

Abstract
The X-ray crystal structures of R304K trichodiene synthase and its complexes with inorganic pyrophosphate (PPi) and aza analogues of the bisabolyl carbocation intermediate are reported. The R304K substitution does not cause large changes in the overall structure in comparison with the wild-type enzyme. The complexes with R- and S-azabisabolenes and PPi bind 3 Mg2+ ions and each undergoes a diphosphate-triggered conformational change that caps the active site cavity. This conformational change is only slightly attenuated compared to that of the wild-type enzyme complexed with Mg2+3-PPi, in which R304 donates hydrogen bonds to PPi and D101. In R304K trichodiene synthase, K304 does not engage in any hydrogen bond interactions in the unliganded state and it donates a hydrogen bond only to PPi in the complex with R-azabisabolene; K304 makes no hydrogen bond contacts in its complex with PPi and S-azabisabolene. Thus, although the R304-D101 hydrogen bond interaction stabilizes diphosphate-triggered active site closure, it is not required for Mg2+3-PPi binding. Nevertheless, since R304K trichodiene synthase generates aberrant cyclic terpenoids with a 5000-fold reduction in kcat/KM, it is clear that a properly formed R304-D101 hydrogen bond is required in the enzyme-substrate complex to stabilize the proper active site contour, which in turn facilitates cyclization of farnesyl diphosphate for the exclusive formation of trichodiene. Structural analysis of the R304K mutant and comparison with the monoterpene cyclase (+)-bornyl diphosphate synthase suggests that the significant loss in activity results from compromised activation of the PPi leaving group.
Abbreviations: FPP, farnesyl diphosphate; GPP, geranyl diphosphate; PPi, inorganic pyrophosphate
Cyclic terpenoids, natural products derived from acyclic isoprenoid precursors such as geranyl diphosphate (C10 monoterpenes), farnesyl diphosphate (C15 sesquiterpenes), and geranylgeranyl diphosphate (C20 diterpenes), are found in myriad life forms where they serve a wide variety of physiological and ecological functions (1, 2, 3). Bacteria and fungi produce numerous cyclic sesquiterpene antimicrobial and antifungal agents that target competing organisms and thereby confer a selective advantage to the host organism. For example, various Streptomyces species secrete terpene antibiotics (4, 5) and certain species of Fusarium secrete the trichothecane class of antibiotics (6). In comparison, plants produce C10, C15, and C20 cyclic terpenoids for defense against fungi and insects. For example, Abies grandis (Grand fir) secretes multiple cyclic terpenoids in response to stem wounds and insect attack (7). Interestingly, cyclic terpenoids contribute to the desirable taste and aroma of plants such as Salvia officinalis (sage) (8), leading to their use as culinary flavorings. A more significant health-related benefit of plant terpenoids is in their potential pharmaceutical utility, e.g., the cancer chemotherapy drug paclitaxel (Taxol)™ (9), antimalarials like artemisinin from Artemisia annua (10), anti-microbials like linalool and citranellal from Melissa officinalis (balm) (11), and antifungal agents such as α- and β-pinenes from tea tree oil (12).
In bacteria, plants, and fungi, cyclic sesquiterpenes (C15) derive from the acyclic, linear precursor farnesyl diphosphate (FPP) through multi-step reactions catalyzed by sesquiterpene synthases (also known as sesquiterpene cyclases) (2, 13, 14). The highly reactive carbocation intermediates of terpenoid cyclization reactions are protected from bulk water during catalysis by a diphosphate-triggered conformational change that caps the cyclase active site. The molecular recognition of the substrate diphosphate group is achieved by metal coordination interactions with divalent metal ions (Mg2+, or sometimes Mn2+) (15, 16) and by multiple hydrogen bonds with enzyme residues. For example, in Fusarium sporotrichiodes trichodiene synthase complexed with inorganic pyrophosphate (PPi), the PPi anion coordinates to 3 Mg2+ ions (liganded by D100, N225, S229, E233, and PPi) and also accepts hydrogen bonds from R182, K232, R304, and Y305 (17). Of these residues, R304 appears to play a key role in the diphosphate-triggered active site closure mechanism: upon PPi binding, R304 breaks a hydrogen bond with T69 in order to form hydrogen bonds with PPi and D101 (which breaks a hydrogen bond interaction with R62) (Figure 1a) (17). These conformational changes, plus those involving the N-terminus (M1-P5), helices 1, D, H, K, and L, and loops 1-A, D-D1, H-α-1, J-K, and K-L (Figure 1b) are likely triggered by the diphosphate group of the FPP substrate as well. The trichodiene synthase mechanism is summarized in Figure 2a (18).
Figure 1.
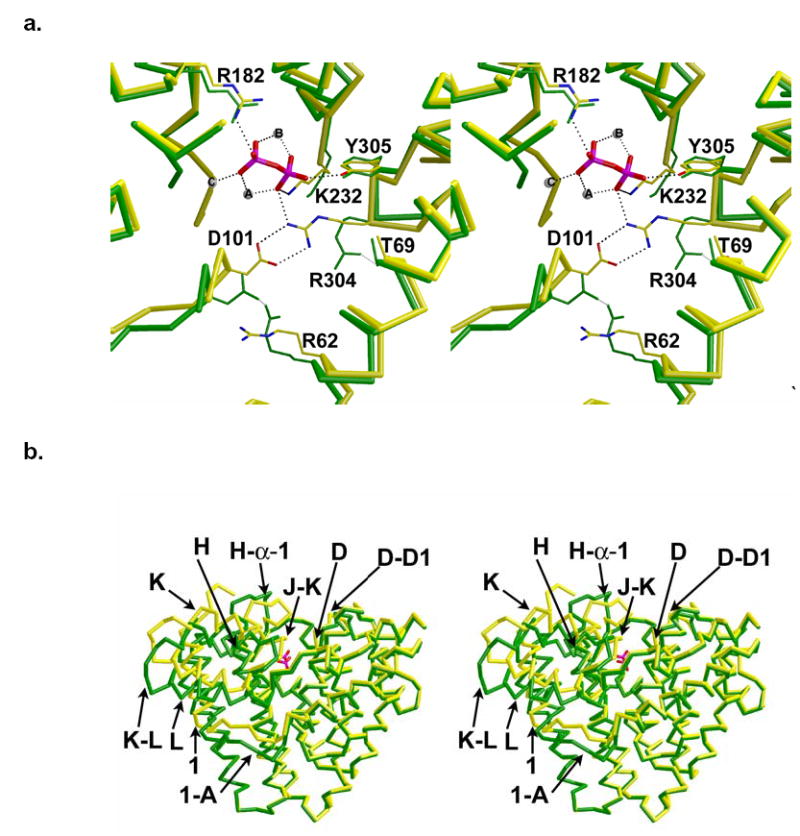
(a) Superposition of the active sites of trichodiene synthase, unliganded (green) and complexed with diphosphate (yellow) reveals that D101 and R304 break hydrogen bonds with R62 and T69, respectively, to form a new salt link with each other, thus capping the active site. Diphosphate is shown in red and magenta. Solvent atoms are omitted for clarity and Mg2+ ions appear as spheres. (b) Superposition of Cα traces of trichodiene synthase: unliganded (green) and Mg2+3-PPi complex (yellow). The PPi anion (red) illustrates the location of the active site; helices and loops that undergo significant diphosphate-induced conformational changes are indicated.
