

Abstract
Estrogen receptors, PPARs, and liver X receptors are members of the nuclear receptor superfamily of ligand-dependent transcription factors that regulate diverse aspects of development and homeostasis. Recent studies of the biologic roles of these receptors and their mechanisms of action have significantly advanced our understanding of transcriptional programs that control lipid and carbohydrate metabolism, immunity and inflammation, and wound repair. These findings provide insights into the therapeutic actions of existing drugs that target nuclear receptors and raise new possibilities for development of safer, more effective drugs for the prevention and treatment of metabolic and cardiovascular diseases. In this introduction to this Review series, underlying mechanisms that enable nuclear receptors to positively and negatively regulate gene expression are presented as background to the focused reviews on estrogen receptors, PPARs, liver X receptors, and the PPARγ coactivator-1 (PGC-1) family of coactivators.
Members of the nuclear receptor superfamily of ligand-dependent transcription factors play a multitude of essential roles in development, homeostasis, reproduction, and immune function (1–4). Several members of this family, including steroid receptors such as the estrogen receptor (ER) and PPARs, are also targets of drugs that are used in a variety of clinical settings. As a group, nuclear receptors have been extraordinarily informative in providing model systems for understanding regulated gene expression. For example, the ability to switch the role of a nuclear receptor from that of a transcriptional repressor to that of a transcriptional activator, by treatment of cells with a synthetic or natural ligand, dramatically accelerated our understanding of DNA regulatory sequences that control gene expression and the elucidation of biochemical events within the cell that ultimately lead to transcriptional activation (5–7). In parallel, the cloning of cDNAs encoding the different members of this family opened up new frontiers in receptor biology and the potential to extend the utility of nuclear receptor–based therapeutics in the prevention and treatment of cardiovascular, metabolic, and inflammatory diseases (2–4).
Recent studies have revealed an extraordinary diversity of coactivator and corepressor protein complexes that interact with nuclear receptors and mediate their transcriptional activities (8, 9). The identification of these proteins, and the understanding of how they interact with nuclear receptors in a manner that can be either positively or negatively regulated by ligands, help to explain the mechanisms of action of existing selective ER modulators (SERMs) (10) and provide insights into how other nuclear receptors might be selectively regulated to achieve specific therapeutic goals. In addition, increasing evidence links the actions of at least some nuclear receptors, including the ER, to signal transduction events at the cell membrane, providing additional avenues for therapeutic intervention (11, 12). Discovery in the nuclear receptor field continues at a breathtaking pace, and emerging findings have many implications for new lines of clinical investigation. In this eight-article Review series, we focus on the roles of ERs, PPARs, liver X receptors (LXRs), and the PPARγ coactivator-1 (PGC-1) gene family in regulating lipid and carbohydrate metabolism and inflammation. These studies are revealing new regulatory circuits that have important implications for understanding the pathogenesis of diseases including obesity, diabetes, and atherosclerosis, and they have the potential to accelerate the development of new, safer treatment strategies.
A nuclear receptor primer
Genomic and cDNA sequencing efforts have defined at least 48 nuclear receptors that are encoded by the human and mouse genomes (13). These include the classical steroid hormone receptors (estrogen, progesterone, androgen, glucocorticoid, and mineralocorticoid receptors); “orphan receptors,” which exhibit conserved features of the nuclear receptor family but have not been linked to endogenous ligands; and so-called “adopted orphan receptors,” which were initially identified as orphan receptors but were subsequently linked to endogenous ligands. The adopted orphan receptors include the thyroid hormone receptors, retinoic acid receptors, PPARs, and LXRs (for an animated review of nuclear receptor family signaling, see the Nuclear Receptor Signaling Atlas; www.nursa.org). Nearly all members of this family contain a highly conserved DNA-binding domain that mediates sequence-specific recognition of target genes and a C-terminal domain that determines the specific ligand-binding properties of each receptor and mediates ligand-regulated transcriptional activation and/or repression functions (1) (Figure 1). The amino terminus is poorly conserved among different nuclear receptors and often contains an important additional transcriptional activation domain.
