

Abstract
Asparagine-linked glycosylation, the co-translational covalent attachment of carbohydrates to asparagine side chains, has a major effect on the folding, stability, and function of many proteins. The carbohydrate composition in mature glycoproteins is heterogeneous due to modification of the initial oligosaccharide by glycosidases and glycosyltransferases during the glycoprotein passage through the endoplasmic reticulum and Golgi apparatus. Despite the diversity of carbohydrate structures, the core β-d-(GlcNAc)2 remains conserved in all N-linked glycoproteins. Previously, results from our laboratory showed that the molecular composition of the core disaccharide has a critical and unique conformational effect on the peptide backbone. Herein, we employ a synergistic experimental and computational approach to study the effect of the stereochemistry of the carbohydrate–peptide linkage on glycopeptide structure. A glycopeptide derived from a hemagglutinin protein fragment was synthesized, with the carbohydrate attached to the peptide with an α-linked stereochemistry. Computational and biophysical analyses reveal that the conformations of the peptide and α- and β-linked glycopeptides are uniquely influenced by the attached saccharide. The value of computational approaches for probing the influence of attached saccharides on polypeptide conformation is highlighted.
Introduction
It is now well appreciated that the modification of proteins with carbohydrates plays a very important role in many biological events. For example, the carbohydrate moieties of glycoproteins can be involved in cell–cell communication, immune response, cell adhesion, intracellular targeting, protease resistance, and many other processes.1–3 The carbohydrates can also impact several physicochemical properties of proteins including hydration, hydrophilicity, and conformational stability.4 Additionally, because N-linked glycosylation occurs co-translationally during biosynthesis on the ribosome, the attachment of the carbohydrates may affect the protein folding pathway. Detailed studies of the impact of carbohydrates on glycoprotein structure have been limited due to the flexibility of the carbohydrates, which complicates crystallographic analyses. Thus, 2D-NMR studies of homogeneous glycopeptides that have been prepared via chemical or chemoenzymatic synthesis have been exploited to afford insight into the molecular details of the effect of carbohydrate peptide and protein conformation.4–6
Previously, we evaluated the effects of specific N-linked glycans on peptide conformation.7,8 It was shown that the two C2 N-acetamido groups of the natural (GlcNAc-β–GlcNAc-β–) core are critical in inducing a well-defined type I β-turn in the peptide backbone. These groups appear to provide a unique conformational effect on the interglycosidic and the glycopeptide linkages, thereby propagating a precise conformation in the peptide backbone. We have recently developed a stereoselective synthesis of the pure α-linked glycopeptide, 1-α (Chart 1). The route implements a building block approach and has been modified to improve the selectivity for the desired α-anomer.9 Access to the pure α-linked glycopeptide, together with biophysical information from the corresponding β-linked and nonglycosylated species,7 provides a unique opportunity to compare the conformational profiles of the three peptides, and further to evaluate whether the emerging computational methods including molecular dynamics (MD) simulations with explicit water solvation support the experimental findings for the family of peptides. MD calculations provide an excellent complement to experimental techniques to probe the specific effects of the saccharides at the molecular level. Such comparisons are not possible with the biophysical measurements alone due to the fluxional nature of the interactions that cannot be studied with the available spectroscopic methods. The comparison of the αand β-linked systems is particularly attractive because these species differ only in a single stereocenter; therefore, predicting similarities or differences between the conformations by computation will present a rigorous test of the current methodology.
Chart 1.
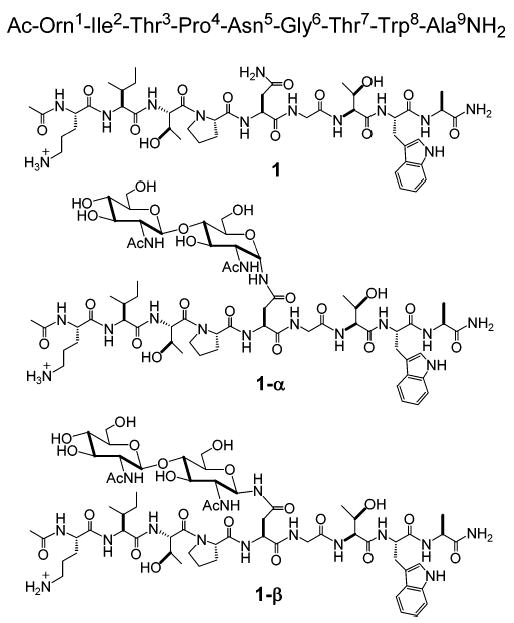
Chemical Structures of 1, 1-α, and 1-β
Results and Discussion
Synthesis.