Figure 2.
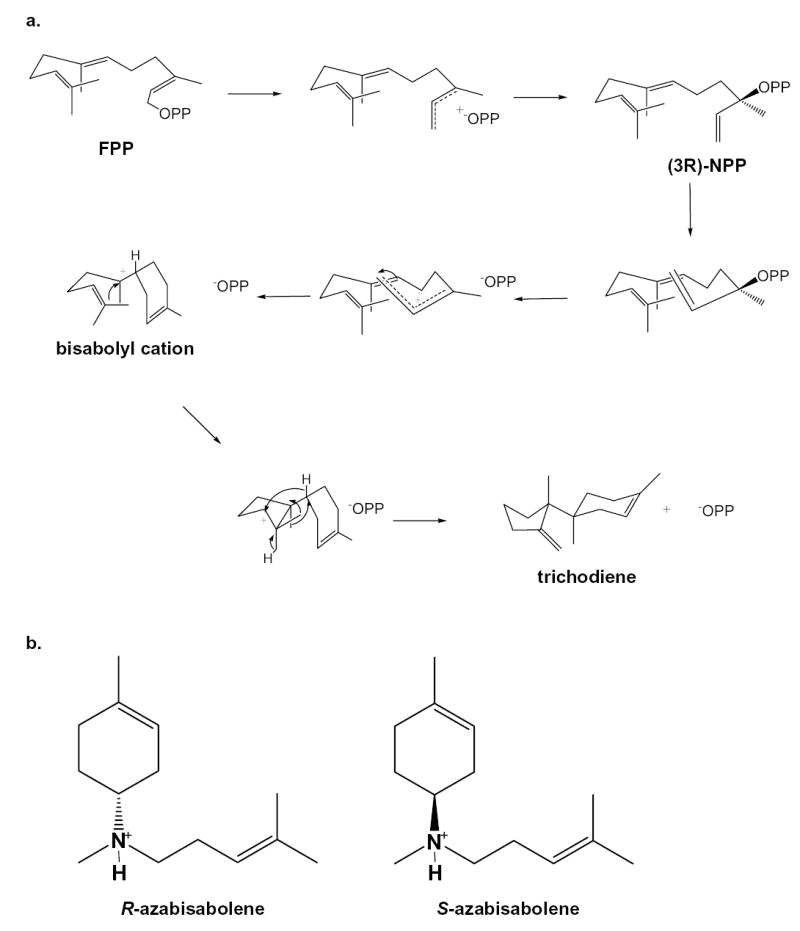
(a) Postulated mechanism for the cyclization of farnesyl diphosphate (FPP) to trichodiene by trichodiene synthase; OPP = diphosphate, NPP = nerolidyl diphosphate (18). (b) R-azabisabolene, a cationic analogue of the bisabolyl cation in the trichodiene synthase mechanism, is a strong competitive inhibitor in the presence of inorganic diphosphate (PPi) with Ki = 0.51 μM (22); similarly, the enantiomer S-azabisabolene binds in the presence of PPi with Ki = 0.47 μM (22), suggesting that the stereochemical discrimination is weak at the corresponding step in catalysis.
Mutagenesis studies indicate that conservative mutations of R304 (loop J-K), D101 (helix D), or D100 (helix D) compromise activity (19, 20). For example, consider D100, which coordinates to Mg2+A and Mg2+C (17). The D100E mutation results in a 22-fold decrease in kcat/KM and the formation of five aberrant products in addition to trichodiene (20). The crystal structure of D100E trichodiene synthase shows that the diphosphate-triggered conformational change is highly attenuated, partly due to compromised metal binding (21). D101E trichodiene synthase shows a 5-fold decrease in kcat/KM and the generation of five aberrant products in addition to trichodiene (20). In comparison, the R304K mutation results in a 5,000-fold decrease in kcat/KM as well as the generation of at least two aberrant products in addition to trichodiene (19).
To probe the importance of R304 as a hydrogen bond donor to diphosphate and its structural role in the diphosphate-triggered conformational change that caps the active site cavity, we now report the X-ray crystal structures of R304K trichodiene synthase and its complexes with both (4R)-7-azabisabolene (designated “R-azabisabolene”), a cationic analogue of the natural bisabolyl carbocation intermediate, and its enantiomer (4S)-7-azabisabolene (designated “S-azabisabolene”) (Figure 2b) (22).
