

Abstract
The horseshoe crab cationic antimicrobial peptide polyphemusin I is highly active in vitro but not protective in mouse models of bacterial and LPS challenge, while a synthetic polyphemusin variant, PV5, was previously shown to be protective in vivo. In this study, we investigated the interaction of these peptides with lipid membranes in an effort to propose a mechanism of interaction. The solution structure of PV5 was determined by proton NMR in the absence and presence of dodecylphosphocholine (DPC) micelles. Like polyphemusin I, PV5 is a β-hairpin but appeared less amphipathic in solution. Upon association with DPC micelles, PV5 underwent side chain rearrangements which resulted in an increased amphipathic conformation. Using fluorescence spectroscopy, both peptides were found to have limited affinity for neutral vesicles composed of phosphatidylcholine (PC). Incorporation of 25 mol % cholesterol or phosphatidylethanolamine into PC vesicles produced little change in the partitioning of either peptide. Incorporation of 25 mol % phosphatidylglycerol (PG) into PC vesicles, a simple prokaryotic model, resulted in a large increase in the affinity for both peptides, but the partition coefficient for PV5 was almost twice that of polyphemusin I. Differential scanning calorimetry studies supported the partitioning data and demonstrated that neither peptide interacted readily with neutral PC vesicles. Both peptides showed affinity for negatively charged membranes incorporating PG. The affinity of PV5 was much greater as the pretransition peak was absent at low peptide to lipid ratios (1:400) and the reduction in enthalpy of the main transition was greater than that produced by polyphemusin I. Both peptides decreased the lamellar to inverted hexagonal phase transition temperature of PE indicating the induction of negative curvature strain. These results, combined with previous findings that polyphemusin I promotes lipid flip-flop but does not induce significant vesicle leakage, ruled out the torroidal pore and carpet mechanisms of antimicrobial action for these polyphemusins.
The polyphemusins are a group of cationic peptides isolated from the American horseshoe crab, Limulus polyphemus (1), and share a great similarity to the tachyplesins from the Japanese horseshoe crab, Tachypleus tridentatus (2). These peptides are 17–18 amino acid residues in length and contain two disulfide bonds which act to constrain the peptide backbone into an antiparallel β-hairpin connected by a β-turn. To date, the solution and micelle-bound structures of tachyplesin I (3) and the solution structure of polyphemusin I (4) as well as the structures of various analogues have been determined by nuclear magnetic resonance. These structures have served as templates for peptide design as well as tools for investigating structure–activity relationships as an approach to determine the mechanism of action of this family of peptides.
The mechanism of action of the polyphemusins and tachyplesins, while not clear, is believed to involve membrane translocation (4–6). Previous studies utilizing model membranes have shown that, at low peptide to lipid ratios, polyphemusin I readily induces lipid flip-flop between membrane leaflets but produces a low degree of entrapped calcein release (5). In addition, a translocation assay has demonstrated that polyphemusin I becomes digested by liposome-entrapped protease (5). Combined, these findings indicate that polyphemusin I is capable of accessing the interior of liposomes and does so without greatly disrupting or permeabilizing the lipid bilayer.
In an effort to improve the high intrinsic antimicrobial activity of the polyphemusins, a series of analogues were designed with increased charge and amphipathic character as indicated by computer modeling (7). Characterization of these analogues revealed a 2–4-fold decrease in antimicrobial activity; however, one analogue in particular, PV5,1 with an additional arginine inserted into the turn region, displayed a 2-fold reduction in hemolytic activity and substantially improved protection in mouse models of endotoxemia (7). Specifically, PV5 produced a 50% survival rate in glactosamine-sensitized mice challenged with Escherichia coli LPS compared to 10% survival produced by polyphemusin I and no survival without peptide treatment. Recently, the related peptide tachyplesin has been demonstrated to function as a secondary secretagogue of LPS-induced hemocyte exocytosis presumably through a G protein mediated pathway (8). While these studies were conducted using horseshoe crab hemocytes, it is conceivable that interaction of these β-hairpin peptides with mammalian hemocytes could elicit the same response, resulting in amplification of the innate immune system. This may explain the ability of the polyphemusins to protect mice challenged with LPS but does not account for the differences observed between polyphemusin I and PV5.
To continue to investigate the mechanism of action of the polyphemusins, we have determined here the three-dimensional solution structure of PV5 in the presence and absence of DPC micelles using two-dimensional proton NMR and compared it to the previously determined structure of polyphemusin I (4). The interactions of both polyphemusin I and PV5 with model membranes, representing eukaryotic and prokaryotic compositions, were investigated using fluorescence spectroscopy and differential scanning calorimetry. A mechanism of membrane translocation for the polyphemusins is proposed, based on the results presented here as well as previous findings.
MATERIALS AND METHODS
Peptide Synthesis
Polyphemusin I (RRWCFRVCYRGF-CYRKCR-NH2) and PV5 (RRWCFRVCYRGRFCYRKCR-NH2) were synthesized by Fmoc solid-phase peptide synthesis using a model 432A peptide synthesizer (Applied Biosystems Inc.) at the University of British Columbia. Both peptides were oxidized using a Tris–DMSO solution (100 mM Tris-HCl, 20% DMSO, pH 8) for 24 h at room temperature to promote disulfide bond formation (9). The correctly folded peptides were then purified by reversed-phase chromatography using a Pharmacia model LKB FPLC. Correct disulfide bond formation (between cysteine residues 4–17 and 8–13 of polyphemusin I and residues 4–18 and 8–14 of PV5) of the purified peptides was confirmed by MALDI mass spectrometry through an observed 4 mass unit difference between the reduced and oxidized forms of the peptides (data not shown) and further verified through the observation of long-range NOEs in the NOESY spectra of PV5 and previously for polyphemusin I (4). For clarity, the primary structures and disulfide connectivity of the synthesized peptides are shown in Figure 1.
Figure 1.

Primary structures of polyphemusin I (PM1) and its analogue PV5. Disulfide linkages are shown as solid lines. The spacing in polyphemusin I is done for sequence alignment and does not represent a break in the peptide backbone.
NMR Spectroscopy
PV5 was dissolved in H2O:D2O (9: 1) at a concentration of 2 mM, with or without 300 mM DPC. The pH* of the final samples were 3.80 and 3.95 in the absence and presence of DPC. NMR spectra of PV5 without DPC were recorded at 25 °C on a Varian Unity 500 NMR spectrometer operating at a 1H frequency of 499.94 MHz. NMR spectra of PV5 containing 300 mM DPC were recorded at 40 °C on a Varian Inova 600 NMR spectrometer operating at a 1H frequency of 599.84 MHz. DQF-COSY (10), TOCSY (11), and NOESY (12) spectra were obtained using standard techniques. Water suppression was achieved using the WATERGATE technique (13, 14) or by presaturation. Spectra were collected with 512 data points in F1 and 2048 data points in F2. TOCSY spectra were acquired using the Malcolm Levitt (MLEV) 17 pulse sequence (15) at a spin-lock time of 60 ms. NOESY spectra were recorded with a mixing time of 150 ms. The NMR data were processed with NMRPIPE (16).