The chemical synthesis of 1-α was carried out using the corresponding tert-butyldimethylsilyl building block. The α-linked building block was incorporated into the target peptide via standard solid-phase peptide synthesis, and the peptide was cleaved from the resin using the conditions noted previously.10 The addition of an extra 0.8% water and additional shaking for 45 min after the standard 3 h of cleavage produced the best results for the removal of the protecting groups. The peptide was purified by HPLC and characterized by ESMS ([M + H+] 1421.6 (obsd); 1421.5 (calcd) and QAA. Detailed procedures for the chemical synthesis of 1-α are presented in the Supporting Information.
Biophysical Characterization. NMR Studies.
The principal biophysical tool that is implemented in these studies is NMR spectroscopy. In contrast to other available solution-state techniques, such as circular dichroism or fluorescence spectroscopy, NMR provides discrete information about peptide conformation at a molecular level. Spin system assignment for glycopeptide 1-α was carried out using TOCSY and ROESY spectra (see Experimental Section). Glycopeptide 1-α displayed mainly random coil characteristics (values of αC–H chemical shift deviations from random coil were close to 0 ppm). However, a detailed analysis of nuclear Overhauser effect (NOE) patterns, 3JNHα coupling constants of Asn5, and variable-temperature (VT) coefficients indicate that the conformation of glycopeptide 1-α is more similar to that of the unglycosylated peptide 1, which has an Asx-turn structure,7 than to that of the β-linked glycopeptide 1-β, which manifests a type I β-turn in the peptide sequence around the carbohydrate moiety.
As depicted in Figure 1, a type I β-turn shows several measurable differences from the Asx-turn in the NMR analysis. For this peptide, the interproton distance a between the Gly6–Thr7 amides [dNN(Gly6,Thr7)] is expected to be shorter for an Asx-turn than for a type I β-turn. On the other hand, the Asn5–Gly6 amide distance b is expected to be shorter for a type I β-turn than for an Asx-turn.7 As summarized in Table 1, the ROESY spectrum of glycopeptide 1-α displays the specific dNN(Gly6,Thr7) NOE that is present in unglycosylated peptide 1, but absent in the β-linked glycopeptide 1-β.7 Furthermore, in contrast to glycopeptide 1-β, the ROESY spectrum for glycopeptide 1-α revealed a weak dNN(Asn5,Gly6) NOE as in the unglycosylated peptide (Table 1).
Figure 1.
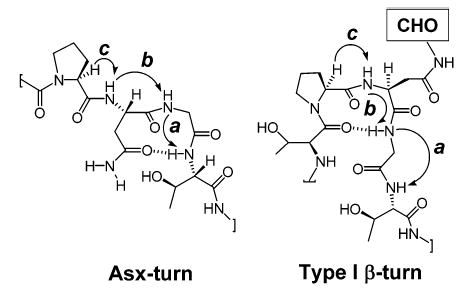
Structure of Asx and Type I β-turn and their NOE patterns.
Table 1.
Comparison of the Experimental Relative Intensities of the Diagnostic ROESY Cross-Peaks in the Turn Region of 1, 1-α, and 1-β
| NOE | 1a | 1-α | 1-βa |
|---|---|---|---|
| a, dNN(Gly6, Thr7) | weak | weak | absent |
| b, dNN(Asn5, Gly6) | weak | weak | strong |
| c, dαN(Pro4, Asn5) | strong | strong | strong |
Data for peptides 1 and 1-β taken from ref 7.