Materials and Methods
Site-Directed Mutagenesis of R304K trichodiene synthase
The R304K mutation was introduced by PCR mutagenesis. The trichodiene synthase expression vector pZW03 was used as the template. The forward primer incorporated two point mutations that altered a codon from Arg (AGG) to Lys (AAA; boldface): 5′-G CAC TTG TGC GAT CGC AAA TAC CGC CTT AGC GAG-3′. The optimal reaction mixture for PCR amplification of the mutagenic insert was as follows: 125 ng each of forward and reverse primers, 1 μL of 10 mM dNTP mix, 2 or 4 μL of 16 ng/μL wild-type plasmid, 10 μL of Stratagene reaction buffer, and 2.5 units of Pfu turbo polymerase, in 50 μL. The lid of the thermal cycler was preheated to 105°C and after initial denaturation of the reaction mixture (without the polymerase) at 95°C for 1 min, 2.5 units of the polymerase were added to the mixture. Sixteen cycles, each consisting of 30 sec denaturation at 95°C, annealing at 55°C for 1 min, followed by a 10 min extension at 68°C, were performed. The reaction program concluded with a 10 min final extension at 68°C and the reaction mixture was held at 4°C until the PCR tubes were removed from the cycler. To each reaction mixture, 1 μL of Dpn1 enzyme was added before incubation for 1 h at 37°C. The reactions were immediately placed on ice and transformed into XL1-Blue supercompetent cells using the protocol recommended by Stratagene and plated on LB-Agar plates containing 50 μg/mL ampicillin. Mini-preps (Qiagen mini-prep kit) from overnight cultures of single colonies of transformants yielded plasmids that were subsequently sequenced (DNA Sequencing Facility, University of Pennsylvania), using a primer ~100 nt upstream of the mutation site, to confirm that the mutation was incorporated.
Expression, purification, and crystallization of R304K trichodiene synthase
The plasmid containing the gene encoding for R304K trichodiene synthase was transformed into Escherichia coli BL21 (DE3), overexpressed, and purified as described for wild-type trichodiene synthase (17, 23), and crystallized by the hanging drop vapor diffusion method described for wild-type and mutant trichodiene synthases (17, 21, 24). The complexes between R304K trichodiene synthase and the azabisabolenes were prepared using the same crystal soaking protocol used for the preparation of the respective complexes with Y305F trichodiene synthase (24). R304K trichodiene synthase crystallized essentially isomorphously with the wild-type enzyme (space group P3121, a = b = 122.2 Å, c = 151.2 Å) (17). Crystals were prepared for data collection by cryoprotection in 25% ethylene glycol and flash cooling in liquid nitrogen.
Diffraction data were collected at the Brookhaven National Laboratory (beamline X12B). Data were indexed and merged using the program HKL2000 (25). The structures were solved by the difference Fourier technique. The programs CNS (26) and O (27) were used in refinement and rebuilding, respectively. Noncrystallographic symmetry constraints were used in the initial stages of refinement and subsequently relaxed into appropriately weighted restraints as judged by Rfree as refinement progressed. Molecular models shown in figures were prepared with Bobscript version 2.4 (28, 29). Data collection and refinement statistics are reported in Table 1.
Table 1.
Data Collection and Refinement Statistics
| Mutant/Complex | R304K | R304K-Mg2+3-PPi-R-azabisabolene | R304K-Mg2+3-PPi-S-azabisabolene |
|---|---|---|---|
| Resolution Range (Å) | 50–2.9 | 80–2.5 | 60–2.75 |
| Reflections (measured/unique) | 194638/29285 | 224114/42643 | 190271/30875 |
| Completeness (%) (overall/outer shell) | 99.2/100 | 91.9/75.8 | 88.6/91.1 |
| Rmerge a (overall/outer shell) | 0.078/0.454 | 0.084/0.343 | 0.138/0.554 |
| <I/σ> (overall/outer shell) | 30.7/5.0 | 15.9/4.1 | 13/3.2 |
| Protein atoms (no.) b | 5792 | 5747 | 5692 |
| Solvent atoms (no.) b | 58 | 195 | 97 |
| Metal ions (no.) b | 2 | 3 | 3 |
| Ligand atoms (no.) b | – | 24 | 9 |
| Reflections used in refinement (work/free) | 27821/1435 | 40318/2130 | 29151/1523 |
| R/Rfreec | 0.213/0.254 | 0.216/0.251 | 0.216/0.256 |
| r.m.s. deviations | |||
| bonds (Å) | 0.007 | 0.007 | 0.007 |
| angles (deg.) | 1.2 | 1.1 | 1.2 |
| proper dihedral | |||
| angles (deg.) | 18.1 | 18.6 | 18.9 |
| improper dihedral | |||
| angles (deg.) | 0.8 | 0.8 | 0.8 |
Rmerge = ∑|Ij−〈Ij〉|/∑Ij, where Ij is the observed intensity for reflection j and 〈Ij〉 is the average intensity calculated for reflection j from replicate data.
per asymmetric unit.
R = ∑||Fo|−|Fc||/∑|Fo|, where R and Rfree are calculated by using the working and test reflection sets, respectively.
Results
Unliganded R304K trichodiene synthase
The structure of unliganded R304K trichodiene synthase (Figure 3) is mostly similar to that of the unliganded wild-type enzyme (17). However, loops D-D1, J-K, and H-α-1, and helix K appear to shift slightly from their positions in the wild-type enzyme. The r.m.s. deviation between the two structures is 0.54 Å for 349 Cα atoms. In the unliganded forms of these enzymes, R304 donates a hydrogen bond to T69 and D101 accepts a hydrogen bond from R62 (17, 21, 24). In contrast, K304 of R304K trichodiene synthase does not form any hydrogen bonds and the nearest polar residues are >4 Å away. Possibly, the amino group of K304 hydrogen bonds with water molecules that are disordered at the modest resolution of this structure determination. The D101-R62 hydrogen bond remains intact in this mutant.
Figure 3.