Figure 1.
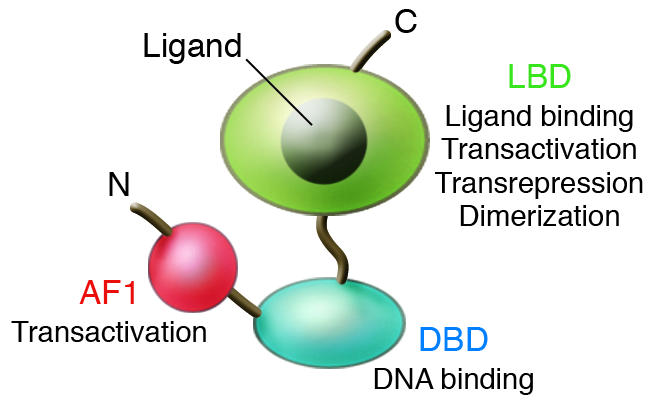
Domain structure of nuclear receptors. N and C represent the amino and carboxyl termini, respectively. AF1 is a variable amino-terminal transactivation domain. The ligand-binding domain (LBD) also mediates dimerization, transcriptional activation, and transcriptional repression functions. DBD, DNA-binding domain.
The activities of steroid hormone and adopted orphan receptors are controlled by small lipophilic molecules. In the case of the steroid and thyroid hormone receptors, the natural ligands are classical hormones that are produced in endocrine organs and are subject to physiologic feedback control circuits (1). After release into the circulation, these hormones ultimately diffuse into cells in target tissues, where they bind to their cognate receptors. The ligand-binding properties of steroid and thyroid hormone receptors are highly specific, and binding constants for their respective hormones are typically in the nanomolar to subnanomolar range. In contrast, many of the adopted orphan receptors are regulated by lipid metabolites that are produced within cells, allowing transcriptional changes in response to alterations in internal lipid homeostasis (14). For example, LXRs are regulated by oxysterols that accumulate under conditions in which total cellular cholesterol content rises. Oxysterol activation of LXRs results in feed-forward upregulation of genes that enable macrophages and other peripheral cells to export cholesterol to extracellular acceptors for reverse cholesterol transport and promote biliary excretion of cholesterol by the liver (15). The ligand-binding properties of adopted orphan receptors are generally less specific and of lower affinity than those of steroid receptors, reflecting their ability to bind a range of lipid metabolites that exhibit physiologic intracellular concentrations in the high-nanomolar to low-micromolar range.
Steroid receptors, such as the ER, are synthesized in inactive forms that associate with heat shock protein complexes in the cytoplasm and/or nucleus (16). As a result of this interaction, unliganded steroid receptors exhibit little or no occupancy of target genes. Hormone binding promotes receptor dissociation from these complexes and nuclear entry. In general, steroid hormone receptors bind to hormone response elements (REs) in positively regulated target genes as homodimers (Figure 2A) (17). Steroid hormone REs consist of variations of palindromic arrangements of 2 core recognition elements that are contacted by the DNA-binding domain in a symmetric manner. Specific bases within these core recognition motifs determine binding specificity for each of the steroid hormone receptors. In contrast, most of the adopted orphan receptors, including PPARs and LXRs, bind to target genes as heterodimers with retinoid X receptors (RXRs) in the presence or absence of ligand (Figure 2B). The REs for these receptors most often consist of variations of 2 core recognition elements arranged as direct repeats. The precise sequence and spacing of these motifs determine the specificity of binding by different homodimeric or heterodimeric pairs (18). For example, PPAR/RXR heterodimers preferentially bind to direct repeats spaced by 1 bp (19), while LXR/RXR heterodimers preferentially bind to direct repeats spaced by 4 bp (20, 21).
Figure 2.

