitivity to oxidative stress generated by iron addition, indicating that autophagocytosed MTs are indeed able to bind low-mass iron and reduce its capacity to catalyse Fenton-type reactions (Figure 8).
Figure 8. Lysosomal stability following exposure to DFO or a hydrated iron phosphate complex.

Cells were exposed for 12 h to 100 μM Zn. Some samples were then incubated for 3 h in normal medium containing 30 μM of an iron phosphate complex or 1 mM DFO. After another 1 h under standard culture conditions, the cells were exposed to a bolus dose of 100 μM H2O2 in PBS for 30 min, followed by another 6 h under standard culture conditions. Ensuing AO assay of preserved acidic vacuoles (late endosomes and lysosomes) by flow cytofluorimetry showed increased lysosomal stability in cells incubated with Zn or DFO, and reduced stability in those incubated with the iron phosphate complex. Results (percentage of cells with reduced number of intact lysosomes) are expressed as mean±S.D. and represent a minimum of four experiments. Comparisons were made with control cells exposed to H2O2 (second bar).
DISCUSSION
The exposure of cells in culture to moderate concentrations of Zn ions is a well-known way to generate up-regulation of MTs [20,21]. In the present study, confirming the results of others, it was found that exposure to 100 μM Zn (in the form of ZnSO4) for 12–24 h significantly and time-dependently increased cellular MT. In parallel, cells became considerably less sensitive to oxidative stress, applied as a bolus dose of H2O2. Interestingly, and probably of great importance for the understanding of how MTs provide resistance to oxidative stress, it was also found that the obtained protection was paralleled by much increased lysosomal stability to oxidative stress. Exposing cells to Zn for just 1 h did not enhance lysosomal stability to oxidative stress, and Zn exposure followed by an additional hour under standard culture conditions did not diminish its protective effect, arguing against a direct lysosome-stabilizing effect of Zn.
As was previously pointed out, cellular damage following oxidative stress for the most part seems to result from lysosomal rupture due to intralysosomal iron-catalysed oxidative reactions causing peroxidation of lysosomal membranes. Lysosomes are very sensitive to oxidative stress and related rupture of these organelles, with release of a host of hydrolytic enzymes, seems to be an early event upstream of mitochondrial damage and ensuing apoptosis/necrosis [2,5,6,11,12,19]. By allowing cells to endocytose apo-ferritin or the potent iron chelator DFO, either in pure form or after conjugation to starch, or exposing them to a lysosomotropic iron chelator, LAP, before the oxidative event, lysosomal rupture and resulting mitochondrial damage/apoptosis/necrosis can be substantially prevented, whereas it is much enhanced if cells have endocytosed iron, for example a hydrated iron phosphate complex, before the oxidative event [5,6,11,12,19].
The lysosomal compartment is a system of vacuoles that constantly fuse and fission, allowing its content to slowly be distributed throughout. From a physiological point of view, the compartment, by fusion with secretory vesicles pinched off from the Golgi apparatus, constantly receives new hydrolytic enzymes and, by fusion with autophagosomes and late endosomes, accepts intracellular and extracellular substrates respectively for degradation into building blocks, such as simple sugars and amino and fatty acids, which can be reutilized for synthesis of complex biomolecules after active or passive transport into the cytosol [35].
Autophagocytotic degradation of cellular structures is an important turnover mechanism that will result in the release of low- mass, redox-active iron intralysosomally when iron-containing metalloproteins are degraded. Consequently, lysosomes constitute a compartment with a high concentration of low-mass iron [5,12,36]. From lysosomes, iron is transported to the cytosol, by mechanisms not yet fully understood, to become part of the cytosolic pool of labile iron. Especially in multiplying cells with active iron uptake, this pool is also augmented from late endosomes, which contain iron internalized by the transferrin receptor-mediated route. The pool is the iron source for various anabolic purposes [37].