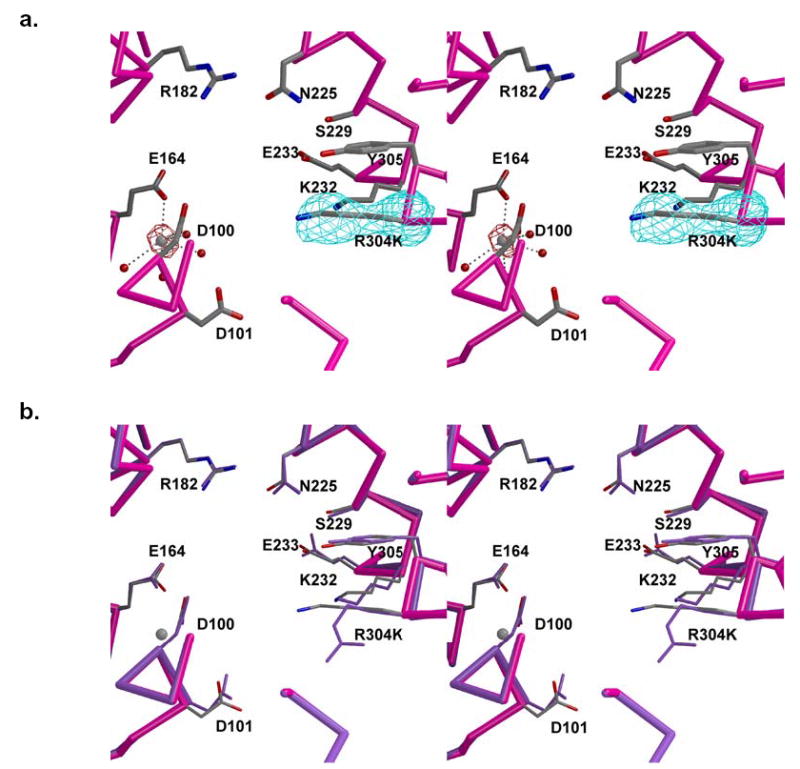
Active site of unliganded R304K trichodiene synthase. (a) Simulated annealing omit maps of K304 and Mg2+ ion are shown in cyan (6.0 σ) and maroon (8.0 σ), respectively. The mutated side chain is well-defined by clear electron density. Metal coordination interactions are represented by black broken lines. (b) Superposition with the active site of unliganded wild-type trichodiene synthase (purple).
Interestingly, in contrast with the unliganded forms of wild-type, D100E, and Y305F trichodiene synthases (17, 21, 24), unliganded R304K trichodiene synthase binds one Mg2+ ion in the active site of monomer B (Figure 3a). This metal ion is coordinated by the side chains of D100 and E164, and 4 water molecules. This metal ion most closely corresponds to Mg2+C in the structure of the wild-type enzyme complexed with PPi (Figure 1a) (17). Metal binding in the absence of diphosphate has previously been observed in the active sites of two other unliganded terpene cyclases, 5-epi aristolochene synthase (30) and (+)-bornyl diphosphate synthase (31). It is not clear how the R304K mutation facilitates Mg2+C coordination by D100 and E164 in the unliganded mutant.
R304K trichodiene synthase-Mg2+3-PPi-R-azabisabolene complex
The Mg2+-PPi cluster binds to the active site of monomer B of the dimer (Figure 4). As established for wild-type (17) and Y305F (24) trichodiene synthases, PPi binding triggers a conformational change in monomer B (ligand binding to monomer A is hindered due to interlattice contacts). The side chain of K304 donates a hydrogen bond to PPi but not to D101. The r.m.s. deviation between the unliganded and liganded forms of R304K trichodiene synthase is 1.2 Å for 353 Cα atoms (in comparison, the r.m.s. deviation between the unliganded and Mg2+3-PPi liganded forms of the wild-type enzyme is 1.4 Å for 349 Cα atoms). The overall conformational changes are, in general, similar to the diphosphate-induced conformational changes described for wild-type trichodiene synthase and involve the N-terminus (M1-P5), helices 1, D, H, K, and L, and loops 1-A, D-D1, H-α-1, J-K, and K-L (17) (Figure 1b). However, conformational changes in helix H, and loops D-D1, H-α-1, and J-K are slightly attenuated. Accordingly, the r.m.s. deviation between the Mg2+3-PPi liganded forms of wild-type and R304K trichodiene synthases is 0.35 Å for 353 Cα atoms.
Figure 4.
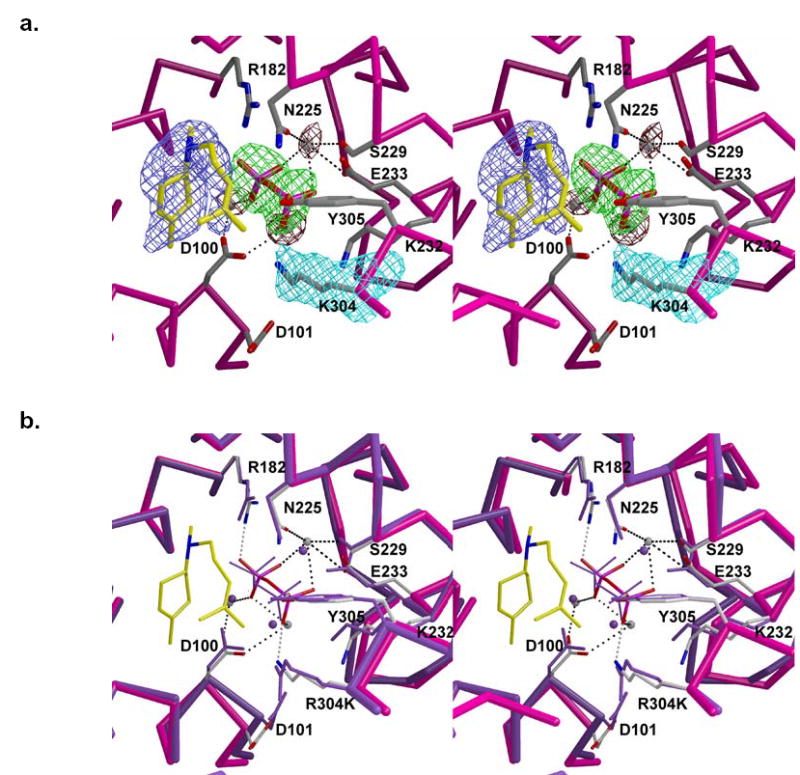
Active site of R304K trichodiene synthase complexed with Mg2+3-PPi and R-azabisabolene (yellow carbon skeleton). (a) Simulated annealing omit maps of K304, PPi, metal ions, and R-azabisabolene are shown in cyan (4.6 σ), green (5.4 σ), maroon (5.2 σ), and blue (4.0 σ), respectively. Metal coordination interactions are shown as black dotted lines. (b) Superposition with the activesite of wild-type trichodiene synthase complexed with Mg2+3-PPi (purple). Hydrogen bonds and metal coordination interactions in the mutant are shown asgray and black dotted lines, respectively.