Information on amide proton variable-temperature (VT) coefficients can provide insight into whether an exchangeable proton is protected from solvent exchange due to hydrogen bonding.11 In general, VT coefficients below 6.0 ppb/K in water are associated with protons involved in hydrogen bonding. A major characteristic of an Asx turn is a hydrogen bond between the carboxyamide carbonyl of the Asn side chain and the backbone amide of the Thr or Ser two residues away from the Asn in the C-terminal direction (in this case Thr7) (Figure 1).7 Therefore, a low VT coefficient will be expected for the Thr7 amide proton for a peptide in an Asx-turn conformation. The calculated VT coefficient for the Thr7 amide of 1-α was 5.9 ppb/K, a value in good agreement with a hydrogen bond. Also, a similarly low VT coefficient was observed for the γNH of the Asn5 side chain (5.8 ppb/K). These values are similar to those observed in 1, which showed values of 6.2 and 5.4 ppb/K for the Thr7 amide and one of the Asn5 γNH2 protons, respectively. On the other hand, 1-β exhibited a higher VT value for the Asn5 γNH2 proton (7.2 ppb/K).7 Although a low VT coefficient for the Thr7 amide was also observed for 1-β, this was previously explained by an unusual and strong dNα(Gly6,Thr7) NOE which indicated that Thr7 may fold back, thus protecting the threonine amide.7
In addition to NOE and VT studies, the earlier evaluation of peptides 1 and 1-β included an analysis of the 3JHNα coupling constant for Asn5.8 For example, a distinct spectral feature supporting the conclusion that 1-β adopts a type I β-turn was the elevated 3JHNα coupling constant of 9.6 Hz for Asn5. The 3JHNα coupling constant is associated with the amino acid φ dihedral angles through the Karplus equation.12 The corresponding observed value for 1-α was 7.5 Hz, a value closer to the 7.0 Hz value observed for 1. This result is also consistent with those previously observed with the cellobiose-glycopeptide which fails to induce the β-turn.8 Together, these results support the proposal that 1-α adopts an extended conformation similar to that of the unglycosylated peptide 1,7 rather than a type I β-turn structure exhibited by the β-linked glycopeptide 1-β.7 These data also reinforce the previous observation that the (GlcNAc-β–GlcNAc-β) moiety is unique, inducing the type I β-turn structure.7,8
Solution-State Structure.
The solution-state structure of 1-α was solved by a simulated annealing protocol using conditions similar to those for 1 and 1-β.7 A total of 100 NOE distance and 21 chiral restraints were included in the simulated annealing protocol. As expected from the αC–H chemical shift deviations, the majority of the peptide was disordered. However, the backbone residues near the glycosylation site adopted a unique, nonrandom conformation (Figure 2). These results are similar to those observed for 1 and 1-β.7 From the analysis, 10/10 structures displayed an extended conformation near the glycosylation site. Eight of the displayed structures are similar to those including the Asx-turn observed for 1, while all are very different from those of glycopeptide 1-β (Figure 2). The more open Asx-turn-like structure is expected for 1-α (in comparison to peptide 1) because the carbohydrate moiety and the organized water molecules associated with the sugar will occupy extra space between the Asn5 side chain and the backbone near Thr7. Similar NOE intensities from the carbohydrate to the Asn5 NHγ resulted in the expected unordered carbohydrate moiety from the simulated annealing.
Figure 2.
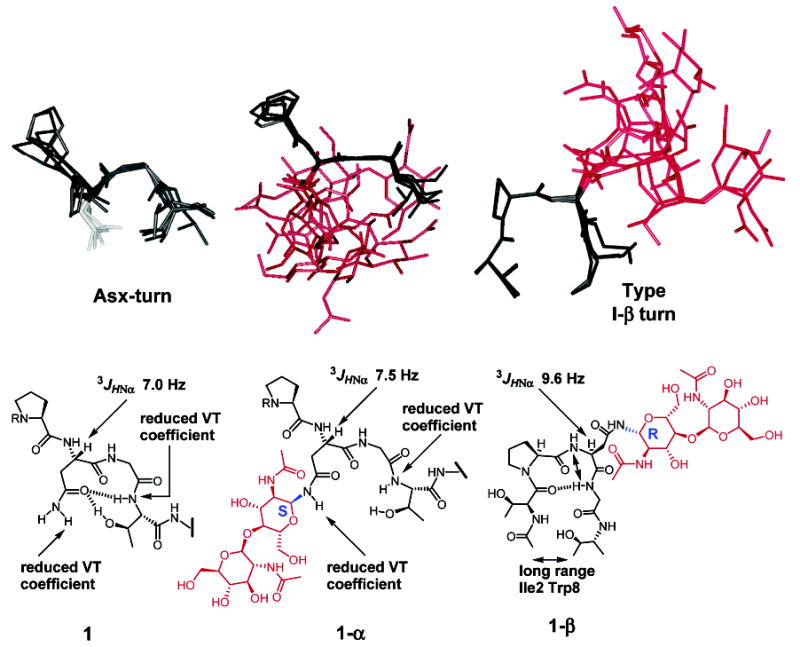
Comparison of the solution-state structure between 1, 1-α, and 1-β. 1 and 1-α are shown from Pro4 to Thr7. 1-β is shown from Thr3 to Thr7. The structures of 1 and 1-β were taken from refs 7 and 8.