Low-mass iron is potentially redox-active. Since the lysosomal compartment is acidic (pH 4.5–5.5) and rich in reducing equivalents such as cysteine, iron would partly exist in ferrous form, capable of catalysing Fenton-type reactions [15,16]. With this in mind, one may postulate that cellular sensitivity to oxidative stress would largely be a consequence of: (i) capacity to degrade H2O2, preventing this oxidant from diffusing into lysosomes in dangerous amounts during oxidative stress, (ii) amounts of intralysosomal low mass iron and (iii) the extent to which such iron is prevented from being in redox-active form. With respect to (i), it is clear that the high degree of sensitivity to oxidative stress shown by cells such as β-cells and insulinoma cells is a consequence of their low amounts of H2O2-degrading enzymes [38]. When it comes to (ii), the high sensitivity to oxidative stress seen following experimental lysosomal iron loading and in primary and secondary haemochromatosis [5,12,19,39] may be an effect of iron-enriched lysosomes. Finally, considering (iii), the presence of intralysosomal iron-binding molecules and their lysosomal turnover may be of great importance.
Endocytosed or autophagocytosed ferritin survives for some time intralysosomally and may bind iron, unless it is not already saturated, thereby reducing lysosomal sensitivity to oxidative stress [19]. It is possible that autophagocytosis of thiol-rich proteins, for instance MTs, will do the same. MTs contain large numbers of -SH groups, which enable them to bind a variety of metals. Zn and Cd are most firmly bound, but Fe also forms MT complexes [24,40–43]. Furthermore, it has been demonstrated that Fe (just like any other metal bound by MT) induces MT [44,45]. Consequently, we may expect that normal autophagocytosis of up-regulated cytosolic MTs would result in protection against oxidative stress by stabilizing lysosomes. This hypothesis is strongly supported by the findings in our study. The idea is also in agreement with results by Mello-Filho et al. [46], who postulated that iron chelation by MTs might eliminate Fenton-mediated radical damage, and with the results of other groups, who showed proteolytic degradation of MTs by lysosomal cathepsins, but not by cytosolic proteolytic enzymes, suggesting that lysosomes are chiefly responsible for the turnover of MTs. It was also shown that apo-MT is quickly degraded by lysosomal proteases, while metal-conjugated MTs are surprisingly resistant, suggesting that autophagocytosed MT may remain intact for some time after binding iron [26,27]. Thus it seems justified to suggest that autophagocytosed MTs should decrease intralysosomal Fenton-type reactions, giving exactly the effects on lysosomal stability that we have found. This idea receives further support from the findings by Pérez and Cederbaum [47] and Viarengo et al. [44] who showed that metal-induced up-regulation of MT does not induce non-specific antioxidative defence systems. This is in agreement with our findings that reduced thiols are not increased and H2O2 is not better degraded following Zn exposure. Therefore the observed stabilization of lysosomes points to an Fe-binding property of MT inside lysosomes.
As shown by our experiments done in vitro, Fenton-type reactions would take place under the acidic and reducing conditions that exist in lysosomes. As pointed out, lysosomes are rich in low mass iron and very sensitive to oxidative stress, indicating that some of their iron must occur in redox-active ferrous form. This is probably related to the presence in lysosomes of reducing equivalents, such as cysteine [16].
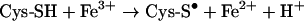 |
We have previously shown that an endocytotic uptake of DFO and DFO conjugated to starch, HMM-DFO, with further transport to the lysosomal compartment, results in lysosomal resistance to oxidative stress [5,6,11] and, following on from these conclusions, DFO was now found to prevent Fenton-type reactions under lysosomal conditions, verifying that DFO is indeed able to bind lysosomal redox-active iron. The finding that MT, at a ratio to iron of 1:2, prevents Fenton-type reactions under acidic and reducing conditions shows that MT apparently is able to chelate iron intralysosomally, something that would occur after MT autophagocytosis.
Oxidative stress during apoptosis, and lysosomal involvement in the apoptotic process, is being increasingly recognized [2,7–9]. Acquisition of resistance to apoptogenic agents seems to be a common event during malignant transformation, and highly malignant, therapy-resistant tumours often overexpress MTs, heat-shock proteins or thioredoxin [8,48–50]. Since these proteins are probably turned over by autophagocytosis, it may be assumed that these and other -SH-rich proteins may express their antiapoptotic capacity by reducing intralysosomal oxidation and lysosomal membrane destabilization. Experiments are under way in our laboratories to further investigate this exciting possibility.