NOE Data Analysis and Structure Calculation
All NMR spectra were analyzed using NMRView version 5.0.3 (17). NOE cross-peaks were assigned and integrated. The NOE volumes were converted to distances and calibrated using intraresidue HN–Hα cross-peaks and the mean distance of 2.8 Å determined by Hyberts et al. (18). The distances were then converted into distance restraints by calculating upper and lower distance bounds using the equations of Hyberts et al. (18). Pseudoatom restraints were corrected as previously described (19) by adding 1 and 1.5 Å to the upper distance bound of unresolved methylene and methyl protons, respectively, and resolved methylene protons were float-corrected by adding 1.7 Å to the upper distance bound. Structure calculations were performed using X-PLOR-NIH version 2.9.0 (20). One hundred structures were generated by the DGSA protocol and further refined. The refinement consisted of simulated annealing, decreasing the temperature from 310 to 10 K over 50000 steps. In the sample without DPC, 49 PV5 structures were calculated with no NOE violations >0.3 Å, and the 16 lowest energy conformers with final energies <28 kcal mol−1 were selected for presentation. In the sample containing DPC, 26 PV5 structures were calculated with no NOE violations >0.2 Å, and the 17 lowest energy conformers with final energies <36 kcal mol−1 were selected for presentation. Structural analysis and visualization were performed using PROCHECK (21, 22) and MOLMOL (23).
Membrane Partitioning
The ability of polyphemusin I and PV5 to associate with and partition into lipid membranes was investigated using fluorescence spectroscopy as previously described (24). Liposomes were made by dissolving lipids at the indicated molar ratios in a chloroform:methanol (2:1) solution. Liquid was removed under a stream of nitrogen, and the lipid film was further dried under vacuum for a minimum of 2 h. The lipid film was suspended in 10 mM HEPES and 150 mM NaCl, pH 7.4, with vortexing, and liposomes were formed by sonicating to clarity. A 1 μM peptide solution in 10 mM HEPES and 150 mM NaCl, pH 7.4, was added to a cuvette, and the tryptophan fluorescence was measured on a PerkinElmer model LS50B luminescence spectrometer (Boston, MA) at an excitation wavelength of 280 nm and emission range of 300–400 nm, giving the 100% unbound peptide spectra. Aliquots of the desired SUV composition at a stock concentration of 13.2 mM lipid in 10 mM HEPES and 150 mM NaCl, pH 7.4, were added, and the fluorescence spectra were recorded as above. In all cases, binding experiments were performed three times.
To determine the binding of peptide to liposomes, binding isotherms were analyzed as partition equilibrium as previously described (24–26) using the formula Xb ) KpCf, where Xb was the molar ratio of bound peptide per total lipid, Kp was the partition coefficient, and Cf was the equilibrium concentration of free peptide in solution. To determine Xb, the fluorescence signal arising from all peptides in the lipid-bound form, F∞, had to be determined. This value was estimated from the fluorescence plateau reached during titration (in the case of POPC:POPG SUVs, Figure 4C) or extrapolated from the Y-intercept of a double reciprocal plot of total peptide fluorescence (F) vs the total concentration of lipid (CL), as previously described by Schwarz et al. (25) (data not shown). Thus, knowing the fluorescence intensities of the free (F0, no lipid added) and bound (F∞) peptide forms, the fraction of membrane-bound peptide, fb, was determined by the formula fb ) (F – F0)/(F∞– F0), where F was the peptide fluorescence after each addition of vesicles. An additional assumption was made that the peptides interact solely with the lipids in the outer leaflet of the vesicles (60% of total lipid) (26), and thus Xb values were corrected according to the formula Xb* ) Xb/0.6.
Figure 4.
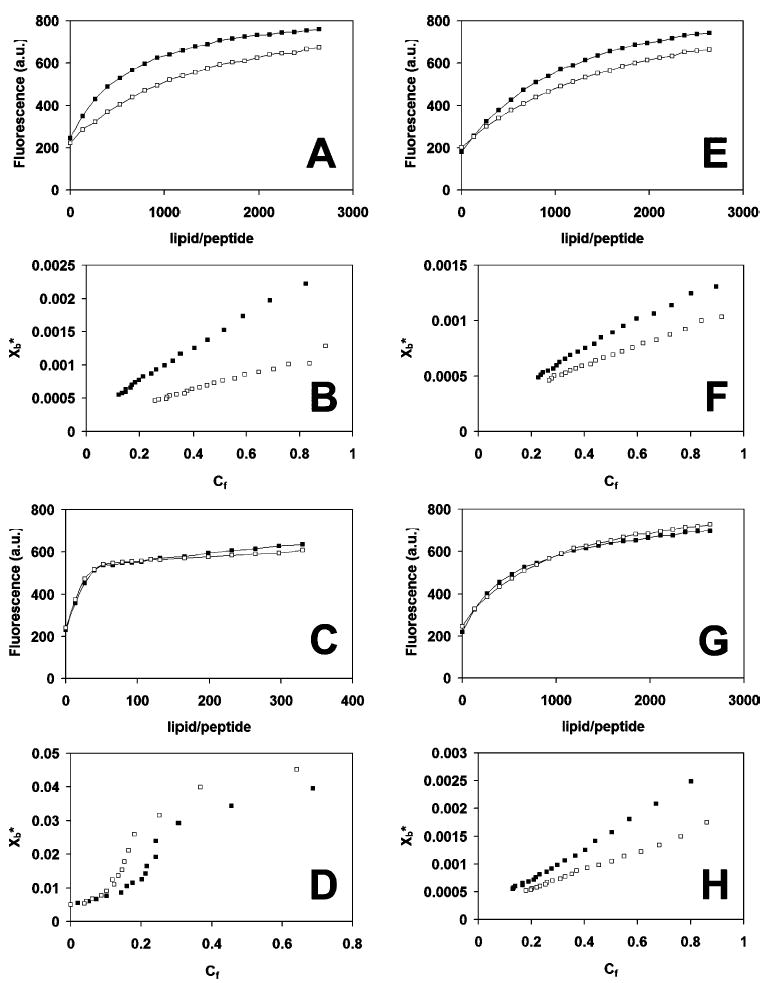
Peptide partitioning into vesicles. (A, C, E, G) Fluorescence increase of polyphemusin I (PM1) and PV5 upon titration with vesicles. Samples contained 1 μM peptide in 10 mM HEPES and 150 mM NaCl, pH 7.4. The abscissa indicates the lipid to peptide molar ratio during titration. Excitation and emission wavelengths were 280 and 335 nm, respectively. (B, D, F, H) Binding isotherms of Xb* (molar ratio of bound peptide per total lipid) versus Cf (concentration of unbound peptide) determined from the fluorescence curves and the equations indicated in the text. (A, B) POPC; (C, D) POPC:POPG (3:1); (E, F) POPC:cholesterol (3:1); (G, H) POPC:POPE (3:1); polyphemusin I, filled squares; PV5, unfilled squares. Brackets indicate mole ratios.