Molecular Dynamics.
Explicitly solvated MD simulations provide a valuable tool for investigating the conformational space populated, within the limits of the force-field and simulation time scale, at an atomic level. The starting conformations utilized were independent of the simulated annealing-derived structures and were based only on the three NOEs observed in the central region of the peptide and the 3JHNα values observed for the peptide backbone. Once the systems reproduced the majority of the restraint data, they were equilibrated for 1 ns, yielding experimentally consistent starting structures. In the case of the unglycosylated peptide, 1, a second simulation was performed in which the hydrogen bond that is characteristic of an Asx-turn was included as an initial constraint to obtain the starting structure. The structure from this simulation is referred to as 1*. In the MD simulation of 1*, the Asx-turn remained stable for the first 2 ns before collapsing to a structure similar to that obtained from the simulation of 1. Therefore, for all computed values for 1*, only those from the first 2 ns were utilized.
Calculation of the root-mean-square deviation (RMSD) with respect to the starting structure provides a guide of the flexibility in the peptide backbone, which is typically between 1.5 and 2.0 Å for an MD simulation of a stable folded protein. Therefore, the RMSDs for the Cα atoms of peptide residues Thr3, Pro4, Asn5, Gly6, and Thr7 were calculated between the experimentally consistent starting structure and 1 ps snapshots from each of the MD trajectories. For 1, 1*, 1-α, and 1-β, the average RMSDs were 1.51 ± 0.35, 1.41 ± 0.31, 2.55 ± 0.41, and 1.26 ± 0.14 Å, respectively. The small standard deviation in 1-β emphasizes the small fluctuation of the backbone in the naturally occurring glycopeptide, relative to either the unglycosylated or the α-linked peptides. Relative to the peptide backbone, the glycan in both 1-α and 1-β shows relatively little internal motion, as seen by the similarity of their RMSDs (0.80 ± 0.23 and 0.78 ± 0.19 Å respectively), computed for all atoms in each disaccharide. Despite this similarity, it is clear that the α-linked chitobiose has a very different effect on the peptide-backbone conformation (Figure 3) as well as on the flexibility of the backbone.
Figure 3.
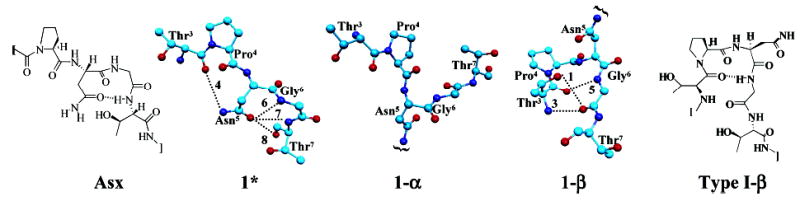
The most representative structures obtained from the MD simulations of 1*, 1-α, and 1-β, with a schematic of their corresponding secondary structure, Thr3–Thr7, shown. The hydrogen bonds are labeled with respect to Table 2.
Notably, 1-β exhibits three relatively long-lived intrapeptide hydrogen bonds, forming the foundation for the β-turn (hydrogen bonds 1, 3, and 5 in Table 2 and Figure 3). In contrast, throughout the MD simulation of 1-α, only two, short-lived intrapeptide hydrogen bonds were observed. In addition, upon altering the peptide–carbohydrate linkage from β- to α- while maintaining the peptide backbone in the β-turn conformation found in 1-β, the structure collapses to one similar to that found in the MD simulation of 1-α, reinforcing the importance of the β-linkage for β-turn induction.