Acknowledgments
The work was supported by the Linköping University Hospital funds to U. T. B. and by the Faculty of Health Sciences, Linköping University, with a postdoctoral stipend to S. K. B.
References
- 1.Dypbukt J. M., Ankarcrona M., Burkitt M., Sjöholm Å., Ström K., Orrenius S., Nicotera P. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. J. Biol. Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 2.Brunk U. T., Neuzil J., Eaton J. W. Lysosomal involvement in apoptosis. Redox Rep. 2001;6:91–97. doi: 10.1179/135100001101536094. [DOI] [PubMed] [Google Scholar]
- 3.Itoh K., Ishii T., Wakabayashi N., Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free Radical Res. 1999;31:319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- 4.Borghesi L. A., Lynes M. A. Stress proteins as agents of immunological change: some lessons from metallothionein. Cell Stress Chaperones. 1996;1:99–108. doi: 10.1379/1466-1268(1996)001<0099:spaaoi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu Z., Persson H. L., Eaton J. W., Brunk U. T. Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radical Biol. Med. 2003;34:1243–1252. doi: 10.1016/s0891-5849(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 6.Kurz T., Leake A., Von Zglinicki T., Brunk U. T. Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem. J. 2004;378:1039–1045. doi: 10.1042/BJ20031029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guicciardi M. E., Leist M., Gores G. J. Lysosomes in cell death. Oncogene. 2004;23:2881–2890. doi: 10.1038/sj.onc.1207512. [DOI] [PubMed] [Google Scholar]
- 8.Nylandsted J., Gyrd-Hansen M., Danielewicz A., Fehrenbacher N., Lademann U., Høyer-Hansen M., Weber E., Multhoff G., Rohde M., Jäättelä M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cirman T., Oresic K., Mazovec G. D., Turk V., Reed J. C., Myers R. M., Salvesen G. S., Turk B. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 2004;279:3578–3587. doi: 10.1074/jbc.M308347200. [DOI] [PubMed] [Google Scholar]
- 10.Li W., Yuan X., Nordgren G., Dalen H., Dubowchik G. M., Firestone R. A., Brunk U. T. Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett. 2000;470:35–39. doi: 10.1016/s0014-5793(00)01286-2. [DOI] [PubMed] [Google Scholar]
- 11.Persson H. L., Kurz T., Eaton J. W., Brunk U. T. Radiation-induced cell death: importance of lysosomal destabilization. Biochem. J. 2005;389:877–884. doi: 10.1042/BJ20050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persson H. L., Yu Z., Tirosh O., Eaton J. W., Brunk U. T. Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radical Biol. Med. 2003;34:1295–1305. doi: 10.1016/s0891-5849(03)00106-0. [DOI] [PubMed] [Google Scholar]
- 13.Yu Z., Li W., Hillman J., Brunk U. T. Human neuroblastoma (SH-SY5Y) cells are highly sensitive to the lysosomotropic aldehyde 3-aminopropanal. Brain Res. 2004;1016:163–169. doi: 10.1016/j.brainres.2004.04.075. [DOI] [PubMed] [Google Scholar]
- 14.Richardson D. R., Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- 15.Schafer F. Q., Buettner G. R. Acidic pH amplifies iron-mediated lipid peroxidation in cells. Free Radicals Biol. Med. 2000;28:1175–1181. doi: 10.1016/s0891-5849(00)00319-1. [DOI] [PubMed] [Google Scholar]
- 16.Pisoni R. L., Acker T. L., Lisowski K. M., Lemons R. M., Thoene J. G. A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: possible role in supporting lysosomal proteolysis. J. Cell Biol. 1990;110:327–335. doi: 10.1083/jcb.110.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klionsky D. J., editor. Georgetown, TX: Eurekah.com/Landes Bioscience; 2004. Autophagy. [Google Scholar]