Differential Scanning Calorimetry
The interaction of polyphemusin I and PV5 with phospholipid membranes was investigated using differential scanning calorimetry. In all cases, synthetic lipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL), and used without further purification, and all buffers and samples were degassed under vacuum for 15 min prior to being loaded into the calorimeter. For experiments involving DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and DMPG (1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)], sodium salt) lipid species, lipids were dissolved in chloroform:methanol (2:1) at the indicated molar ratios, dried under a stream of N2, and held under vacuum overnight. The lipid film was then suspended in 10 mM Tris-HCl and 150 mM NaCl, pH 7.4, to yield multilamellar vesicles (MLVs) at a working concentration of 1 mg/mL. DSC scans were recorded on a CSC Nano II differential scanning calorimeter (Lindon, UT). Scans were performed from 5 to 40 °C with a temperature increase of 1 ° C/min and a 10 min equilibration period before each scan. Between scans, peptide was added from a concentrated stock solution to give the indicated peptide to lipid mole ratios. In all cases, the thermogram of buffer alone was subtracted prior to plotting and analysis. The raw data were converted to molar heat capacity using the CPCalc program using the corresponding lipid concentrations and molecular weights and the partial specific volume of 0.988 mL/g (27).
For experiments involving DiPoPE (1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine), lipid and peptide were codissolved in chloroform:methanol (2:1) at the indicated mole ratios, dried under a stream of N2, and held under vacuum overnight. The peptide–lipid film was suspended in 10 mM Tris-HCl, 100 mM NaCl, and 2 mM EDTA, pH 7.4, to yield multilamellar vesicles (MLVs) at a working lipid concentration of 7.5 mg/mL. The fluid lamellar (Lα) to inverted hexagonal (HII) phase transition temperature of the lipids was measured with a MicroCal VP-DSC differential scanning calorimeter (Northampton, MA). A minimum of three scans were performed from 10 to 60 °C with a temperature increase of 1 °C/min and a 15 min equilibration period before each scan. In all cases, the thermogram of buffer alone was subtracted prior to plotting and analysis using MicroCal Origin 7.0.
RESULTS
NMR Spectroscopy
DQF-COSY, TOCSY, and NOESY spectra were collected for PV5 at 25 °C, pH* 3.8, and at 40 °C, pH* 3.95, in the absence and presence of 300 mM DPC, respectively. Proton resonances were assigned sequentially, and the chemical shift assignments are recorded in the Supporting Information and deposited at the BMRB. The TOCSY spectra indicated good separation of spin systems, with minor overlap occurring between residues Arg-6 and Arg-16 (data not shown). It was possible to assign the complete or partial spin systems of all residues with the exception of the N-terminal arginine for which the amide resonance was not observed. The NOESY spectra indicated strong dαN(i,i + 1) contacts throughout the molecule and are characteristic of a β-sheet structure (28). In addition, several long-range connectivities, separated by as many as 13 residues, were further confirmation the disulfide-constrained antiparallel β-hairpin structure of PV5.
NOE Analysis and Structure Generation
The solution structure of PV5 was calculated using 129 total NOE restraints of which 67 were intraresidue and 62 were interresidue restraints (Table 1). The structure of PV5 in the presence of DPC micelles was calculated using 137 total NOE restraints of which 68 were intraresidue and 69 were interresidue restraints (Table 1). In both the solution and micelle-associated samples, NOE restraints were distributed evenly throughout the molecule, and complete lists of NOE-derived distance restraints have been deposited in the PDB. The ensemble of the 16 lowest energy calculated PV5 structures is shown in Figure 2A. The 16 structures aligned well over the β-sheet region (residues 7, 8, 14, and 15) with an average pairwise RMSD of 0.21 ± 0.05 and 0.79 ± 0.09 Å for backbone and heavy atoms, respectively. The 17 lowest energy structures of PV5 in the presence of 300 mM DPC (Figure 2C) micelles showed modest differences to the structures determined in the absence of DPC. These 17 structures also aligned well over the β-sheet region with average pairwise RMSDs of 0.18 ± 0.09 and 1.18 ± 0.18 Å for backbone and heavy atoms, respectively.
Table 1.
Structural Statistics of PV5 Determined by 1H NMR in H2O:D2O (9:1) in the Presence or Absence of 300 mM DPC Micelles
| NOE restraints | H2O:D2O | DPC micelles |
|---|---|---|
| total | 129 | 137 |
| intraresidue | 67 | 68 |
| interresidue | 62 | 69 |
| mean total energy (kcal mol−1) | 26.2 ± 0.5 | 34.3 ± 1.2 |
| mean pairwise RMSD | ||
| turn (8–14) | ||
| backbone | 0.41 ± 0.10 | 0.18 ± 0.09 |
| heavy | 1.43 ± 0.28 | 1.18 ± 0.18 |
| sheet (7, 8, 14, 15) | ||
| backbone | 0.21 ± 0.05 | 0.23 ± 0.04 |
| heavy | 0.79 ± 0.09 | 0.98 ± 0.27 |
Figure 2.
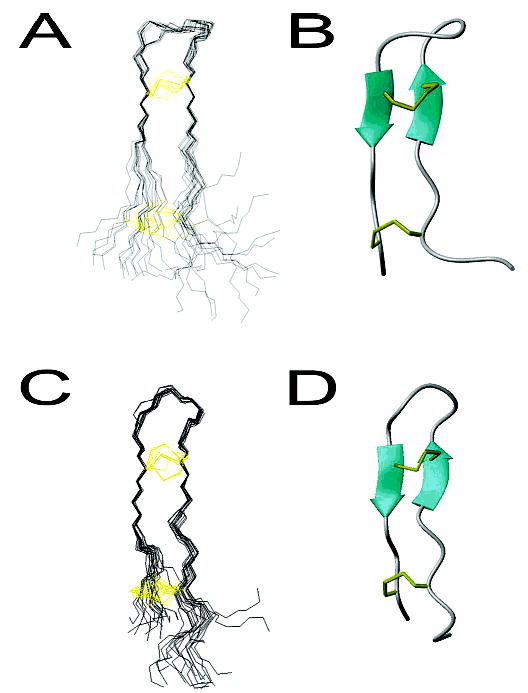
Three-dimensional solution structure of PV5 in the absence and presence of DPC micelles. (A, C) The set of structures calculated for PV5. 16 structures are presented for PV5 in aqueous medium (A), and 17 structures are presented for PV5 in DPC micelles (C). The backbone is colored black and the cysteine side chains are indicated in yellow. Structures are aligned over β-sheet residues 7, 8, 14, and 15. (B, D) Ribbon diagram of the representative PV5 structures in the absence or presence of DPC micelles, respectively. Figures were prepared with MOLMOL (23).
For clarity, the structure with the lowest average pairwise RMSD to the mean has been chosen as a representative structure for both the aqueous and DPC-associated samples (panels B and D of Figure 2, respectively). The structure of PV5 was found to be an antiparallel β-sheet connected by a turn region. In both DPC and non-DPC samples, the sheet region was relatively flat with the turn and tail regions projecting out of this plane although the degree of projection appeared less in the DPC environment. Figure 3A shows the contact surface of the representative structure of PV5 painted according to the electrostatic potential of the molecule. PV5 did not appear to be a highly amphipathic molecule in solution but rather was more amphiphilic, with its cationic loop and termini regions separated by a hydrophobic midsection. However, due to the dynamic termini, as evidenced by the poor overlay of these regions (Figure 2A), it appeared to demonstrate substantial conformational flexibility. This appeared to be reduced in the micelle-bound structure in which the variation in lowest energy structures was substantially reduced, possibly reflecting a requirement for the PV5 molecule to adopt a more amphipathic conformation upon interaction with a hydrophobic environment. Indeed, the structure of PV5 determined in the presence of DPC micelles indicated an amphipathic conformation as indicated in Figure 3B. Reorganization of the side chains of the molecule results in a hydrophobic face and an area of cationic charge due to the clustering of the arginine residues. The amphipathic conformation of PV5 in lipid environments was in fact similar to the inherent amphipathic character of the parent peptide, polyphemusin I, in solution alone (Figure 3C).