Mechanisms of sequence-specific DNA binding by steroid hormone and heterodimeric nuclear receptors. (A) Steroid hormone receptors, such as the ER and glucocorticoid receptor (GR), generally bind as homodimers in a ligand-dependent manner to hormone REs that consist of palindromic arrangements of core recognition motifs (arrows). (B) RXR heterodimers bind in the presence or absence of ligands (shown here in the presence of ligand) to similar core recognition elements arranged as direct repeats. Both the specific sequence of the core recognition motif and the spacing between motifs are determinants of DNA-binding specificity. PPAR/RXR heterodimers preferentially bind to direct repeats spaced by 1 bp, while LXR/RXR heterodimers preferentially bind to direct repeats spaced by 4 bp.
Nuclear receptors can positively or negatively regulate gene expression by several mechanisms. First, steroid hormone and adopted orphan receptors can activate transcription of direct target genes after their binding to REs in promoter or enhancer regions by recruiting coactivator complexes in a ligand-dependent manner (Figure 3A). These complexes function as intermediary factors that are required for the ultimate recruitment of RNA polymerase II (8, 9). Recent studies have indicated that many coactivator complexes are involved in ligand-dependent activation of nuclear receptor target genes, and that they are recruited to the promoter in a specific sequence that cycles repetitively over time scales of minutes (22). These observations reflect the multiple steps that must be accomplished in order for a gene to be switched from an inactive or repressed state to an actively transcribed state, including nucleosome remodeling, execution of specific histone modifications (e.g., acetylation of specific histone tail residues), and recruitment of basal transcription factors. Coactivator complexes appear to function in a combinatorial and gene-specific manner to integrate information provided by DNA-bound transcription factors and signaling cascades that impact the regulatory regions of genes.
Figure 3.

Mechanisms of transcriptional activation and repression by steroid hormone and heterodimeric nuclear receptors. (A) Ligand-dependent transactivation. The binding of hormones or synthetic agonists causes the recruitment of coactivator complexes to the ligand-binding domain. Ligand-dependent recruitment of these factors is similar for heterodimeric receptors and steroid hormone receptors (not shown). For simplicity, only a single generic complex is illustrated. In general, ligand-dependent transcription of nuclear receptor target genes is associated with the recruitment of numerous coactivator complexes that act in a combinatorial or sequential manner. These complexes are associated with a number of enzymatic activities, including histone acetyltransferase (HAT), histone methyltransferase (HMT), and nucleosome remodeling (NRM) activities. Structurally distinct ligands may alter the pattern of recruitment of these factors, resulting in altered patterns of gene activation. (B) Active repression. A subset of heterodimeric nuclear receptors, including PPAR/RXR and LXR/RXR heterodimers, are capable of binding to REs in the absence of ligand and recruiting corepressor complexes that actively repress transcription. A number of corepressor complexes are associated with histone deacetylase (HDAC) activities, as well as histone methyltransferase and nucleosome remodeling activities that are generally distinct from those associated with coactivator complexes. HDM, histone demethylase. (C) SERM/antagonist–dependent repression. Some SERMs (shown here in gray), such as tamoxifen, and PPAR antagonists promote corepressor interactions with the ligand-binding domain, resulting in active repression in permissive cell types. (D) Ligand-dependent trans-repression. Many nuclear receptors, including glucocorticoid receptors, ERs, PPARs, and LXRs (indicated in blue), are capable of antagonizing signal-dependent activation of inflammatory response genes by transcription factors such as NF-κB.