Metal ions in the active site of R304K trichodiene synthase interact with D100, N225, S229, E233, and PPi in the same fashion observed in the Mg2+3-PPi complex with the wild-type enzyme (Figure 4). Mg2+C also makes a long-range electrostatic interaction with E164 (2.9 Å). The PPi anion appears to rotate slightly toward the mouth of the active site and is stabilized by metal coordination interactions and hydrogen bonds with R182 and K304. In wild-type, D100E, and Y305F trichodiene synthases, PPi also receives a hydrogen bond from K232 (17, 21, 24). Surprisingly, this interaction lengthens to ~4 Å in the R304K mutant. Although there is no charge difference between the side chains of arginine and lysine, the R304K mutation results in the loss of the R304-D101 hydrogen bond interaction, and D101 appears to move away from the mutated side chain (Figure 4b). The side chain of D101 retains its hydrogen bond interaction with R62. The side chain of K232 in the mutant moves a little closer to the former position of the R304 guanidinium group.
R304K trichodiene synthase-Mg2+3-PPi-S-azabisabolene complex
The Mg2+3-PPi cluster binds to the active site of monomer B only and interacts with the enzyme in a manner similar to that outlined above in the complex with R-azabisabolene. Additionally, the diphosphate-induced conformational changes in monomer B are similar to those described above. The r.m.s. deviations of this complex with unliganded R304K trichodiene synthase and with wild-type trichodiene synthase complexed with Mg2+3-PPi cluster are 1.2 Å and 0.37 Å, for 353 Cα atoms, respectively. Interestingly, S-azabisabolene binds to the active sites of both monomers. However, electron density for the ligand is ambiguous in both active sites and precludes confident interpretation of the orientation and conformation of S-azabisabolene (Figure 5). Although both enantiomers of azabisabolene have been shown to be equally effective inhibitors of trichodiene synthase in the presence of PPi (22), it is conceivable that S-azabisabolene, the aza-analogue of the enantiomer of the actual bisabolyl carbocation intermediate, may be bound in more than one orientation. Thus, we have not included S-azabisabolene in the final model.
Figure 5.
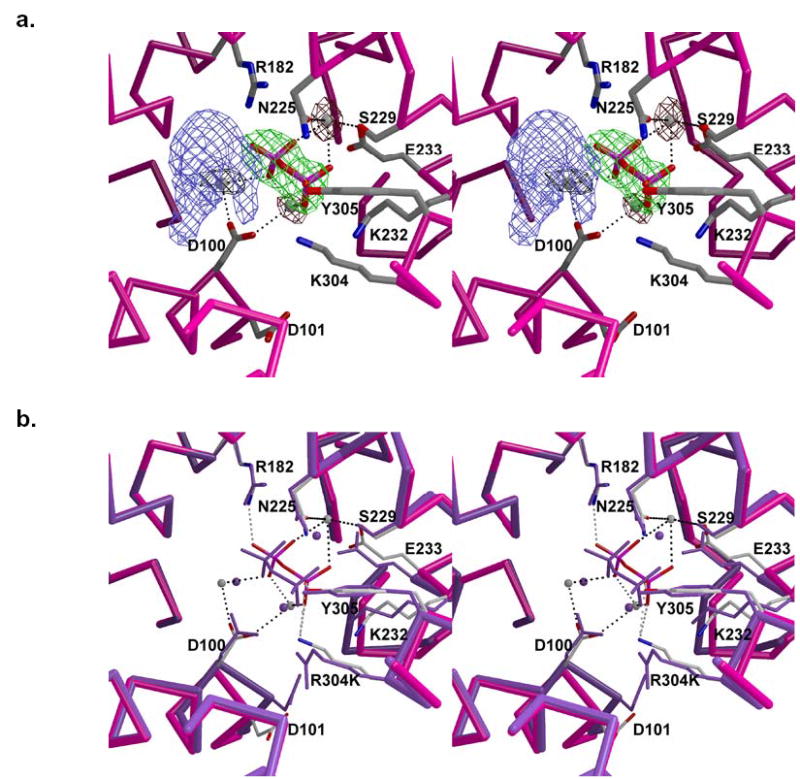
Active site of R304K trichodiene synthase complexed with Mg2+3-PPi and S-azabisabolene. (a) Simulated annealing omit maps of PPi, metal ions and S-azabisabolene are shown in green (7.0 σ), maroon (4.6 σ), and blue (4.5 σ), respectively. Note that the electron density is sufficiently ambiguous that S-azabisabolene cannot be modelled into the density. Metal coordination interactions are shown as black dotted lines. (b) Superposition with the active site of wild-type trichodiene synthase complexed with Mg2+3-PPi (purple). Hydrogen bonds and metal coordination interactions in the mutant are shown as gray and black dotted lines, respectively.
Discussion
The R304K mutation results in a structural change in the active site that apparently facilitates the binding of a magnesium ion not previously observed in unliganded trichodiene synthase structures. The ion is coordinated by D100 in the D100DSKD motif, E164, and four water molecules. This metal ion most closely corresponds to Mg2+C in the wild-type trichodiene synthase-Mg2+3-PPi complex (17). Mg2+C in trichodiene synthase corresponds to Mg2+A in 5-epi aristolochene synthase (30) and (+)-bornyl diphosphate synthase (31), and these enzymes contain glutamate residues corresponding to E164 of trichodiene synthase: 5-epi aristolochene synthase has E379 and (+)-bornyl diphosphate synthase has E429 (30, 31). However, these glutamate residues do not coordinate to metal ions in unliganded or liganded enzyme structures. Since the E164-Mg2+C interaction in R304K trichodiene synthase lengthens to 2.9 Å upon the binding of R-azabisabolene and Mg2+3-PPi, we cannot conclude any direct role in metal binding or catalysis for this residue in wild-type terpenoid cyclases.