Figure 3.
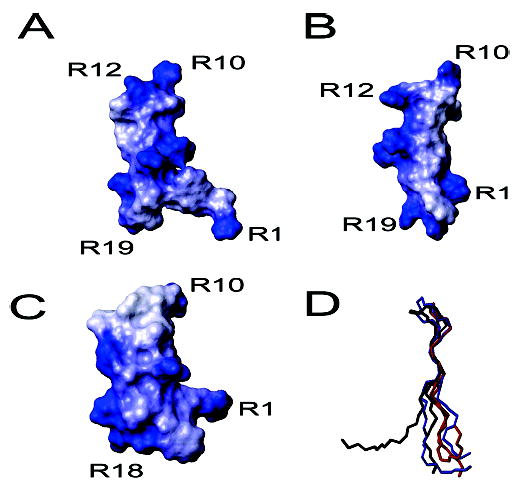
Contact surfaces and backbone overlay of the representative structures of PV5 and polyphemusin I. (A–C) Contact surfaces of the representative structures of PV5, in the absence or presence of DPC micelles, and polyphemusin I, respectively, painted with their corresponding electrostatic potentials. The additional arginine (R12) in PV5 is indicated while other residues are labeled for orientation. (D) Backbone overlay of the representative structures of PV5 (black), PV5 in DPC micelles (blue), and polyphemusin I (red). Figures were prepared with MOLMOL (23).
A backbone overlay of the representative structures of PV5, in both the absence and presence of DPC micelles, and polyphemusin I (4) revealed only minor differences between the peptides (Figure 3C). The head and tail regions of solution PV5 projected further out of the plane formed by the backbone compared to those of polyphemusin I, due to the insertion of an arginine residue at position 12 of PV5. This insertion disrupted the classical four-member β-turn region of polyphemusin and was the only sequence difference. Addition of DPC micelles reduced the nonplanar nature of the PV5 backbone and resulted in a structure similar to that of polyphemusin I.
Membrane Partitioning
The ability of polyphemusin I and PV5 to associate with and partition into lipid bilayers was determined using fluorescent spectroscopy, with the single tryptophan residue in each peptide serving as an intrinsic fluorophore. Tryptophan fluorescence is a common and useful method to determine the influence of the polarity of the local environment. In a polar environment, excited tryptophan residues interact with polar solvent molecules, suppressing their mobility and thus decreasing the energy of the excited state (29). This decrease in energy can be observed by the minimal fluorescence intensity. As the polarity of the environment decreases, the tryptophan fluorescence shifts to a lower wavelength (a blue shift) with a corresponding increase in intensity. As liposomes were titrated into a cuvette containing either polyphemusin I or PV5, the fluorescence signal, monitored at 335 nm, was observed to increase. Representative fluorescence spectra are included in Figure 4A,C,E,G.
Peptide partition coefficients (Kp) were determined from the slope of the initial, linear portion of the binding isotherms of bound peptide per total lipid (Xb*) vs the equilibrium concentration of free lipid (Cf) (see Figure 4B,D,F,H for representative binding isotherms). Both peptides had low affinity for neutral POPC vesicles with partition coefficients (means ± standard deviations of three separate experiments) of (2.7 ± 0.5) × 103 and (1.3 ± 0.2) × 103 M−1 for polyphemusin I and PV5, respectively (Table 2). Incorporation of 25 mol % of the negative lipid, POPG, which served as a simple model of an anionic prokaryotic membrane, increased the affinity of both peptides but had a greater effect on PV5 [(53 ± 2) × 103 M−1], which had a partition coefficient almost double that of polyphemusin I [(31 ± 9) × 103 M−1]. Incorporation of 25 mol % cholesterol, which served as a model of a eukaryotic membrane, showed little effect on the affinity of both peptides compared to that of POPC alone with partition coefficients of (1.4 ± 0.2) × 103 M−1 for polyphemusin I and (1.0 ± 0.1) × 103 M−1 for PV5. Incorporation of 25 mol % zwitterionic POPE resulted in similar partition coefficients for both polyphemusin I and PV5 of (2.2 ± 0.3) × 103 M−1 and (2.0 ± 0.4) × 103 M−1, respectively.
Table 2.
Partition Coefficients Indicating the Affinity of Polyphemusin I (PM1) and PV5 for Liposomes of Various Lipid Composition
| partition coefficient (×103 M−1)
|
||
|---|---|---|
| liposomes (mol/mol) | PM1 | PV5 |
| POPC | 2.7 ± 0.5 | 1.3 ± 0.2 |
| POPC:POPG (3:1) | 31 ± 9 | 53 ± 2 |
| POPC:POPE (3:1) | 2.2 ± 0.3 | 2.0 ± 0.4 |
| POPC:cholesterol (3:1) | 1.4 ± 0.2 | 1.0 ± 0.1 |
Differential Scanning Calorimetry
In this study, the effects of polyphemusin I and PV5 on the thermotrophic phase behavior of zwitterionic DMPC and anionic DMPC:DMPG multilamellar vesicles were observed by DSC. DSC is a useful tool for characterizing the interaction of compounds with lipid bilayers. Since the phase transition temperatures and transition enthalpies of phospholipid bilayers, particularly those incorporating phosphatidylcholine and phosphatidylg-lycerol (30), have been extensively studied and are well understood, the effects of exogenously added compounds can be determined by monitoring the changes in these values. Indeed, DSC has been used in a variety of peptide–lipid studies (31–34). The effect of added peptide on the pretransition and main gel to liquid-crystalline transition serves as an indicator of the ability of the peptide to associate with lipid headgroups and disrupt the lipid acyl chain packing, respectively. In addition, the effect of peptide on the lamellar (Lα) to inverted hexagonal (HII) phase transition temperature (TH) serves as an indicator of induced curvature strain and is often used to provide insights into the mechanism of action of a particular peptide (35, 36). Together, these data provide an insight to the overall mechanism of the interaction of the peptide with the lipid bilayer studied.
DSC thermograms indicating the pretransition (lamellar gel, Lβ′, to rippled gel, Pβ ′) and main gel to liquid-crystalline transition (Lα) are shown in Figure 5 for DMPC and in Figure 6 for DMPC:DMPG vesicles. The DSC thermograms of pure DMPC MLVs (Figure 5, for a peptide:lipid ratio of 0:800) indicated a small, broad pretransition peak at 15.1 °C and a tall, narrow Lα peak centered at 24.2 °C. Addition of increasing concentrations of either polyphemusin I or PV5 had only minor effects, indicating that neither peptide readily associated with or disrupted the acyl chain packing of net-neutral (zwitterionic) DMPC bilayers. For example, at low to moderate peptide concentrations (peptide:lipid ratios <1: 200) the Lα transition peak at 24.2 °C was only slightly reduced in amplitude compared to the untreated DMPC control. The pretransition peak at 15.1 °C was still observed, even at high peptide concentrations (peptide:lipid ratios >1: 100), indicating that the interaction of the peptide with the lipid headgroups was minimal. At all peptide concentrations, the enthalpy of both the pretransition and main transition peaks, as revealed by the peak heights and areas, was somewhat less in the presence of polyphemusin I than in the presence of PV5, indicating that polyphemusin I interacted more strongly with zwitterionic DMPC vesicles than did PV5.