- 18.Shintani T., Klionsky D. J. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garner B., Roberg K., Brunk U. T. Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radical Res. 1998;29:103–114. doi: 10.1080/10715769800300121. [DOI] [PubMed] [Google Scholar]
- 20.Andrews G. K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000;59:95–104. doi: 10.1016/s0006-2952(99)00301-9. [DOI] [PubMed] [Google Scholar]
- 21.Hamer D. H. Metallothionein. Annu. Rev. Biochem. 1986;55:913–951. doi: 10.1146/annurev.bi.55.070186.004405. [DOI] [PubMed] [Google Scholar]
- 22.Klaassen C. D., Liu J. Metallothionein transgenic and knock-out mouse models in the study of cadmium toxicity. J. Toxicol. Sci. 1998;23:97–102. doi: 10.2131/jts.23.supplementii_97. [DOI] [PubMed] [Google Scholar]
- 23.Cai L., Iskander S., Cherian M. G., Hammond R. R. Zinc- or cadmium-pre-induced metallothionein protects human central nervous system cells and astrocytes from radiation-induced apoptosis. Toxicol. Lett. 2004;146:217–226. doi: 10.1016/j.toxlet.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Chimienti F., Jourdan E., Favier A., Seve M. Zinc resistance impairs sensitivity to oxidative stress in HeLa cells: protection through metallothioneins expression. Free Radicals Biol. Med. 2001;31:1179–1190. doi: 10.1016/s0891-5849(01)00701-8. [DOI] [PubMed] [Google Scholar]
- 25.Wang G. W., Klein J. B., Kang Y. J. Metallothionein inhibits doxorubicininduced mitochondrial cytochrome c release and caspase-3 activation in cardiomyocytes. J. Pharmacol. Exp. Ther. 2001;298:461–468. [PubMed] [Google Scholar]
- 26.Hahn S. H., Yoo O. J., Gahl W. A. Effect of metal ions on the stability of metallothionein in the degradation by cellular fractions in vitro. Exp. Mol. Med. 2001;33:32–36. doi: 10.1038/emm.2001.7. [DOI] [PubMed] [Google Scholar]
- 27.Feldman S. L., Failla M. L., Cousins R. J. Degradation of rat liver metallothioneins in vitro. Biochim. Biophys. Acta. 1978;544:638–646. doi: 10.1016/0304-4165(78)90338-0. [DOI] [PubMed] [Google Scholar]
- 28.Mizzen C. A., Cartel N. J., Yu W. H., Fraser P. E., McLachlan D. R. Sensitive detection of metallothioneins-1, -2 and -3 in tissue homogenates by immunoblotting: a method for enhanced membrane transfer and retention. J. Biochem. Biophys. Methods. 1996;32:77–83. doi: 10.1016/0165-022x(95)00044-r. [DOI] [PubMed] [Google Scholar]
- 29.Panus P. C., Radi R., Chumley P. H., Lillard R. H., Freeman B. A. Detection of H2O2 release from vascular endothelial cells. Free Radicals Biol. Med. 1993;14:217–223. doi: 10.1016/0891-5849(93)90013-k. [DOI] [PubMed] [Google Scholar]
- 30.Boyne A. F., Ellman G. L. A methodology for analysis of tissue sulfhydryl components. Anal. Biochem. 1972;46:639–653. doi: 10.1016/0003-2697(72)90335-1. [DOI] [PubMed] [Google Scholar]
- 31.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 32.Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 33.Myhre O., Andersen J. M., Aarnes H., Fonnum F. Evaluation of the probes 2′,7′-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem. Pharmacol. 2003;65:1575–1582. doi: 10.1016/s0006-2952(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 34.Zdolsek J. M., Roberg K., Brunk U. T. Visualization of iron in cultured macrophages: a cytochemical light and electron microscopic study using autometallography. Free Radicals Biol. Med. 1993;15:1–11. doi: 10.1016/0891-5849(93)90120-j. [DOI] [PubMed] [Google Scholar]