In R304K trichodiene synthase complexed with R-azabisabolene and Mg2+3-PPi, the PPi anion shifts 0.7 Å towards the mouth of the active site relative to its position in complex with the wild-type enzyme. This results in a larger active site cavity with volume 512 Å3, which is 17% larger than the active site volume of 437 Å3 measured for the wild-type enzyme in its complex with Mg2+3-PPi (17) using the CastP server (24, 32, 33). The larger active site volume in R304K trichodiene synthase confers additional degrees of freedom to the orientations and conformations of the substrate and carbocation intermediates. Thus, a significantly compromised active site template is likely responsible for the formation of aberrant products (19).
The 5000-fold loss of activity measured for R304K trichodiene synthase appears to result from compromised molecular recognition of the substrate diphosphate group. The side chain of D101 in the complex does not move appreciably from its position in the unliganded enzyme and it retains the hydrogen bond interaction with R62 as observed in the unliganded enzyme. Thus, the loss of the R304-D101 hydrogen bond in the R304K trichodiene synthase-Mg2+3-PPi-R-azabisabolene complex compromises some of the diphosphate-induced conformational changes in the vicinity of D101. In particular, the D100-Mg2+B interaction lengthens from 2.3 Å in wild-type and Y305F trichodiene synthase complexes (17, 24) to 3.1 Å in the R304K trichodiene synthase-Mg2+3-PPi-R-azabisabolene complex. Additionally, Mg2+B moves 0.9 Å away from its position in the wild-type and Y305F trichodiene synthase complexes due to the movement of PPi that results from the R304K mutation.
Metal coordination interactions with PPi are longer in the R304K trichodiene synthase-Mg2+3-PPi-R-azabisabolene complex (average Mg2+-O distance = 2.7 Å) than in the wild-type trichodiene synthase-Mg2+3-PPi complex (average Mg2+-O distance = 2.2 Å), thereby weakening diphosphate stabilization. Presuming that the enzyme-substrate complex is similarly destabilized, we conclude that less efficient activation of the substrate diphosphate group accounts for the significant loss of activity measured for this mutant.
Structural and kinetic results with other trichodiene synthase mutants help support the preceding conclusion. Pre-steady state kinetic studies have shown that the ionization of FPP in the active site is the rate-limiting chemical step in trichodiene synthase catalysis and that product release is rate-limiting overall (34). In mutants with compromised binding of the substrate diphosphate group or PPi (e.g., D100E trichodiene synthase (21)), it is expected that the free energy barrier for FPP ionization should increase and the free energy barrier for PPi release should decrease. Single-turnover experiments with D101E trichodiene synthase indeed reveal that the rate of FPP consumption is decreased 100-fold, consistent with compromised FPP ionization (34).
The binding of R-azabisabolene to R304K trichodiene synthase is similar to that observed in its complex with D100E trichodiene synthase, which reflects an unproductive binding conformation and orientation (24). Thus, as we have noted, there are limitations in the use of positively charged aza analogues of carbocation intermediates to elucidate structural aspects of terpene cyclase reaction mechanisms (24, 31). However, it is notable that the positively charged azabisabolenes nevertheless influence the binding of PPi to R304K trichodiene synthase. This mutant appears to have lower affinity for Mg2+3-PPi since crystals soaked in buffer solutions lacking azabisabolene inhibitors reveal an empty active site (data not shown); upon the addition of R- or S-azabisabolene, the binding of both azabisabolene and Mg2+3-PPi is clearly confirmed (Figures 4 and 5). Thus, Mg2+3-PPi binding appears to be enhanced by the positively charged azabisabolene ammonium ion, which may compensate for the compromised hydrogen bond interactions with PPi in R304K trichodiene synthase. Indeed, both azabisabolene enantiomers synergistically lower the apparent Ki for inhibition of wild-type trichodiene synthase by PPi (22).
Finally, it is particularly informative to compare the molecular recognition of PPi and/or alkyl diphosphate groups in the array of crystal structures now available for complexes with trichodiene synthase (17, 21, 24, this work) and the monoterpene synthase (+)-bornyl diphosphate synthase (31). Each cyclase binds PPi through a partly conserved array of hydrogen bonds and coordination interactions with 3 Mg2+ ions, and the PPi ion in each complex orients two oxygen atoms – one from each phosphate moiety – toward the hydrophobic active site cavity that accommodates the terpene substrate/product (Figure 6). One of these oxygen atoms in each complex corresponds to the original phosphoester oxygen of the substrates FPP or GPP. The known lack of positional isotope exchange in the conversion of GPP to bornyl diphosphate allows for the direct identification of the precise PPi oxygen that corresponds to the phosphoester linkage of both the product bornyl diphosphate and the substrate geranyl diphosphate (35, 36), based on comparison of the crystal structures of (+)-bornyl diphosphate synthase complexed with PPi and with product (+)-bornyl diphosphate (Figure 6a) (31).
Figure 6.