Table 2.
Percent Occupanciesa of Intrapeptide Hydrogen Bonds from MD Simulation of 1, 1*, 1-α, and 1-β
| hydrogen bond
|
||||||
|---|---|---|---|---|---|---|
| donor | acceptor | 1 | 1* | 1-α | 1-β | |
| 1 | Thr3 HOβ | Gly6 CO | 38 | |||
| 2 | Thr3 HOβ | Asn5 CO | 10 | |||
| 3 | Thr3 HN | Gly6 CO | 86 | |||
| 4 | Asn5 HNγ | Thr3 CO | 23 | |||
| 5 | Gly6 HN | Thr3 CO | <10 | <10 | 52 | |
| 6 | Gly6 HN | Asn5 Oγ | 35 | 13 | <10 | |
| 7 | Thr7 HN | Asn5 Oγ | 63 | |||
| 8 | Thr7 HOβ | Asn5 Oγ | 10 | |||
Occupancy was fulfilled when the proton–heavy atom distance was less than 2.4 Å; a dash indicates an occupancy of less than 1%.
Over the course of the 1-β MD simulation, no hydrogen bonds were detected between β-GlcNAc1 and the central region of the peptide; however, several short-lived hydrogen bonds, specifically between α-GlcNAc1(O6)–Thr7(CO, N, Oγ) and α-GlcNAc1(O6, O5)–Trp8(N), were observed in the 1-α MD simulation. In addition, during the MD simulation of 1-α, initiated in the β-turn conformation, formation of a hydrogen bond between α-GlcNAc1(N2) and Asn5(CO) appeared to trigger the decay of the β-turn conformation. Because this interaction involves the backbone of the glycosylated asparagine, it could form regardless of peptide sequence. Therefore, it appears that the formation of hydrogen bonds between α-GlcNAc1 and the central region of the peptide, independent of its sequence, disrupts the β-turn conformation.
While the hydrogen bond between the Thr7 NH proton and the Asn5 Oγ, which is characteristic of an Asx-turn, was not observed during the initial MD simulation of the free peptide 1, over two-thirds of the experimental data were nevertheless well reproduced (Table 2 and Figure 4). Despite the use of the hydrogen-bond distance restraint in 1*, the computed 3JHNα values for the first 2 ns are not in better agreement with experimental values than those derived from 1. However, taking into account the conformations from both 1 and 1* yields better agreement overall with the 3JHNα values (Table 3). Once the Asx-turn conformation collapses, it does not re-form, which may be an artifact of the time scale used in these simulations or may point to weaknesses in the peptide force field.13,14
Figure 4.
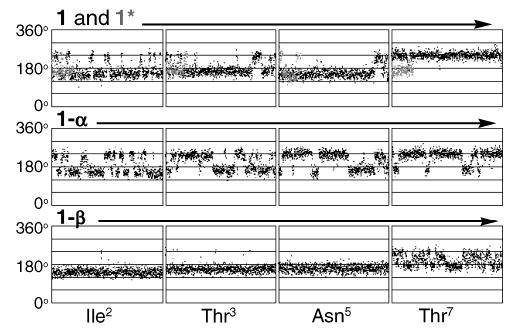
Plot of the φ backbone torsion angle for Ile2, Thr3, Asn5, and Thr7 (left to right) for the 10 ns MD simulation of 1, 1*, 1-α, and 1-β with 1 (black) and 1* (gray) superimposed.
Table 3.
Comparison of the Experimental and MD-Deriveda 3JHNα Couplings for 1, 1*, 1-α, and 1-β
| 1 | 1* | 1-α | 1-β | |
|---|---|---|---|---|
| Ile2 | 6.6 (7.4 ± 1.9) | (8.3 ± 1.7) | 7.9 (7.1 ± 2.0) | 6.6 (7.0 ± 1.6) |
| Thr3 | 7.0 (8.3 ± 1.7) | (6.4 ± 2.0) | 7.4 (7.0 ± 2.0) | 6.5 (8.3 ± 1.2) |
| Asn5 | 7.0 (7.6 ± 1.6) | (8.9 ± 1.3) | 7.5 (6.2 ± 1.3) | 9.6 (8.6 ± 1.2) |
| Thr7 | 7.0 (4.5 ± 1.7) | (8.9 ± 1.3) | 7.5 (5.3 ± 1.3) | 6.3 (7.0 ± 2.0) |
In Hz, computed value with standard deviations in parentheses. Experimental values for 1 and 1-β obtained from ref 15.