- 34a.Timm F. Zur histochemie der schwermetalle. Das sulfid-silberverfahren. Dtsch. Z. Ges. Med. 1958;46:706–711. [PubMed] [Google Scholar]
- 35.de Duve C., Wattiaux R. Functions of lysosomes. Annu. Rev. Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 36.Petrat F., de Groot H., Rauen U. Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem. J. 2001;356:61–69. doi: 10.1042/0264-6021:3560061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat. Res. 2003;531:81–92. doi: 10.1016/j.mrfmmm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 38.Olejnicka B. T., Andersson A., Tyrberg B., Dalen H., Brunk U. T. β-Cells, oxidative stress, lysosomal stability, and apoptotic/necrotic cell death. Antioxid. Redox Signal. 1999;1:305–315. doi: 10.1089/ars.1999.1.3-305. [DOI] [PubMed] [Google Scholar]
- 39.Eaton J. W., Qian M. Molecular bases of cellular iron toxicity. Free Radicals Biol. Med. 2002;32:833–840. doi: 10.1016/s0891-5849(02)00772-4. [DOI] [PubMed] [Google Scholar]
- 40.Stillman M. J. Metallothioneins. Coord. Chem. Rev. 1995;144:461–511. [Google Scholar]
- 41.Good M., Vašák M. Iron(II)-substituted metallothionein: evidence for the existence of iron-thiolate clusters. Biochemistry. 1986;25:8353–8356. doi: 10.1021/bi00374a003. [DOI] [PubMed] [Google Scholar]
- 42.Ding X., Bill E., Good M., Trautwein A. X., Vašák M. Mossbauer studies on the metal-thiolate cluster formation in Fe(II)-metallothionein. Eur. J. Biochem. 1988;171:711–714. doi: 10.1111/j.1432-1033.1988.tb13843.x. [DOI] [PubMed] [Google Scholar]
- 43.Werth M. T., Johnson M. K. Magnetic circular dichroism and electron paramagnetic resonance studies of iron(II)-metallothionein. Biochemistry. 1989;28:3982–3988. doi: 10.1021/bi00435a053. [DOI] [PubMed] [Google Scholar]
- 44.Viarengo A., Burlando B., Cavaletto M., Marchi B., Ponzano E., Blasco J. Role of metallothionein against oxidative stress in the mussel Mytilus galloprovincialis. Am. J. Physiol. 1999;277:R1612–R1619. doi: 10.1152/ajpregu.1999.277.6.R1612. [DOI] [PubMed] [Google Scholar]
- 45.Fleet J. C., Andrews G. K., McCormick C. C. Iron-induced metallothionein in chick liver: a rapid, route-dependent effect independent of zinc status. J. Nutr. 1990;120:1214–1222. doi: 10.1093/jn/120.10.1214. [DOI] [PubMed] [Google Scholar]
- 46.Mello-Filho A. C., Chubatsu L. S., Meneghini R. V79 Chinese-hamster cells rendered resistant to high cadmium concentration also become resistant to oxidative stress. Biochem. J. 1988;256:475–479. doi: 10.1042/bj2560475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pérez M. J., Cederbaum A. I. Metallothionein 2A induction by zinc protects HEPG2 cells against CYP2E1-dependent toxicity. Free Radicals Biol. Med. 2003;34:443–455. doi: 10.1016/s0891-5849(02)01302-3. [DOI] [PubMed] [Google Scholar]
- 48.Janssen A. M., van Duijn W., Kubben F. J., Griffioen G., Lamers C. B., van Krieken J. H., van de Velde C. J., Verspaget H. W. Prognostic significance of metallothionein in human gastrointestinal cancer. Clin. Cancer Res. 2002;8:1889–1896. [PubMed] [Google Scholar]
- 49.Joseph M. G., Banerjee D., Kocha W., Feld R., Stitt L. W., Cherian M. G. Metallothionein expression in patients with small cell carcinoma of the lung: correlation with other molecular markers and clinical outcome. Cancer. 2001;92:836–842. [PubMed] [Google Scholar]
- 50.Kakolyris S., Giatromanolaki A., Koukourakis M., Powis G., Souglakos J., Sivridis E., Georgoulias V., Gatter K. C., Harris A. L. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin. Cancer Res. 2001;7:3087–3091. [PubMed] [Google Scholar]