Figure 5.
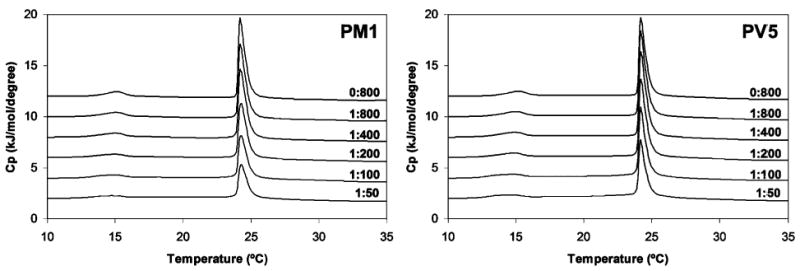
Differential scanning calorimetry of the pretransition and main gel to liquid-crystalline phase transition of DMPC vesicles at the indicated peptide to lipid molar ratios. Lipid was dissolved in chloroform, dried, and suspended at a concentration of 1 mg/mL.
Figure 6.
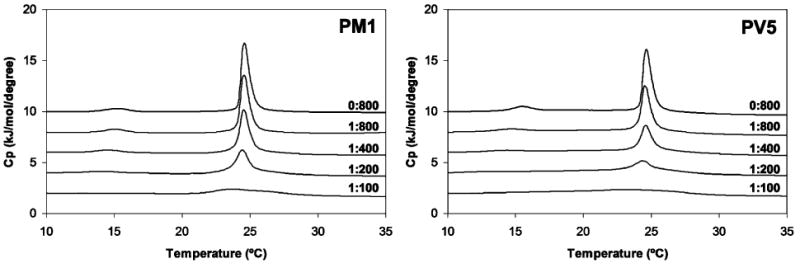
Differential scanning calorimetry of the pretransition and main gel to liquid-crystalline phase transition of DMPC:DMPG (3:1) vesicles at the indicated peptide to lipid molar ratios. Lipids were dissolved in chloroform at a molar ratio of 3:1, dried, and suspended at a concentration of 1 mg/mL.
While eukaryotic membranes contain predominantly zwitterionic lipids, bacterial membranes contain substantial (up to 30 mol %) amounts of negatively charged lipids such as phosphatidylglycerol (PG) and cardiolipin. The DSC thermograms of anionic DMPC:DMPG (3:1 molar ratio) multilamellar vesicles (Figure 6, peptide:lipid ratios of 0:800) appeared to be similar to those of pure DMPC, with a small, broad pretransition peak at 15.2 °C and a tall, narrow Lα peak centered at 24.6 °C. The addition of increasing concentrations of polyphemusin I or PV5 caused much more prominent changes than in the case of zwitterionic DMPC vesicles, indicating that both peptides readily associated with and disrupted the acyl chain packing of the negatively charged vesicles. At low peptide concentrations (peptide: lipid ratios <1:400), the main transition peak at 24.6 °C was considerably reduced in magnitude compared to untreated control DMPC:DMPG vesicles. At moderate concentrations (peptide:lipid ratios of 1:100), this peak was almost entirely abolished, indicating the near absence of a phase transition. The pretransition peak at 15.2 °C was greatly reduced at low peptide concentrations (peptide:lipid ratios <1:800) and abolished almost entirely at a PV5:lipid ratio of 1:400 and a polyphemusin I:lipid concentration of 1:200, indicating there were very significant interactions of both peptides with the lipid headgroups. At all peptide concentrations, the enthalpies of both the pretransition and Lα transitions were less in the presence of PV5 than in the presence of polyphemusin I, indicating that, opposite to the results for zwitterionic DMPC vesicles, PV5 interacted more strongly with negative DMPC: DMPG vesicles than polyphemusin I.
DSC was also used to determine the effects of polyphemusin I and PV5 on membrane curvature by monitoring the temperature (TH) of the phase transition of DiPoPE vesicles from the liquid-crystalline (Lα) to inverted hexagonal (HII) phase. The DSC thermogram of pure DiPoPE indicated a TH of 43.8 °C as indicated by the vertical line (Figure 7). At very low concentrations of PV5 (peptide:lipid ratio of 1:1000) the TH was reduced, indicating that PV5 induced negative curvature. An increase in peptide concentration (peptide:lipid ratio of 1:500) further reduced the TH and led to a slight broadening of the transition peak. Very low concentrations of polyphemusin I (peptide:lipid ratio of 1:1000) also caused a reduction in TH compared to untreated DiPoPE vesicles, indicating negative curvature strain, and similarly resulted in a broadening in peak width. This peak was also asymmetric in shape, indicating a reduction in lipid cooperativity of the transition most likely due to the presence in the membrane of peptide-rich regions. Further increasing the polyphemusin I concentration (peptide:lipid ratio of 1:500) actually increased the TH slightly above that observed at the lower polyphemusin I. While this suggests that polyphemusin I stabilizes the lamellar form, it should be noted that the TH observed for both concentrations are reduced compared to the untreated lipid control. Thus, the DSC thermograms indicated that both polyphemusin I and PV5 induced negative membrane curvature strain and promoted the formation of inverted hexagonal phases by decreasing the phase transition temperature. In addition, the different effects on phase transition peak width indicate that polyphemusin I had a much greater effect on lipid cooperativity, as the phase transition peaks were much broader with polyphemusin I compared to the transition peaks of DiPoPE with or without PV5.
Figure 7.
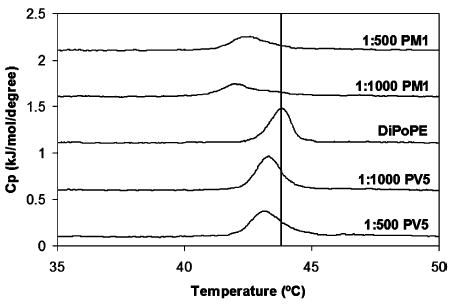
Differential scanning calorimetry of the lamellar (Lα) to inverted hexagonal (HII) phase transition of DiPoPE at the indicated peptide to lipid molar ratios. Peptide and lipid were codissolved in a chloroform–methanol solution, dried, and suspended in 10 mM HEPES and 150 mM NaCl, pH 7.4, to yield multilamellar vesicles at a working concentration of 7.5 mg/mL lipid. Peptide to lipid molar ratios are indicated. The vertical line indicates the transition temperature of pure DiPoPE (TH ) 43.8 °C) with no added peptide.
DISCUSSION
Due to the excellent antimicrobial activity of polyphemusin I (<0.2 μM for both Gram-negative and Gram-positive organisms), but disappointing ability to protect in animal models of infection and sepsis, a series of polyphemusin I analogues were previously designed to improve amphipathic character and/or increase cationic charge (7). One analogue, PV5, was found to be quite protective in mice models of bacterial challenge and endotoxemia while retaining effective in vitro activity (less than 0.4 μM for both Gram-negative and Gram-positive organisms) (7). In our ongoing effort to characterize the antimicrobial mechanism of the polyphemusins, these two peptides were used as representatives of this family. To investigate the structural components involved in their activity, the three-dimensional solution structure of PV5 was determined by 1H NMR, in the absence and presence of DPC micelles, and compared to the previously determined structure of polyphemusin I (4). In addition, the interaction of both peptides with lipid membranes was investigated using fluorescence spectroscopy and differential scanning calorimetry.