It is clear that the conformational preference observed for the central backbone region of 1-β is different from that of 1, 1*, and 1-α (Figure 3). The rapid conformational transitions observed in the simulations of 1, 1*, and 1-α generally contrast with the behavior of 1-β, which displays relatively stable conformations for the peptide-backbone φ torsion angles (Figure 4). The MD simulations provide an excellent indication of the conformations sampled, but even over 10 ns may not have reached statistical convergence.16 Differences in relative conformer populations could alter the values for 3JHNα significantly.
From both the RMSD values and the hydrogen bond analyses, it is clear that the β-linkage is necessary to preserve the β-turn conformation and that it leads to a decrease in flexibility of the peptide. The rigid behavior of the Ile2, Thr3, and Asn5 residues, observed during the MD simulation of 1-β, reaffirms the stability of this system in the β-turn conformation. The contrasting behavior of 1-α reinforces the conclusions derived from the NMR data that the α-linked chitobiose has a distinctly different effect on peptide conformation relative to 1-β.
Conclusions
Herein, we have reported a detailed comparison of biophysical and computational data for an α- and a β-linked glycopeptide and the corresponding unglycosylated peptide. Two-dimensional NMR studies were used to evaluate the conformational properties of the new α-linked glycopeptide to compare these with the properties of the unglycosylated peptide, as well as the β-linked glycopeptide, reported previously. The NMR studies revealed that the stereochemistry at the anomeric center of the N-linked carbohydrate dramatically affects the backbone conformation of the glycopeptide, and, indeed, only the β-linked glycopeptide adopts a compact β-turn conformation. The three systems have been simultaneously subjected to computational analysis involving MD simulations utilizing explicit water solvation. The findings of these studies are in excellent agreement with the experimental solution-state conformational analysis. Specifically, the simulations illustrate the role played by Thr3 in the β-turn induction, which proceeds via formation of an intrapeptide hydrogen-bond network between both the backbone and side chain of Thr3 and the backbone of Gly6, which only occurred during the simulation of 1-β. In addition, the formation of hydrogen bonds between α-GlcNAc1 and the central region of the peptide leads to destabilization of the β-turn conformation. Taken together, these findings provide a tested computational framework that may be used to predict the conformational impact of carbohydrates on peptide and protein structure and suggest future directions for evaluating the synergistic application of experimental and computational methods in the evaluation of glycoconjugate structures. Furthermore, the conformational consequences of the stereochemistry of the anomeric center in the carbohydrate–peptide linkage reported in this study may also provide valuable information for the design of neoglycopeptides and glycopeptide mimetics that may be useful as therapeutic agents.17,18
Experimental Section
NMR Studies.
NMR spectra were acquired using the same conditions reported for 1 and 1-β.7 One to four mM peptide solutions in 90:10 H2O:D2O at pH 4.5 were used with DMSO as internal reference. NMR TOCSY, ROESY, and DQF COSY spectra were recorded on a 600 MHz Bruker Advance spectrometer. TOCSY spectra recorded at 7, 12, 17, 22, and 27 °C were used to calculate the amide proton VT coefficients. ROESY spectra were acquired using 400 ms mixing times with WATERGATE gradient suppressions. The NMR data were processed using Felix 97 software.
Simulated Annealing.
The solution-state structure of glycopeptide 1-α was solved by a simulated annealing protocol using similar conditions as for peptide 1 and glycopeptide 1-β.7 The structure was generated using the Biopolymer option of Insight II. ROESY cross-peak volumes were measured and sorted into strong and weak bins. The strong peaks were assigned a distance range of 1–3 Å, and the weak peaks were assigned a distance range of 2–4 Å. An additional distance restraint of 2.0–5.0 Å between Thr7 amide and the Asn5 carboxyamide was justified by the decreased VT coefficient observed for Thr7 and the Asn NHγ.7 A total of 100 NOE distance and 21 chiral restraints were included in the simulated annealing protocol.