Second, many of the adopted orphan receptors, including PPARs and LXRs, can bind to target genes in the absence of ligand and actively repress transcription (Figure 3B). This repressive function results from the recruitment of corepressor complexes, that act in an antagonistic manner, to coactivators (7). In the presence of activating ligands, corepressor complexes are replaced by coactivator complexes, resulting in a switch from repression to gene activation (Figure 3A). This exchange process is a key target of synthetic compounds that regulate nuclear receptor activity. Nuclear receptor agonists function in a manner analogous to that of endogenous hormones to promote coactivator binding. In contrast, antagonists function to inhibit coactivator binding and, in many cases, promote the binding of corepressors (23, 24). These properties can result in complex effects on nuclear receptor function. For example, tamoxifen and raloxifene are SERMs that promote dissociation of the ER from heat shock protein complexes and allow it to bind to target genes. Intriguingly, these compounds are antiestrogenic/antagonistic in the breast, but estrogenic/agonistic in liver and bone. Current evidence suggests that estrogen antagonism in the breast results from the ability of tamoxifen and raloxifene to inhibit coactivator binding and promote corepressor binding to the ligand-binding domain, converting the ER into an active repressor in this tissue (Figure 3C). In contrast, it is hypothesized that, because of tissue-specific differences in corepressor expression or activity, corepressors are not recruited to tamoxifen- or raloxifene-bound ERs in liver or bone, while coactivators can still be recruited to the activation domain present in the amino terminus. As a result, ER target genes become activated, although not as effectively as they would be by estrogen itself.
Third, many nuclear receptors can inhibit signal-dependent activation of promoters by other classes of transcription factors in a ligand-dependent manner. The prototypic example of this is the ability of the glucocorticoid receptor to inhibit activation of inflammatory response genes by interfering with the activities of signal-dependent transcription factors such as activator protein-1 (AP-1) and NF-κB (Figure 3D) (25). This activity, referred to as trans-repression, is promoter-specific but does not appear to involve sequence-specific DNA binding. Instead, the nuclear receptors such as the glucocorticoid receptor appear to be tethered to target promoters through protein-protein interactions. Trans-repression most likely accounts for a majority of the antiinflammatory actions of clinically used synthetic glucocorticoids (26), such as dexamethasone, and may be an important mechanism by which other nuclear receptor ligands, such as the PPAR agonists, exert therapeutic effects (27). Recent studies indicate that multiple molecular mechanisms may be used by different members of the nuclear receptor family to inhibit signal-dependent gene activation, and that these mechanisms may be subject to selective modulation by structurally different ligands (28). A major therapeutic goal will be to exploit these findings to identify ligands that differentially modulate the activation, active repression, and trans-repression functions of nuclear receptors in order to achieve safer, more specific targeting of pathogenic processes.
Guide to the Reviews
This Review series begins with the Review by Deroo and Korach on ERs and human disease (29). Although ERs are among the most extensively studied nuclear receptors and have been targets of pharmacologic intervention for decades, they continue to be the basis for new mechanistic insights and have provided valuable lessons for translation of basic research findings into clinical application. Deroo and Korach review the roles of estrogens and ERs in a diverse range of human diseases, including hormone-sensitive cancers, osteoporosis, neurodegenerative diseases, cardiovascular disease, and obesity. While many lines of evidence support the view that maintenance of estrogen action after menopause should be beneficial in bone, the cardiovascular system, and the CNS, increased risk of breast cancer and the recent unexpected observation (30) of an increase in risk of thromboembolism and stroke in postmenopausal women have significantly complicated clinical management decisions. Development of improved tissue-specific SERMs might alleviate some of the risks associated with conventional hormone replacement therapy.
Lefebvre, Chinetti, Fruchart, and Staels lead off a “miniseries” within the series on PPARs, with a review of PPARα and metabolic disease (31). PPARα regulates lipid and glucose metabolism in liver and muscle and is the molecular target of the fibrate class of drugs that are used in the treatment of hypertriglyceridemia. Lefebvre and colleagues discuss the metabolic roles of PPARα in these tissues as well as recent studies that have revealed pleiotropic antiinflammatory and antiproliferative effects of PPARα agonists in macrophages and other cell types within the artery wall. These activities are likely to contribute to the antiatherogenic effects of fibrates that are observed in humans and animal models.