An ensemble of the lowest energy structures of PV5 in the absence and presence of DPC micelles was determined in this study (Figure 2A). The structure of PV5 was that of an antiparallel β-hairpin. Comparison of the backbone structures of PV5 with the previously determined structure of polyphemusin I (4) revealed only small differences between the two peptides (Figure 3D). The head and tail regions of PV5 projected further out of the plane formed by the backbone than those of polyphemusin I. The insertion of an arginine at position 12 of PV5 disrupted the four-member β-turn region accounting for the change in structure of the head region when compared to polyphemusin I. While there were conformational differences between the tail regions of the representative structures, the large degree of conformational flexibility in this region suggested that these differences likely do not account for differential membrane interactions of these peptides. The structure of PV5 determined in the presence of DPC micelles revealed that the side chains of the molecule undergo a reorganization which resulted in an increased amphipathic conformation (Figure 3B). Unfortunately, we were unable to study the peptide in the presence of negatively charged SDS micelles as this led to precipitation, presumably due to peptide-induced micelle aggregation.
Laederach et al. have identified a potential hinge region at the center of the related peptide tachyplesin and have postulated that this allows the peptide to adopt a more surface hydrophobic character when present in a membrane environment (3). This hinge egion, proposed as a result of only two long-range NOE restraints, was not observed in our study of PV5. It is conceivable that such a hinge could also act to bring the cationic turn and termini regions in closer proximity, to facilitate a larger cationic patch to interact with anionic lipids. If this mechanism of binding were applied, for instance, to peptide–LPS interactions, where the cationic head and termini of the peptide bind to the anionic phosphate groups present in the LPS, the additional cationic charge imparted by arginine-12 may aid in this function (Figure 3A–C). This oriented binding and additional cationic charge could also explain why the interaction of PV5 with LPS was less inhibited by the presence of added Mg2+ ions than was polyphemusin I (7).
Polyphemusin I and PV5 exhibited only minor interactions with neutral membranes composed of PC, PC:cholesterol, or PC:PE. The partition coefficient of polyphemusin I was 2-fold greater than that of PV5 for PC vesicles; however, both peptides partitioned relatively poorly into neutral membranes compared to the negative PC:PG vesicles (Table 2). The DSC thermograms of PC correlated with the partitioning data and indicated that, even at quite high peptide concentration, pretransition and main transition peaks were still present (Figure 5). Thus, both polyphemusin I and PV5 interacted weakly with the lipid headgroups and acyl chains of neutral membranes, although polyphemusin I displayed a slightly increased membrane interaction as evidenced by the greater reduction in enthalpy of the main gel to liquid-crystalline transition compared to PV5.
The addition of biologically significant lipids was also studied to determine their effects on membrane partitioning. Addition of 25 mol % of the eukaryotic lipid cholesterol to produce PC:cholesterol vesicles showed little effect on the affinity of both peptides compared to that of POPC alone (Table 2). Phosphatidylethanolamine was also used in partition studies due to its relatively high content in prokaryotic cells (>70%) as well as its presence in eukaryotic membranes, albeit to a much lesser extent. In addition, this type II, nonlamellar phase forming lipid was of interest to study when considering membrane translocation, as E. coli mutants deficient in PE synthesis lack the ability to transport proteins across their plasma membrane (37). Incorporation of 25 mol % PE to produce PC:PE vesicles resulted in similar partition coefficients for both peptides which differed little compared to their partitioning into POPC alone. This indicated that the presence of PE alone does not promote the partitioning of either peptide.
Both peptides showed considerably increased affinities for negatively charged membranes composed of PC:PG (Table 2), on the order of 10–40-fold greater for the negatively charged PC:PG vesicles compared to the neutral PC vesicles. Interestingly, however, the partition coefficient of PV5 for PC:PG vesicles was almost double that of polyphemusin I. The DSC thermograms for negatively charged membranes were consistent with these partitioning data and indicated that, even at a low peptide concentration, the pretransition and main transition peaks were greatly reduced in magnitude for both peptides, suggesting a large interaction with the negatively charged lipid headgroups and disruption of acyl chain packing (Figure 6). The affinity of PV5 appeared to be significantly greater as evidenced by the larger reduction in enthalpy of the main thermal transition peak compared to that induced by polyphemusin I. Since the initial step in peptide–membrane association is thought to be due to electrostatic interaction, the additional arginine in PV5 likely accounts for the observed increase in partitioning into PG-containing membranes when compared to polyphemusin I. While the effect of charge modification has not been well studied for the polyphemusins, studies have been conducted on other cationic peptides. Increasing the charge of the α-helical peptide magainin II has been shown to increase the permeabilizing ability of negatively charged PG-containing vesicles while no relationship was observed for neutral PC membranes (38). This also highlights the limitations in using model membranes to study biological phenomena as the lack of partitioning into neutral PC membranes cannot explain the cytotoxic effects observed with some peptides. Certainly, the various proteins and receptors found in eukaryotic membranes interact with peptides to some extent. Tachyplesin, which, due to its similarity to the polyphemusins, would not be expected to partition into zwitterionic membranes, has been demonstrated to act as a secretagogue upon hemocytes (8). This finding suggests that the polyphemusins are capable of interacting with eukaryotic cells in some manner that partitioning alone does not explain.
Both polyphemusin I and PV5 promoted negative membrane curvature strain as indicated by a reduction in the hexagonal phase transition temperature of DiPoPE (Figure 7). Transient, nonbilayer formation may play a role in the membrane translocation of proteins and peptides, and non-bilayer-forming lipids are required for protein folding (39) and protein transport across membranes (37). The promotion of nonbilayer transitions by these peptides may thus explain part of their antimicrobial mechanism.
The effects of polyphemusin, on the lamellar to inverted hexagonal phase transition temperature of pure DiPoPE, contrast with those observed for other cationic peptides. Addition of the cationic peptides LL-37 (40), magainin 2 (41), and an analogue, MSI-78 (42), increases the TH, indicating the induction of positive curvature strain as more energy is required to drive the formation of the inverted hexagonal phase (cf. polyphemusins that lowered the TH and induced negative curvature strain, thus promoting the formation of the inverted hexagonal phase). The induction of positive membrane curvature strain is consistent with a mechanism involving the torroidalpore model in which peptide-induced positive membrane curvature would lead to the formation of a torus-like pore, which, upon collapse, may disperse peptide on either side of the lipid bilayer. Due to the induction of negative membrane curvature observed here for polyphemusin I and PV5, it is clear that the polyphemusin family of peptides does not function through this mechanism. This is similar of pardaxin which induces negative curvature strain even at very low concentrations (peptide:lipid mole ratio of 1:50000) which also does not function through a torroidal mechanism (35).