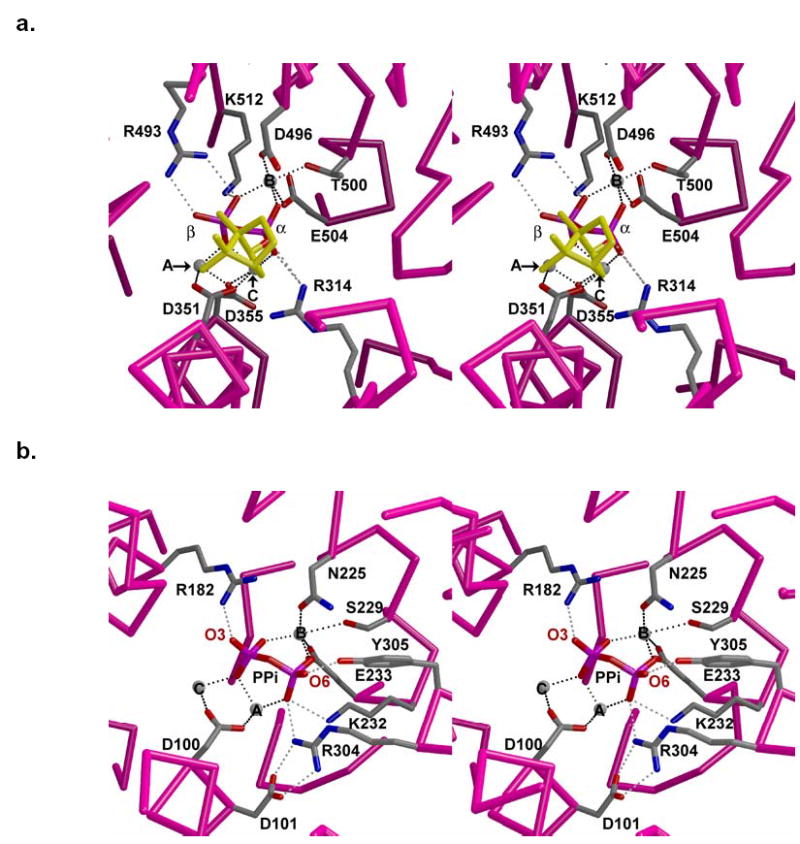
(a) Active site of (+)-bornyl diphosphate synthase from Salvia officinalis complexed with Mg2+3 and product bornyl diphosphate (yellow hydrocarbon moiety). Metal ions are represented by gray spheres. Hydrogen bonds and metal coordination interactions are shown as gray and black dotted lines, respectively. (b) Active site of wild-type trichodiene synthase complexed with Mg2+3 and co-product PPi. Metal ions are represented by gray spheres. Hydrogen bonds and metal coordination interactions are shown as gray and black dotted lines, respectively. The view corresponds to that in (a), from within the active site.
In the structure of the trichodiene synthase-Mg2+3-PPi complex, the O6 atom of PPi corresponds to the phosphoester oxygen of (+)-bornyl diphosphate in its complex with (+)-bornyl diphosphate synthase upon alignment of the two structures (Figure 6). The phosphoester oxygen of (+)-bornyl diphosphate corresponds to the original phosphoester oxygen of the substrate, geranyl diphosphate (35, 36). Therefore, by the analogy evident in the alignment of the two structures in Figure 6, we propose that the O6 atom of PPi in the trichodiene synthase-Mg2+3-PPi complex corresponds to the original phosphoester oxygen of the substrate FPP. This O6 atom of PPi interacts weakly with Nɛ -H of R304 (3.3 Å) and strongly with Y305 (2.7 Å) in the complex with PPi (17). Additional interactions with this phosphate group include hydrogen bonds with R304 and K232, as well as metal coordination interactions with Mg2+A and Mg2+B. We expect that the substrate diphosphate group makes similar interactions in the enzyme-substrate complex. It is notable that interactions with the substrate α-phosphate moiety would be significantly perturbed in R304K trichodiene synthase through loss of the K232 hydrogen bond and lengthening of metal coordination interactions. Therefore, we suggest that the massive loss of activity measured for R304K trichodiene synthase arises from compromised activation of the α-phosphate of the PPi leaving group of FPP. This hypothesis is consistent with the total loss of catalytic activity observed for R304A trichodiene synthase (unpublished results), in which there is no compensation for the lost positive charge at position 304 required for substrate activation. Future studies will allow us to further dissect the catalytic contribution of other intermolecular interactions with the substrate α-phosphate group and their importance for substrate activation.
Acknowledgments
We thank the Brookhaven National Laboratory Beamline X12B for access to X-ray crystallographic data collection facilities. We thank Professor Robert Coates of the University of Illinois for the generous gift of azabisabolene inhibitors, and we thank Dr. Michael J. Rynkiewicz for numerous scientific discussions.
Footnotes
This work was supported by National Institutes of Health Grants GM56838 (D.W.C.) and GM30301 (D.E.C.).
Atomic coordinates of R304K trichodiene synthase and its complexes with R-azabisabolene-Mg2+3-PPi and S-azabisabolene-Mg2+3-PPi have been deposited in the Protein Data Bank with accession codes 2AEK, 2AEL, and 2AET, respectively.
References
- 1.Wise, M. L., and Croteau, R. In Comprehensive Natural Products Chemistry (Vol. 2) Isoprenoids Including Carotenoids, and Steroids, ed. Cane, D. E., pp 97–153. (Elsevier, Oxford, U. K., 1999).
- 2.Cane, D. E. In Comprehensive natural products chemistry (Vol. 2) Isoprenoids including carotenoids, and steroids, ed. Cane, D. E., pp 155–200. (Elsevier, Oxford, U. K., 1999).
- 3.MacMillan, J., and Beale, M. H. In Comprehensive natural products chemistry (Vol. 2) Isoprenoids including carotenoids, and steroids, ed. Cane, D. E., pp 217–243. (Elsevier, Oxford, U. K., 1999).
- 4.Kawada SZ, Yamashita Y, Ochiai K, Ando K, Iwasaki T, Takaguchi T, Nakano H. Terpentecin and UCT4B, new family of topoisomerase II targeting antitumor antibiotics produced by Streptomyces: producing organism, fermentation and large scale purification. J Antibiot. 1995;48:211–216. doi: 10.7164/antibiotics.48.211. [DOI] [PubMed] [Google Scholar]
- 5.Okazaki T, Enokita R, Torikata A, Inukai M, Takeuchi M, Takahashi S, Arai M. Studies on Actinomycetes Producing Pentalenolactone and its New Related Compounds, . Ann Rep Sankyo Res Lab. 1979;31:94–103. [Google Scholar]
- 6.Hohn, T. M., McCormick, S. P., Alexander, N. J., Desjardins, A. E., and Proctor, R. H. In Molecular Genetics of Host-Specific Toxins in Plant Disease, eds. Kohmoto, K., and Yoder, O. C. (Kluwer Academic Publishers, Dodrecht, The Netherlands, 1998) pp 17–24.