The next installment of the PPAR miniseries, by Semple, Chatterjee, and O’Rahilly (32), focuses on PPARγ and human metabolic disease. PPARγ is essential for fat cell development and regulates glucose and lipid homeostasis in adipose tissue, liver, and skeletal muscle. Among the 3 members of the PPAR subfamily, the physiologic roles of PPARγ in humans are probably best understood, because of the combination of the clinical use of PPARγ agonists as insulin-sensitizing drugs in the treatment of type 2 diabetes, the identification of individuals with rare dominant-negative mutations in PPARγ, and the analysis of more common polymorphisms that have quantitative effects on receptor function. Semple and colleagues synthesize the basic science and clinical literature to provide an integrated view of PPARγ function in humans and explain the antidiabetic actions of clinically used PPARγ agonists. While these drugs are effective insulin sensitizers, their clinical application is currently limited by side effects that include fluid retention, hemodilution, and heart failure, underscoring the importance of identifying safer, selective modulators for this receptor.
Barish, Narkar, and Evans (33) tackle the third member of the PPAR subfamily, PPAR&Dgr; (also referred to as PPARβ). PPAR&Dgr; is the least understood member of the group but is now the subject of intense investigation by many laboratories. Recent studies have uncovered roles of PPAR&Dgr; in controlling fatty acid catabolism and energy uncoupling in adipose tissue and muscle and in negatively regulating inflammation in the macrophage. Barish, Narkar, and Evans discuss the potential implications of these findings with respect to exercise capacity, energy homeostasis, the metabolic syndrome, and atherosclerosis and the development of novel classes of drugs that target PPAR&Dgr;.
Michalik and Wahli (34) complete the PPAR miniseries with a consideration of emerging findings regarding the roles of the PPAR subfamily in tissue protection and repair. Tissue injury initiates a complex repair program that is designed to restore tissue homeostasis. This program is largely based on transcriptional responses to local factors produced in response to an insult. These factors include lipid mediators that can potentially serve as endogenous ligands for PPARs. Michalik and Wahli review recent advances that reveal new biologic roles of PPARs in the control of the program of gene expression required for efficient wound repair.
The series transitions to LXRs with the Review by Zelcer and Tontonoz (35). LXRs have more recently emerged as adopted orphan nuclear receptors that play significant roles in the regulation of lipid metabolism and inflammation. As noted above, LXRs serve as sensors of cellular cholesterol and positively regulate the expression of genes involved in the maintenance of cholesterol homeostasis and fatty acid biosynthesis. In addition, LXRs can potently inhibit inflammatory responses in a ligand-dependent manner and have recently been found to be required for immunity to the intracellular pathogen Listeria monocytogenes. Activation of the LXR pathway by synthetic agonists has been shown to inhibit macrophage foam cell formation and the development of atherosclerosis in animal models, suggesting that LXRs are attractive targets for novel antiatherogenic drugs.
Our series concludes with a Review by Finck and Kelly (36) on the PGC-1 family of coactivators. PGC-1α, initially identified as a PPARγ coactivator, has been the most intensively studied member of this subfamily and has proven to serve coactivator functions not only for nuclear receptors, but for other classes of transcription factors as well. The general theme to emerge from studies of PGC-1α, as well as PGC-1β, is that they serve as inducible coactivators that are dedicated to the transcriptional control of cellular energy balance, including mitochondrial biogenesis and function. PGC-1α and PGC-1β are notable because their expression and transcriptional activities are influenced by environmental, dietary, and metabolic inputs. For example, PGC-1α is highly induced in muscle during cold exposure, coactivating transcriptional programs that lead to enhanced fatty acid oxidation and energy production required for adaptive thermogenesis.
In concert, this Review series provides an up-to-date view of recent progress and future challenges in areas of nuclear receptor research that impact on tissue homeostasis, inflammation, and metabolic and cardiovascular disease. A major goal of the pharmaceutical industry will be to exploit the ability to differentially modulate nuclear receptors with designer ligands in order to develop safer, more effective drugs. While this has proven to be a challenging task, in part because it is still difficult to predict how specific ligand-dependent effects on programs of gene expression in cells will translate to overall effects on physiologic/pathophysiologic processes in vivo, there remains significant optimism that going nuclear will have a substantial impact on the treatment of a broad range of human diseases.