Additional insight into the mechanism of action of the polyphemusins may also be inferred from previous studies focused on lipid flip-flop and dye release. It was shown that at low peptide:lipid ratios (<0.005) significant lipid exchange between membrane leaflets is observed (~60%) but very little calcein release occurs (~5%) (5). To maintain membrane integrity and prevent entrapped contents from leaking out of the vesicles, the peptide must interact with the leaflets sequentially so one will always be intact and impermeable to the dye, or alternatively the disturbance produced must be too small to allow appreciable dye release. Since the degree of flip-flop is so large, the latter explanation seems unlikely. This is because the degree of disturbance necessary for lipid molecules (POPC MW 760) to switch leaflets should be sufficiently large as to cause appreciable calcein (MW 623) dye release. From these data and the results reported here we can propose a model for the translocation of the polyphemusins (Figure 8). It is hypothesized that the peptides translocate the membrane through the formation of a transient, nonbilayer peptide–lipid intermediates. Peptides initially encounter the net negatively charged membrane bilayer (step 1) and interact with negatively charged lipid headgroups in the outer leaflet (step 2). This leads to partial membrane insertion and peptide aggregation within the bilayer, causing negative curvature strain (step 3). Peptide aggregation and induced curvature strain drive the formation of a nonbilayer intermediate (step 4). Upon formation of this state, the outer leaflet regains integrity and reforms a permeability barrier. Collapse of this intermediate structure leads to the redistribution of the peptide in both leaflets, during which a small amount of dye may be released (step 5). While the data presented in this paper support only steps 1 and 2 of this mechanism, it serves to further define the aggregate model of membrane translocation previously proposed by our group (43). It should also be noted that the aggregate model is similar in nature to the sinking-raft model proposed by Pokorny et al. (44, 45).
Figure 8.
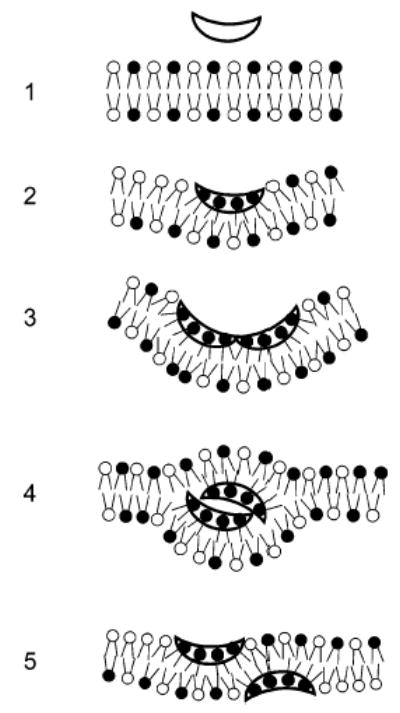
Mechanism of membrane translocation of the polyphemusins. Steps: (1) Peptides initially encounter the negatively charged lipid bilayer. (2) Electrostatic interaction of the peptide (hollow) with the negatively charged lipid headgroups (black) and partial membrane insertion. (3) Aggregation of the bound peptide within the outer leaflet of the bilayer and induction of negative membrane curvature. (4) Transient nonbilayer formation occurs due to the combination of negative curvature and peptide aggregation. (5) Collapse of the nonbilayer intermediate and corresponding peptide translocation to the membrane inner leaflet. It should be noted that the figure is for clarification purposes and is not intended to infer scale or stoichiometry.
The findings presented here, combined with previous observations that polyphemusin I promotes lipid flip-flop between membrane leaflets but does not induce significant vesicle leakage (5), rule out the torroidal pore (46) and carpet mechanisms of action (47).
Supplementary Material
Acknowledgments
We thank Dr. Mark Okon for acquisition of the NMR spectra, Mrs. Suzanne Perry-Riehm for acquisition of the mass spectra, and Dr. Louise Creagh and the UBC Centre for Biological Calorimetry for use of the VP-DSC. We also thank Dr. Sathiah Thennarasu (University of Michigan), Joseph McPhee, David Jung (University of British Columbia), and Howard Powers for helpful discussions.
Footnotes
We acknowledge funding from the Canadian Institutes of Health Research and the Applied Food and Materials Network to R.E.W.H. and NIH funding support to A.R. (AI054515). R.E.W.H. is the recipient of a Canada Research Chair, J.-P.S.P. is the recipient of a UBC postgraduate fellowship, and A.T. was supported by NIH funds (AI054515 to A.R.).
The structures of PV5 have been deposited in the PDB (http://www.rcsb.org/pdb/) under accession codes 1X7K and 2B5K. The proton chemical shifts of PV5 have been deposited in the BMRB (http://www.bmrb.wisc.edu/) under accession codes BMRB-6608 and BMRB-6839.
Abbreviations: D2O, deuterium oxide; DGSA, distance geometry simulated annealing; DiPoPE, 1,2-dipalmitoleoyl-sn-glycero-3-phosphoethanolamine; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DMPG, 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt); DMSO, dimethyl sulfoxide; DPC, dodecylphosphocholine (D38, 98%); DQF-COSY, double quantum filtered correlated spectroscopy; DSC, differential scanning calorimetry; Tris, tris(hydroxyethyl)aminomethane; EDTA, ethylenediaminetetraacetic acid; FPLC, fast purification liquid chromatography; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; LPS, lipopolysaccharide; MLV, multilamellar vesicle; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser enhancement; NOESY, nuclear Overhauser effect spectroscopy; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; pH*, pH in D2O; PM1, polyphemusin I; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPE, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; POPG, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt); PV5, polyphemusin I analogue; SUV, small unilamellar vesicle; TOCSY, total correlated spectroscopy.