- 7.Steele CL, Katoh S, Bohlmann J, Croteau R. Regulation of oleoresinosis in grand fir (Abies grandis). Differential transcriptional control of monoterpene, sesquiterpene, and diterpene synthase genes in response to wounding. Plant Physiol. 1998;116:1497–1504. doi: 10.1104/pp.116.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wise ML, Savage TJ, Katahira E, Croteau R. Monoterpene synthases from common sage (Salvia officinalis). cDNA isolation, characterization, and functional expression of (+)-sabinene synthase, 1,8-cineole synthase, and (+)-bornyl diphosphate synthase. J Biol Chem. 1998;273:14891–14899. doi: 10.1074/jbc.273.24.14891. [DOI] [PubMed] [Google Scholar]
- 9.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. Plant antitumor agents. VI The isolation and structure of Taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 10.Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 11.Harrewijn, P., van Oosten, A. M., and Piron, P. G. M., In Natural Terpenoids as Messengers: A Multidisciplinary Study of their Production, Biological Functions and Practical Applications, pp 253–296 (Kluwer Academic Publishers, Dodrecht, The Netherlands, 2001).
- 12.Hammer KA, Carson CF, Riley TV. Antifungal activity of the components of Melaleuca alternifolia (tea tree) oil. J Appl Microbiol. 2003;95:853–860. doi: 10.1046/j.1365-2672.2003.02059.x. [DOI] [PubMed] [Google Scholar]
- 13.Lesburg CA, Caruthers JM, Paschall CM, Christianson DW. Managing and manipulating carbocations in biology: terpenoid cyclase structure and mechanism. Curr Opin Struct Biol. 1998;8:695–703. doi: 10.1016/s0959-440x(98)80088-2. [DOI] [PubMed] [Google Scholar]
- 14.Wendt KU, Schulz GE. Isoprenoid biosynthesis: manifold chemistry catalyzed by similar enzymes. Structure. 1998;6:127–133. doi: 10.1016/s0969-2126(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 15.Ashby MN, Edwards PA. Elucidation of the deficiency in two yeast coenzyme Q mutants. Characterization of the structural gene encoding hexaprenyl pyrophosphate synthetase. J Biol Chem. 1990;265:13157–13164. [PubMed] [Google Scholar]
- 16.Hohn TM, Plattner RD. Purification and characterization of the sesquiterpene cyclase aristolochene synthase from Penicillum roqueforti. Arch Biochem Biophys. 1989;272:137–143. doi: 10.1016/0003-9861(89)90204-x. [DOI] [PubMed] [Google Scholar]
- 17.Rynkiewicz MJ, Cane DE, Christianson DW. Structure of trichodiene synthase from Fusarium sporotrichioides provides mechanistic inferences on the terpene cyclization cascade. Proc Natl Acad Sci USA. 2001;98:13543–13548. doi: 10.1073/pnas.231313098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cane DE, Swanson S, Murthy PPN. Trichodiene biosynthesis and the enzymatic cyclization of farnesyl pyrophosphate. J Am Chem Soc. 1981;103:2136–2138. [Google Scholar]
- 19.Cane DE, Shim JH, Xue Q, Fitzsimons BC, Hohn TM. Trichodiene synthase. Identification of active site residues by site-directed mutagenesis. Biochemistry. 1995;34:2480–2488. doi: 10.1021/bi00008a011. [DOI] [PubMed] [Google Scholar]
- 20.Cane DE, Xue Q, Fitzsimons BC. Trichodiene synthase. Probing the role of the highly conserved aspartate-rich region by site-directed mutagenesis. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 21.Rynkiewicz MJ, Cane DE, Christianson DW. X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 22.Cane DE, Yang G, Coates RM, Pyun HJ, Hohn TM. Trichodiene synthase. Synergistic inhibition by inorganic pyrophosphate and aza analogs of the bisabolyl cation. J Org Chem. 1992;57:3454–3462. [Google Scholar]
- 23.Cane DE, Wu Z, Oliver JS, Hohn TM. Overproduction of soluble trichodiene synthase from Fusarium sporotrichiodes in Escherichia coli. Arch Biochem Biophys. 1993;300:416–422. doi: 10.1006/abbi.1993.1056. [DOI] [PubMed] [Google Scholar]
- 24.Vedula LS, Rynkiewicz MJ, Pyun HJ, Coates RM, Cane DE, Christianson DW. Molecular recognition of the substrate diphosphate group governs product diversity in trichodiene synthase mutants. Biochemistry. 2005;44:6153–6163. doi: 10.1021/bi050059o. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski, Z., and Minor, W. (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods in Enzymology. Macromolecular Crystallography (Part A) Vol. 276, pp 307–326, Academic Press, San Diego, CA. [DOI] [PubMed]
- 26.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 28.Bacon D, Anderson WF. A fast algorithm for rendering space-filling molecule pictures. J Mol Graphics. 1988;6:219–220. [Google Scholar]
- 29.Meritt EA, Murphy MEP. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 30.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 31.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Bornyl diphosphate synthase: structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci USA. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cane DE, Chiu HT, Liang PH, Anderson KS. Pre-steady-state kinetic analysis of the trichodiene synthase reaction pathway. Biochemistry. 1997;36:8332–8339. doi: 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
- 35.Cane DE, Saito A, Croteau R, Shaskus J, Felton M. Enzymic cyclization of geranyl pyrophosphate to bornyl pyrophosphate. Role of the pyrophosphate moiety. J Am Chem Soc. 1982;104:5831–5833. [Google Scholar]
- 36.Croteau RB, Shaskus JJ, Renstrom B, Felton NM, Cane DE, Saito A, Chang C. Mechanism of the pyrophosphate migration in the enzymic cyclization of geranyl and linalyl pyrophosphates to (+)- and (−)-bornyl pyrophosphates. Biochemistry. 1985;24:7077–7085. doi: 10.1021/bi00346a009. [DOI] [PubMed] [Google Scholar]