Footnotes
Nonstandard abbreviations used: ER, estrogen receptor; LXR, liver X receptor; PGC-1, PPARγ coactivator-1; RE, response element; RXR, retinoid X receptor; SERM, selective ER modulator.
Conflict of interest: The author has declared that no conflict of interest exists.
References
- 1.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mangelsdorf DJ, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 4.Giguere V. Orphan nuclear receptors: from gene to function. Endocr. Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu. Rev. Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 6.McKenna NJ, Lanz RB, O’Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr. Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 7.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 8.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 9.Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 10.McDonnell DP. The molecular pharmacology of SERMs. Trends Endocrinol. Metab. 1999;10:301–311. doi: 10.1016/s1043-2760(99)00177-0. [DOI] [PubMed] [Google Scholar]
- 11.Manavathi, B., and Kumar, R. 2005. Steering estrogen signals from the plasma membrane to the nucleus: two sides of the coin. J. Cell. Physiol. doi:10.1002/jcp.20551. [DOI] [PubMed]
- 12.Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am. J. Cardiol. 2002;90:3F–6F. doi: 10.1016/s0002-9149(02)02418-9. [DOI] [PubMed] [Google Scholar]
- 13.Duarte J, Perriere G, Laudet V, Robinson-Rechavi M. NUREBASE: database of nuclear hormone receptors. Nucleic Acids Res. 2002;30:364–368. doi: 10.1093/nar/30.1.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chawla A, Repa J, Evans R, Mangelsdorf D. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 15.Tontonoz P, Mangelsdorf DJ. Liver x receptor signaling pathways in cardiovascular disease. Mol. Endocrinol. 2003;17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 16.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 17.Glass CK. Differential recognition of target genes by nuclear receptor monomers, dimers and heterodimers. Endocr. Rev. 1994;15:1503–1519. doi: 10.1210/edrv-15-3-391. [DOI] [PubMed] [Google Scholar]
- 18.Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoonjans K, et al. Induction of the acyl-coenzyme A synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J. Biol. Chem. 1995;270:19269–19276. doi: 10.1074/jbc.270.33.19269. [DOI] [PubMed] [Google Scholar]
- 20.Willy PJ, et al. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 21.Willy PJ, Mangelsdorf DJ. Unique requirements for retinoid-dependent transcriptional activation by the orphan receptor LXR. Genes Dev. 1997;11:289–298. doi: 10.1101/gad.11.3.289. [DOI] [PubMed] [Google Scholar]
- 22.Metivier R, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 23.Shiau AK, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 24.Xu HE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 25.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 26.Reichardt HM, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pascual G, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogawa S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deroo BJ, Korach KS. Estrogen receptors and human disease. J. Clin. Invest. 2006;116:561–570. doi:10.1172/JCI27987. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farquhar, C.M., Marjoribanks, J., Lethaby, A., Lamberts, Q., and Suckling, J.A. 2005. Long term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst. Rev. Issue 3, article no. CD004143. doi:10.1002/14651858.CD004143.pub2. [DOI] [PubMed]
- 31.Lefebvre P, Chinetti G, Fruchart J-C, Staels B. Correcting metabolic and cardiovascular disorders throughout PPARα. J. Clin. Invest. 2006;116:571–580. doi:10.1172/JCI27989. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semple RK, Chatterjee VKK, O’Rahilly S. PPARγ and human metabolic disease. J. Clin. Invest. 2006;116:581–589. doi:10.1172/JCI28003. doi: 10.1172/JCI28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barish GD, Narkar VA, Evans RM. PPAR&Dgr;: a dagger in the heart of the metabolic syndrome. J. Clin. Invest. 2006;116:590–597. doi:10.1172/JCI27955. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michalik L, Wahli W. PPARs: nuclear receptors ensuring efficient tissue protection and repair. J. Clin. Invest. 2006;116:598–606. doi:10.1172/JCI27958. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zelcer N, Tontonoz P. The liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 2006;116:607–614. doi:10.1172/JCI27883. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 2006;116:615–622. doi:10.1172/JCI27794. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]