SUPPORTING INFORMATION AVAILABLE
Tables of chemical shift assignments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Miyata T, Tokunaga F, Yoneya T, Yoshikawa K, Iwanaga S, Niwa M, Takao T, Shimonishi Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: chemical structures and biological activity. J Biochem (Tokyo) 1989;106:663–668. doi: 10.1093/oxfordjournals.jbchem.a122913. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T, Furunaka H, Miyata T, Tokunaga F, Muta T, Iwanaga S, Niwa M, Takao T, Shimonishi Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J Biol Chem. 1988;263:16709–16713. [PubMed] [Google Scholar]
- 3.Laederach A, Andreotti AH, Fulton DB. Solution and micelle-bound structures of tachyplesin I and its active aromatic linear derivatives. Biochemistry. 2002;41:12359–12368. doi: 10.1021/bi026185z. [DOI] [PubMed] [Google Scholar]
- 4.Powers JPS, Rozek A, Hancock REW. Structure–activity relationships for the beta-hairpin cationic antimicrobial peptide polyphemusin I. Biochim Biophys Acta. 2004;1698:239–250. doi: 10.1016/j.bbapap.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Rozek A, Hancock REW. Interaction of cationic antimicrobial peptides with model membranes. J Biol Chem. 2001;276:35714–35722. doi: 10.1074/jbc.M104925200. [DOI] [PubMed] [Google Scholar]
- 6.Matsuzaki K, Yoneyama S, Fujii N, Miyajima K, Yamada K, Kirino Y, Anzai K. Membrane permeabilization mechanisms of a cyclic antimicrobial peptide, tachyplesin I, and its linear analog. Biochemistry. 1997;36:9799–9806. doi: 10.1021/bi970588v. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Scott MG, Yan H, Mayer LD, Hancock REW. Interaction of polyphemusin I and structural analogs with bacterial membranes, lipopolysaccharide, and lipid monolayers. Biochemistry. 2000;39:14504–14514. doi: 10.1021/bi0011173. [DOI] [PubMed] [Google Scholar]
- 8.Ozaki A, Ariki S, Kawabata S. An antimicrobial peptide tachyplesin acts as a secondary secretagogue and amplifies lipopolysaccharide-induced hemocyte exocytosis. FEBS J. 2005;272:3863–3871. doi: 10.1111/j.1742-4658.2005.04800.x. [DOI] [PubMed] [Google Scholar]
- 9.Tam J, Wu C, Liu W, Zhang J. Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J Am Chem Soc. 1991;113:6657–6662. [Google Scholar]
- 10.Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wuthrich K. Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Commun. 1983;117:479–485. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- 11.Braunschweiler L, Ernst RR. Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J Magn Res. 1983;53:521–528. [Google Scholar]
- 12.Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 13.Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- 14.Sklenar V, Piotto M, Leppik R, Saudek V. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J Magn Reson, Ser A. 1993;102:241–245. [Google Scholar]
- 15.Bax A, Davis DG. MLEV-17 based two-dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 16.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 17.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 18.Hyberts SG, Goldberg MS, Havel TF, Wagner G. The solution structure of eglin c based on measurements of many NOEs and coupling constants and its comparison with X-ray structures. Protein Sci. 1992;1:736–751. doi: 10.1002/pro.5560010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedrich CL, Rozek A, Patrzykat A, Hancock REW. Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J Biol Chem. 2001;276:24015–24022. doi: 10.1074/jbc.M009691200. [DOI] [PubMed] [Google Scholar]
- 20.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The X-PLOR-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 21.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 23.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- 24.Rapaport D, Shai Y. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J Biol Chem. 1991;266:23769–23775. [PubMed] [Google Scholar]
- 25.Schwarz G, Stankowski S, Rizzo V. Thermodynamic analysis of incorporation and aggregation in a membrane: application to the pore-forming peptide alamethicin. Biochim Biophys Acta. 1986;861:141–151. doi: 10.1016/0005-2736(86)90573-0. [DOI] [PubMed] [Google Scholar]
- 26.Beschiaschvili G, Seelig J. Melittin binding to mixed phosphatidylglycerol/phosphatidylcholine membranes. Biochemistry. 1990;29:52–58. doi: 10.1021/bi00453a007. [DOI] [PubMed] [Google Scholar]
- 27.Marsh, D. (1990) CRC Handbook of Lipid Bilayers, CRC Press, Boca Raton, FL.
- 28.Wuthrich, K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, Toronto.
- 29.Campbell, I. D., and Dwek, R. A. (1984) Biological spectroscopy, Benjamin/Cummings, Menlo Park, CA.
- 30.Findlay EJ, Barton PG. Phase behavior of synthetic phosphatidylglycerols and binary mixtures with phosphatidylcho-lines in the presence and absence of calcium ions. Biochemistry. 1978;17:2400–2405. doi: 10.1021/bi00605a023. [DOI] [PubMed] [Google Scholar]
- 31.Prenner EJ, Lewis RN, Kondejewski LH, Hodges RS, McElhaney RN. Differential scanning calorimetric study of the effect of the antimicrobial peptide gramicidin S on the thermotropic phase behavior of phosphatidylcholine, phosphatidylethanolamine and phosphatidylglycerol lipid bilayer membranes. Biochim Biophys Acta. 1999;1417:211–223. doi: 10.1016/s0005-2736(99)00004-8. [DOI] [PubMed] [Google Scholar]
- 32.Tournois H, de Kruijff B. Polymorphic phospholipid phase transitions as tools to understand peptide-lipid interactions. Chem Phys Lipids. 1991;57:327–340. doi: 10.1016/0009-3084(91)90084-o. [DOI] [PubMed] [Google Scholar]
- 33.van Kan EJ, Ganchev DN, Snel MM, Chupin V, van der Bent A, de Kruijff B. The peptide antibiotic clavanin A interacts strongly and specifically with lipid bilayers. Biochemistry. 2003;42:11366–11372. doi: 10.1021/bi0349017. [DOI] [PubMed] [Google Scholar]
- 34.Lohner K, Staudegger E, Prenner EJ, Lewis RN, Kriechbaum M, Degovics G, McElhaney RN. Effect of staphylococcal deltalysin on the thermotropic phase behavior and vesicle morphology of dimyristoylphosphatidylcholine lipid bilayer model membranes. Differential scanning calorimetric, 31P nuclear magnetic resonance and Fourier transform infrared spectroscopic, and X-ray diffraction studies. Biochemistry. 1999;38:16514–16528. doi: 10.1021/bi9913101. [DOI] [PubMed] [Google Scholar]
- 35.Hallock KJ, Lee DK, Omnaas J, Mosberg HI, Ramamoorthy A. Membrane composition determines pardaxin’s mechanism of lipid bilayer disruption. Biophys J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies SM, Epand RF, Bradshaw JP, Epand RM. Modulation of lipid polymorphism by the feline leukemia virus fusion peptide: implications for the fusion mechanism. Biochemistry. 1998;37:5720–5729. doi: 10.1021/bi980227v. [DOI] [PubMed] [Google Scholar]
- 37.Rietveld AG, Koorengevel MC, de Kruijff B. Non-bilayer lipids are required for efficient protein transport across the plasma membrane of Escherichia coli. EMBO J. 1995;14:5506–5513. doi: 10.1002/j.1460-2075.1995.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dathe M, Nikolenko H, Meyer J, Beyermann M, Bienert M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001;501:146–150. doi: 10.1016/s0014-5793(01)02648-5. [DOI] [PubMed] [Google Scholar]
- 39.Bogdanov M, Sun J, Kaback HR, Dowhan W. A phospholipid acts as a chaperone in assembly of a membrane transport protein. J Biol Chem. 1996;271:11615–11618. doi: 10.1074/jbc.271.20.11615. [DOI] [PubMed] [Google Scholar]
- 40.Henzler Wildman KA, Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry. 2003;42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 41.Matsuzaki K, Sugishita K, Ishibe N, Ueha M, Nakata S, Miyajima K, Epand RM. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 1998;37:11856–11863. doi: 10.1021/bi980539y. [DOI] [PubMed] [Google Scholar]
- 42.Hallock KJ, Lee DK, Ramamoorthy A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys J. 2003;84:3052–3060. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hancock REW, Chapple DS. Peptide antibiotics. Antimicrob Agents Chemother. 1999;43:1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pokorny A, Birkbeck TH, Almeida PF. Mechanism and kinetics of delta-lysin interaction with phospholipid vesicles. Biochemistry. 2002;41:11044–11056. doi: 10.1021/bi020244r. [DOI] [PubMed] [Google Scholar]
- 45.Pokorny A, Almeida PF. Kinetics of dye efflux and lipid flip-flop induced by delta-lysin in phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, alpha-helical peptides. Biochemistry. 2004;43:8846–8857. doi: 10.1021/bi0497087. [DOI] [PubMed] [Google Scholar]
- 46.Matsuzaki K, Mitani Y, Akada KY, Murase O, Yoneyama S, Zasloff M, Miyajima K. Mechanism of synergism between antimicrobial peptides magainin 2 and PGLa. Biochemistry. 1998;37:15144–15153. doi: 10.1021/bi9811617. [DOI] [PubMed] [Google Scholar]
- 47.Oren Z, Shai Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers. 1998;47:451–463. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.