Molecular Dynamics.
The SANDER module19 of AMBER720 was utilized in conjunction with the PARM9921 parameter set for proteins and the GLYCAM22 parameter set for glycosides and glycoproteins. All MD simulations utilized explicit solvation, with an average of 3000 TIP3P waters.23 Simulated annealing was followed by MD simulations with minimal 3JHNα and distance restraints.24 Restraints were included until agreement with experimental NMR data was achieved, and then equilibration was continued for 1 ns. All restraints were removed for the 10 ns production runs. A control MD simulation of 1-α was performed with the same starting structure as 1-β. During the simulation, the structure quickly decayed leading to behavior similar to that observed in the original 1-α simulation. All MD simulations employed an integration time step of 2 fs, a dielectric of unity, scaling of 1–4 electrostatic and van der Waals interactions by the standard values of 1/1.2 and 1/2.0, restraint of all hydrogen-containing bonds through the SHAKE algorithm, a cutoff of 10 Å for all nonbonded interactions, and particle mesh Ewald treatment of long-range electrostatics.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (RR05357 and GM55230 to R.J.W. and GM39334 to B.I.) for financial support.
Footnotes
Supporting Information Available: Glycopeptide synthesis and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Varki A. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 3.Reuter G, Gabius HJ. Cell Mol Life Sci. 1999;55:368–422. doi: 10.1007/s000180050298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imperiali B. Acc Chem Res. 1997;30:452–459. [Google Scholar]
- 5.Imperiali B, O’Connor SE. Curr Opin Chem Biol. 1999;3:643–649. doi: 10.1016/s1367-5931(99)00021-6. [DOI] [PubMed] [Google Scholar]
- 6.O’Connor SE, Imperiali B. Chem Biol. 1996;3:803–812. doi: 10.1016/s1074-5521(96)90064-2. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor SE, Imperiali B. J Am Chem Soc. 1997;119:2295–2296. [Google Scholar]
- 8.O’Connor SE, Imperiali B. Chem Biol. 1998;5:427–437. doi: 10.1016/s1074-5521(98)90159-4. [DOI] [PubMed] [Google Scholar]
- 9.Holm B, Linse S, Kihlberg J. Tetrahedron. 1998;54:11995–12006. [Google Scholar]
- 10.Bosques CJ, Tai VWF, Imperiali B. Tetrahedron Lett. 2001;42:7207–7210. [Google Scholar]
- 11.Ohnishi OM, Urry DW. Biochem Biophys Res Commun. 1969;36:194–202. doi: 10.1016/0006-291x(69)90314-3. [DOI] [PubMed] [Google Scholar]
- 12.Pardi A, Billeter M, Wüthrich K. J Mol Biol. 1984;180:741–751. doi: 10.1016/0022-2836(84)90035-4. [DOI] [PubMed] [Google Scholar]
- 13.Gnanakaran S, Garcia AE. J Phys Chem B. 2003;107:12555–12557. [Google Scholar]
- 14.Simmerling C, Strockbine B, Roitberg AE. J Am Chem Soc. 2002;124:11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 15.O’Connor, S. E.; Imperiali, B. Ph.D. Thesis; Department of Chemistry, Massachusetts Institute of Technology: Cambridge, MA, 2001; p 302.
- 16.Kirschner KN, Woods RJ. Proc Natl Acad Sci USA. 2001;98:10541–10545. doi: 10.1073/pnas.191362798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcaurelle LA, Bertozzi CR. Chem-Eur J. 1999;5:1384–1390. [Google Scholar]
- 18.Hang HC, Bertozzi CR. Acc Chem Res. 2001;34:727–736. doi: 10.1021/ar9901570. [DOI] [PubMed] [Google Scholar]
- 19.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 20.Case, D. A.; Pearlman, D. A.; Caldwell, J. W.; Cheatham, T. E., III; Wang, J.; Ross, W.; et al. University of California, San Francisco, 2002.
- 21.Wang J, Cieplak P, Kollman PA. J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 22.Woods RJ, Dwek RA, Edge CJ, Fraser-Reid B. J Phys Chem B. 1995;99:3832–3846. [Google Scholar]
- 23.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 24.Vuister GW, Bax A. J Am Chem Soc. 1993;115:7772–7777. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.